
Abstract
Compared to the UL51 gene of other alphaherpesviruses, the duck enteritis virus (DEV) UL51 gene contains ten conserved motifs and has a close evolutionary relationship with members of the genus Mardivirus. The DEV UL51 gene product was identified using a rabbit polyclonal antiserum raised against a 6-His-UL51 fusion protein expressed in Escherichia coli as a 34-kDa protein. Western blotting and RT-(real time) PCR analysis of DEV-infected cells showed that the protein was produced at the late stage of infection and that its production was highly dependent on viral DNA synthesis, suggesting that the gene should be classified as γ2 class. Analysis of extracellular virions revealed that the protein was a component of extracellular mature DEV virions. Indirect immunofluorescence studies localized most of the protein to the juxtanuclear region. These results will provide a basis for further functional analysis of the gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Duck enteritis virus (DEV) is an alphaherpesvirus that causes an acute, contagious and fatal disease in waterfowl (ducks, geese, and swans). DEV results in heavy economic losses to the commercial duck industry due to its high mortality rate and decreased duck egg production [43]. Recently, an increasing number of DEV genes, such as UL5 [40], UL6 [41], UL22 [18], UL23 [29], UL24 [22, 24], UL25–UL30 [31], UL31–UL35 [2, 48], UL44 [30], UL50 [51], US3–US5 [53], US8 [5], US2 and US10 [52], have been identified. In 2006, a DEV genomic library was successfully constructed in our laboratory [7]. Sequence analysis showed that DEV encoded several structural proteins, one of which was identified as a viral phosphoprotein encoded by the UL51 gene with a size of 759 bp (GenBank NO. DQ072725), which is located at the right end of the unique long segment (UL). The UL51 genes [1, 3, 6, 10, 13, 14, 44] are conserved in the herpesvirus family. Recent research has shown that the product of the herpes simplex virus (HSV-1) UL51 gene is a membrane-associated protein that is incorporated into virions, localizing primarily to the inside of the viral envelope [38]. Moreover, the UL51 protein (pUL51) has been found to be palmitoylated at the N-terminal cysteine at position 9 of the protein, which is important for targeting to the Golgi apparatus [37]. Mutational analyses of HSV-1 and pseudorabies virus (PRV) have revealed that the UL51 gene plays a role in virion maturation [25, 26, 38, 39].
In the present study, based on sequence analysis, we have identified the DEV UL51 gene. The gene was subsequently cloned into the prokaryotic expression vector pET-28a (+) and expressed in E. coli. RT-(real time) PCR, western blot, and immunofluorescence analyses were performed to determine the gene transcription/translation time course and intracellular localization of pUL51 in DEV-infected cells, providing a basis for further functional analysis of this gene.
Materials and methods
Viruses and cells
Throughout this study, we used the DEV CHv strain [15, 42, 50] (a high-virulence field strain isolated in our laboratory) grown in duck embryo fibroblasts (DEFs) as described previously [47]. In brief, DEFs were cultured in minimal essential medium (MEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) at 37°C. MEM medium supplemented with 2–3% FBS was used for virus infection.
Sequence analysis of the DEV UL51 gene
Comparison and analysis of the deduced amino acid sequence of the DEV UL51 gene with those of ten other alphaherpesviruses were performed using the MEGALIGN program in LASERGENE (DNAStar 6.0) with CLUSTALV (PAM250) multiple alignment [20]. Next, phylogenetic analysis was carried out using deduced amino acids sequences of 18 UL51 genes (Table 1) using the MEGALIGN program [4].
PCR amplification and plasmid construction
A pair of primers, UL51f (5′-CCGGAATTCATGTTAGCTTTTATCTCCAG-3′, as the forward primer) and UL51r (5′-TCCCTCGAGTTAGACGGCTACCAACG-3′, as the reverse primer), was designed based on the DEV UL51 gene sequence. EcoRI and XhoI sites were incorporated into the forward and reverse primers, respectively, to facilitate cloning. The amplified products were subcloned into the PMD18-T vector (TaKaRa, Japan) and sequenced (TaKaRa). Then, the correct fragment was cloned into the E. coli expression vector pET28a (+) (Novagen, Madison, WI) to yield the plasmid pET28a-UL51. The recombinant plasmid was subsequently confirmed by PCR, restriction enzyme digestion, and DNA sequencing (TaKaRa).
Prokaryotic expression and purification of the DEV UL51 gene
Escherichia coli strain BL21 (DE3) was transformed with plasmid pET28a-UL51. After induction with isopropyl-β-d-thiogalactopyranoside (IPTG), these cells expressed large quantities of the 6-His-tagged UL51 fusion protein with an apparent molecular mass of 34 kDa. The UL51 fusion protein was analyzed by SDS-PAGE followed by staining with Coomassie brilliant blue. According to the manufacturer’s instructions (Bio-Rad), the proteins were purified using Ni2+ affinity resins under denaturing conditions. The purified protein was analyzed using SDS-PAGE and the Bradford assay.
Preparation and purification of UL51 polyclonal antiserum from immunized rabbits
Four adult male rabbits were immunized at subcutaneous sites with 1.0 mg of the purified UL51 fusion protein conjugates in Freund’s complete adjuvant (FCA) to initiate antibody production. Three weeks later, 0.5 mg of the purified UL51 fusion protein with Freund’s incomplete adjuvant (FIA) was used for subsequent boosts. Three booster injections were given at 2-week intervals after the primary injection. Two weeks after the last immunization, the rabbits were exsanguinated, and the sera were collected. Control pre-immune serum was obtained before the first injection. The purified UL51 antiserum was subsequently obtained by purification using caprylic acid and ammonium sulfate precipitation and High-Q anion-exchange chromatography [34]. Western blotting analysis was conducted to examine the reactivity and specificity of the UL51 antiserum.
Preparation of total RNA and RT-(real time) PCR
Duck embryo fibroblasts, grown in 6-well plates, were either mock-infected or infected with the DEV CHv strain at a multiplicity of 5 PFU per cell. To examine the time course of UL51 transcription in infected cells, total RNA was isolated from mock- or DEV-infected cells at 1, 2, 4, 6, 12, 24, 36, 48, and 60 h p.i. using the Total RNA Isolation System (TaKaRa) and detected by 1.0% agarose gel electrophoresis. Next, a cell-volume-equivalent amount of total RNA (15 μl) was digested using RNase-free DNase I (TaKaRa). Based on the nucleotide sequence of the DEV UL51 gene, the forward and reverse primer sequences designed using the software IQ5 (Bio-Rad) were UL51f′ (5′-TCTCCAGCATGTGTGGTCTAAGGC-3′) and UL51r′ (5′-GGCGATGGTAGCATAGCGTTGAC-3′), respectively. Based on the nucleotide sequence of duck β-actin from GenBank, the forward and reverse primer sequences designed were β-actin-f (5′-CCGGGCATCGCTGACA-3′) and β-actin-r (5′-GGATTCATCATACTCCTGCTTGCT-3′), respectively. According to the manufacturer’s instructions (TaKaRa), the RT reaction was performed in a 10-μl reaction volume. Real-time PCR was performed in a volume of 25 μl containing 1.0 μl of the forward primer (10 pmol/L), 1.0 μl of the reverse primer (10 pmol/L), 1.0 μl cDNA template, 12.5 μl real-time PCR Master Mix SYBR Green I, and 9.5 μl water (all reagents were purchased from TaKaRa). All reactions were performed in triplicate and in at least two independent reactions. Using duck β-actin as the reference gene, the average relative content of DEV UL51 gene transcripts was calculated using the 2−ΔΔCt method [28, 32].
Western blotting
Duck embryo fibroblasts were either mock-infected or infected with DEV as described above and harvested at 2, 4, 6, 8, 24, 36, 48 and 60 h p.i. Cells were lysed in SDS sample buffer, electrophoretically separated by SDS-PAGE, and electrically transfered to polyvinylidene difluoride (PVDF) membranes (Amersham, Japan). Nonspecific protein binding was blocked by treating membranes with TBST (25 mmol Tris–HCl, 150 mmol NaCl, pH 7.4, and 0.05% Tween-20) containing 5% bovine serum albumin (BSA). The membranes were incubated with the purified UL51 antiserum. After washing three times with TBST, the membranes were incubated with goat anti-rabbit peroxidase-labeled antibody (Sino-American Biotechnology Co., Shanghai, China) and then developed in diaminobenzidine (DAB) substrate buffer (Beijing Zhong Shan-Golden Bridge Biological Technology Co., Ltd., China). Finally, photos of the membranes were taken with a digital camera.
Immunofluorescence and confocal laser microscopy
Duck embryo fibroblasts were either mock-infected or infected with DEV as described above. At 2, 4, 6, 8, 9, 10, 12, 24, 36, 48 and 60 h p.i., the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and then washed once with phosphate-buffered saline (PBS) and blocked in PBS containing 10% BSA. They were then incubated with the purified UL51 antiserum, washed three times in PBS, and treated with FITC-conjugated goat anti-rabbit IgG (Sino-American Biotechnology Co.). As described by Miller et al. [36], the cell nuclei were visualized by 4′,6-diamidino-2-phenylindole (DAPI) counter-staining (5 μg/ml, Beyotime Institute of Biotechnology, Shanghai, China). Fluorescent images were examined using confocal laser microscopy (Bio-Rad).
Virion purification
Duck embryo fibroblasts, cultured in bottles, were infected with the DEV CHv strain at a multiplicity of 5 PFU per cell. After 1 h adsorption at 37°C, maintenance medium containing 2–3% serum was added. DEV virions were harvested from the extracellular medium at 48 h p.i. After removal of cell debris by low-speed centrifugation, virions were pelleted from the supernatant by centrifugation at 87,000g for 1 h. The virus suspension was layered onto a continuous 10–50% sucrose gradient and centrifuged at 20,000 rpm for 1 h at 4°C. The peak virion-containing fractions were collected as described previously [15, 46], diluted in PBS and pelleted again by centrifugation at 87,000g for 1 h. Purified virions were lysed in SDS sample buffer and then analyzed by western blotting with the purified UL51 antiserum.
Dependence of DEV UL51 protein production on viral DNA synthesis
DEFs, cultured in six-well plates, were infected with the DEV CHv strain at a multiplicity of 5 PFU per cell. After a 1-h adsorption period in the presence of 300 mg/ml acyclovir (ACV) (Glaxo SmithKline) at 37°C, maintenance medium containing 2–3% serum was added. Infected cells were harvested at various times and then analyzed by western blotting with the purified UL51 antiserum.
Results
Sequence analysis of DEV UL51 gene
As shown in Fig. 1, ten conserved sequence motifs of the DEV UL51 protein were predicted by multiple alignments of the other ten reference virus strains. In order to analyze the phylogenetic relationship of UL51 with other alphaherpesviruses (Table 1), we constructed a phylogenetic tree using the putative UL51 protein sequences. A representative minimal tree for UL51 is shown in Fig. 2. In the tree, the 18 alphaherpesviruses were separated into four genera (Simplexvirus, Varicellovirus, Mardivirus, and Iltovirus) with moderate bootstrap scores. The analysis revealed that DEV might have a close evolutionary relationship with the mardiviruses, such as MeHV-1, MDV-1 and MDV-2, which infect meleagrid and gallid birds.

Ten sequence motifs in the deduced amino acid sequences of UL51 among 11 alphaherpesviruses. Highly conserved sites are shown

Phylogenetic tree of the amino acid sequences of the UL51 genes of DEV and 17 other members of the Alphaherpesvirinae (see Table 1) obtained using the MEGALIGN program in LASERGENE (DNAStar 6.0)
Preparation and specificity of UL51 rabbit antiserum
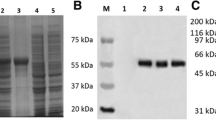
To characterize the UL51 gene product, we first generated the purified UL51 antiserum. Western blotting analysis showed that the UL51 antiserum reacted to a protein with an apparent molecular mass of 34 kDa in the lysates of DEV-infected cells at 24 h p.i (Fig. 3). However, the UL51 antiserum did not react with any proteins in the lysates of mock-infected cells (Fig. 3). In addition, the pre-immune serum did not react with any proteins in the lysates of mock-infected or DEV-infected cells (data not shown). The results indicate that the UL51 antiserum detected the pUL51 specifically in DEV-infected cells; therefore, we used the UL51 antiserum for further experiments to characterize the UL51 gene product of DEV.

Western blotting to analyze protein expression of the UL51 gene product in DEV-infected DEFs. DEFs were mock-infected or infected with DEV. The cells were harvested at 2, 4, 6, 8, 24, 36, 48, or 60 h p.i. Proteins were separated by SDS-PAGE and analyzed by western blotting using the UL51 antiserum. Molecular mass markers are shown on the left
Transcription analysis of the DEV UL51 gene in DEV-infected cells
To study the transcription kinetics of the DEV UL51 gene during viral infection, real-time quantitative RT-PCR with SYBR Green I was conducted. The integrity of total RNA isolated from mock- and DEV-infected cells was verified by 1.0% agarose gel electrophoresis (Fig. 4a). The average relative content of DEV UL51 gene transcripts was calculated using the 2−ΔΔCt method. As shown in Fig. 4b, the DEV UL51 gene transcripts appeared as early as 2 h p.i., and then the content of transcripts increased steadily and reached a peak at 48 h p.i., declining slowly thereafter. In addition, the average relative content of DEV UL51 gene transcripts at 48 h p.i. was approximately 50,000 times that of the transcript at 2 h p.i.

a Total RNA isolated from mock- and DEV-infected cells at 12 h p.i. was analysed using 1.0% agarose gel electrophoresis. b Kinetics of DEV UL51 gene transcription. The average relative content of the DEV UL51 gene transcripts was calculated at 1, 2, 4, 6, 12, 24, 36, 48, and 60 h p.i. using the 2−ΔΔCt method. The data are presented as the fold change in the DEV UL51 gene transcriptional expression normalized to a reference gene (β-actin) and relative to the mock-infected control
Kinetics of pUL51 expression in DEV-infected cells
The kinetics of UL51 protein expression in DEV-infected DEFs was analyzed by western blotting. At various times p.i., cell lysates were subjected to electrophoresis, transfered to PVDF membranes, and reacted with the purified UL51 antiserum. As shown in Fig. 3, pUL51 was first detected at 8 h p.i. as a protein band with a molecular mass of 34 kDa, which increased in amount until 48 h p.i., after which the protein started to decrease at 60 h p.i.
Intracellular localization of the UL51 protein in DEV-infected cells
The intracellular distribution of the DEV UL51 protein was examined by indirect immunofluorescence staining. Specific fluorescence first became detectable in the cytoplasm of infected cells at 8 h p.i. At 12 h p.i., specific fluorescence was found to be distributed in the cytoplasm and especially in the juxtanuclear region. A typical pattern of staining is shown in Fig. 5a2–c2. It is important to note the strong immunofluorescence of the juxtanuclear region (large arrow) and the punctate cytoplasmic staining (small arrow). This pattern of cytoplasmic staining continued throughout the course of infection (Fig. 5a3–c3, a4–d4), although later during infection, some infected cells also contained some punctate staining in the nucleus. No specific fluorescence could be detected with the UL51 antiserum in mock-infected cells (Fig. 5a1–c1).

Localization of pUL51 in DEV-infected cells. Mock-infected (a1–c1) and DEV-infected (a2–4, b2–4 and c2–4) DEF cells were fixed with 4% paraformaldehyde at 12 (a2–c2), 36 (a3–c3) and 60 h (a4–c4) p.i. and incubated with the UL51 antiserum. Cells were then stained with FITC-conjugated goat anti-rabbit immunoglobulin and DAPI as described in “Materials and methods”. Fluorescent images were examined using a confocal laser imaging system and obtained with 488- and 364-nm band-pass filters for excitation of FITC (a1–4) and DAPI (b1–4), respectively. The merged images are shown on the right (c1–4). Note the strong immunofluorescence of the juxtanuclear region (large arrows) and the punctate cytoplasmic staining (small arrows)
The UL51 protein is a component of DEV virions
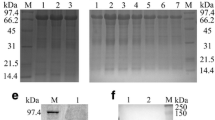
To determine whether the UL51 protein was a component of DEV virions, extracellular virions were collected from culture media harvested at 48 h p.i. Virus particles were purified by sucrose density-gradient centrifugation. When purified virions were subjected to western blot analysis, only a 34-kDa protein was detected in the fractions corresponding to the peak of DEV virions (Fig. 6a, lanes 1, 2). This result suggests that the UL51 protein is a component of DEV virions.

a The association of DEV pUL51 with purified virions. Virus particles were collected from culture medium harvested at 48 h p.i. and purified as described in “Virion purification”. Purified virions were lysed in SDS sample buffer, separated by SDS-PAGE, stained with Coomassie brilliant blue (lane 1), and then analyzed by western blotting with the UL51 antiserum (lane 2). Molecular mass marker sizes are shown on the left. b The dependence of DEV pUL51 production on viral DNA synthesis. The cells were cultured in the presence of 300 mg/ml ACV and harvested at 24 (lanes 1, 3) and 48 (lanes 2, 4) h p.i. Proteins were separated by SDS-PAGE, stained with Coomassie brilliant blue (lanes 1, 2), and analyzed by western blotting with the UL51 antiserum (lanes 3, 4). Molecular mass markers are shown on the left
The dependence of DEV pUL51 production on viral DNA synthesis
To determine whether the production of the DEV UL51 protein is dependent on viral DNA synthesis, infected cells were maintained for various times after a 1-h adsorption period in the presence of ACV. pUL51 production was not detectable in the presence of ACV, even at 24 and 48 h p.i. (Fig. 6b, lanes 1–4), indicating that pUL51 synthesis was highly dependent on viral DNA synthesis and thus indicated that pUL51 is a late gene product.
Discussion
Many herpesvirus UL51 genes have been cloned and sequenced [1, 3, 6, 10, 13, 14, 44], but the DEV UL51 gene sequence and analysis of its expression have not been reported until now.
The results of amino acid sequence comparison showed that the DEV pUL51 has a high degree of similarity to the pUL51 homologues of other alphaherpesviruses (Fig. 1), which are conserved among members of the subfamily Alphaherpesvirinae [9, 17, 23, 27]. Therefore, we infer that the DEV UL51 protein is a member of the tegument family and that it may play role in viral replication.
The subfamily Alphaherpesvirinae includes the genera Simplexvirus, Varicellovirus, Mardivirus, and Iltovirus [12]. Although DEV is an alphaherpesvirus, the genus has not yet been determined [12]. The data from the phylogenetic tree of the DEV UL51 gene indicated that DEV had a closer genetic relationship to MeHV-1, MDV-1 and MDV-2 than to other alphaherpesviruses. That is, DEV had a close evolutionary relationship with members of the genus Mardivirus. This happens to coincide with the fact that their natural host is poultry. Analysis of the evolutionary relationships of other known genes of DEV, such as UL27, UL28, UL30 [31], UL22 and UL23 [29], resulted in a similar conclusion. This suggests that MeHV-1, MDV-1, MDV-2 and DEV may have originated from a common ancestor.
In the present study, we generated a polyclonal antiserum specific for the UL51 gene product by using the 6-His-tagged UL51 protein as antigen. The antibodies were found to react strongly with a 34-kDa protein produced in DEV-infected cells at 24 h p.i., suggesting that this protein is the UL51 gene product. However, nucleotide sequence analysis of the coding sequence of UL51 predicts an acidic protein with a molecular mass of 27.1 kDa. Thus, the apparent molecular mass of the protein is considerably larger than the predicted molecular mass. Such a discrepancy between the predicted molecular mass and the apparent molecular mass on SDS-PAGE could be due either to post-translational modification (such as phosphorylation and/or palmitoylation) or to an unusual amino acid composition [46]. On one hand, the DEV UL51 protein has a relatively high content of hydrophobic amino acids such as alanine (13.9%), leucine (7.9%), and proline (6.7%). On the other hand, many tegument proteins, such as HSV-1 UL51 [38], HSV-1 UL11 [33] and PRV US2 [8], have been found to be modified with acyl or prenyl groups, modifications that are thought to be important for their localization to membranes. These could account for the discrepancy between the apparent and predicted molecular masses of the UL51 protein. Furthermore, previous studies have demonstrated that the apparent molecular masses of the proteins generated from the UL51 gene of other alphaherpesviruses (HSV-1 [9], BoHV-1 [17], and PRV [27]) are also much larger than their predicted molecular masses, correlating with the results for the DEV UL51 gene.
Recent studies have demonstrated that the tegument of herpesvirions is an amorphous protein layer that contains about 20 virus-encoded proteins, including VP1/2 (UL36), VP11/12 (UL46), VP13/14 (UL47), VP16 (UL48), VP22 (UL49), ICP0, ICP4, and the virion host shutoff protein (UL41). It also contains the products of genes US2, US3, US10, US11, UL11, UL13, UL14, UL16, UL17, UL21, UL37, UL51, and UL56 [21, 35, 45, 49]. The tegument proteins serve a variety of essential functions. Early in infection, they regulate viral and cellular gene expression; later, they assemble with the capsid and envelope to form mature progeny virions [37]. Analysis using RT-(real time) PCR and western blotting demonstrated that the accumulation of the UL51 protein occurred at the late stage of infection, suggesting that the protein may be a late viral gene that takes part in assembly with the capsid and envelope to form mature DEV virions. Furthermore, in contrast to BoHV-1 UL51, which is classified as a γ1 gene, DEV UL51 belongs to the γ2 class of viral genes, since expression of its transcript is highly dependent on viral DNA synthesis. This result correlates well with results obtained with the HSV-1 UL51 gene.
Different intracellular localizations may reflect different functions of tegument proteins, e.g., the transactivating function of UL48 in the nucleus and its structural function as a tegument protein in the cytoplasm [11, 16, 35]. The intracellular localization of tegument proteins may also vary at different times after infection; e.g., in HSV-1-infected cells, the juxtanuclear localization pattern of pUL51 predominated from 6 to 12 h p.i., but it localized to the vicinity of the plasma membrane at 24 h p.i. Moreover, homologous proteins may differ in intracellular localization in different herpesviruses [19]. In order to pinpoint the subcellular compartment in which tegumentation takes place, the assessment of the localization of tegument proteins, including the intracellular distribution of the DEV Ul51 protein, has been analyzed primarily by immunofluorescence and confocal laser scanning microscopy. We found that DEV UL51 proteins predominantly localized to the cytoplasm, especially to the juxtanuclear region. These results are in line with those obtained with the homologous proteins of HSV-1, BoHV-1, and PRV, which were detected exclusively or predominantly in the cytoplasm [9, 17, 25]. That is to say, the HSV-1, BoHV-1, PRV, and DEV UL51 proteins share a cytoplasmic location in infected cells. Whether or not the similar cytoplasmic location has any functional implications remains to be established.
In conclusion, we have presented the identification of the DEV UL51 gene and described the basic characteristics of the DEV pUL51, a 34 kDa protein. The protein, a component of extracellular mature virions, was produced at the late stage of infection, and its synthesis was highly dependent on viral DNA synthesis. Thus, we suggest that the gene belongs to the γ2 class. In addition, immunofluorescence studies localized pUL51 mainly to the juxtanuclear region of the cytoplasm in infected cells. Further studies involving construction of DEV UL51 gene mutants are required to study the function of the UL51 protein.
References
Albrecht JC, Nicholas J, Biller D et al (1992) Primary structure of the herpesvirus saimiri genome. J Virol 66:5047–5058
An R, Li HX, Han ZX, Shao YH, Liu SW, Kong XG (2008) The ul31 to ul35 gene sequences of duck enteritis virus correspond to their homologs in herpes simplex virus 1. Acta Virol 52:23–30
Baer R, Bankier AT, Biggin MD et al (1984) DNA sequence and expression of the B95–8 Epstein-Barr virus genome. Nature 310:207–211
Burland TG (2000) DNASTAR’s lasergene sequence analysis software. Methods Mol Biol 132:71–91
Chang H, Cheng AC, Wang MS, Guo YF, Xie W, Lou KP (2009) Complete nucleotide sequence of the duck plague virus gE gene. Arch Virol 154:163–165
Chee MS, Bankier AT, Beck S et al (1990) Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol 154:125–169
Cheng AC, Wang MS, Wen M, Zhou WG, Guo YF, Jia RY, Xu C, Yuan GP, Liu YC (2006) Construction of duck enteritis virus gene libraries and discovery, cloning and identification of viral nucleocapsid protein gene. High Technol Lett 16:948–953
Clase AC, Lyman MG, del Rio T, Randall JA, Calton CM, Enquist LW, Banfield BW (2003) The pseudorabies virus Us2 protein, a virion tegument component, is prenylated in infected cells. J Virol 77:12285–12298
Daikoku T, Ikenoya K, Yamada H, Goshima F, Nishiyama Y (1998) Identification and characterization of the herpes simplex virus type 1 UL51 gene product. J Gen Virol 79:3027–3031
Davison AJ, Scott JE (1986) The complete DNA sequence of varicella-zoster virus. J Gen Virol 67:1759–1816
Elliott G, O’Hare P (2000) Cytoplasm-to-nucleus translocation of a herpesvirus tegument protein during cell division. J Virol 74:2131–2141
Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (2005) Virus taxonomy: classification and nomenclature of viruses: eighth report of the International Committee on the Taxonomy of Viruses. Academic Press, San Diego
Fuchs W, Ziemann K, Teifke JP, Werner O, Mettenleiter TC (2000) The non-essential UL50 gene of avian infectious laryngotracheitis virus encodes a functional dUTPase which is not a virulence factor. J Gen Virol 81:627–638
Gompels UA, Nicholas J, Lawrence G, Jones M, Thomson BJ, Martin ME, Efstathiou S, Craxton M, Macaulay HA (1995) The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology 209:29–51
Guo YF, Cheng AC, Wang MS, Zhou Y (2009) Purification of anatid herpesvirus 1 particles by tangential-flow ultrafiltration and sucrose gradient ultracentrifugation. J Virol Meth doi:10.1016/j.jviromet.2008.12.017
Hafezi W, Bernard E, Cook R, Elliott G (2005) Herpes simplex virus tegument protein VP22 contains an internal VP16 interaction domain and a C-terminal domain that are both required for VP22 assembly into the virus particle. J Virol 79:13082–13093
Hamel F, Boucher H, Simard C (2002) Transcriptional and translational expression kinetics of the bovine herpesvirus 1 UL51 homologue gene. Virus Res 84:125–134
Han XJ, Wang JW, Ma B (2006) Cloning and sequence of glycoprotein H gene of duck plague virus. Agric Sci China 5:397–402
Harms JS, Ren X, Oliveira SC, Splitter GA (2000) Distinctions between bovine herpesvirus 1 and herpes simplex virus type 1 VP22 tegument protein subcellular associations. J Virol 74:3301–3312
Higgins DG, Bleasby AJ, Fuchs R (1992) CLUSTAL V: improved software for multiple sequence alignment. Comput Appl Biosci 8:189–191
Homa FL, Brown JC (1997) Capsid assembly and DNA packaging in herpes simplex virus. Rev Med Virol 7:107–122
Hong-Yan Z, Murata T, Goshima F, Takakuwa H, Koshizuka T, Yamauchi Y, Nishiyama Y (2001) Identification and characterization of the UL24 gene product of herpes simplex virus type 2. Virus genes 22:321–327
Izumiya Y, Jang HK, Kashiwase H, Cai JS, Nishimura Y, Tsushima Y, Kato K, Miyazawa T, Kai C, Mikami T (1998) Identification and transcriptional analysis of the homologues of the herpes simplex virus type 1 UL41 to UL51 genes in the genome of nononcogenic Marek’s disease virus serotype 2. J Gen Virol 79:1997–2001
Jia RY, Cheng AC, Wang MS, Xin HY, Guo YF, Zhu DK, Qi XF, Zhao LC, Ge H, Chen X (2009) Analysis of synonymous codon usage in the UL24 gene of duck enteritis virus. Virus genes 38:96–103
Klupp BG, Granzow H, Klopfleisch R, Fuchs W, Kopp M, Lenk M, Mettenleiter TC (2005) Functional analysis of the pseudorabies virus UL51 protein. J Virol 79:3831–3840
Koshizuka T, Kawaguchi Y, Nozawa N, Mori I, Nishiyama Y (2007) Herpes simplex virus protein UL11 but not UL51 is associated with lipid rafts. Virus Genes 35:571–575
Lenk M, Visser N, Mettenleiter TC (1997) The pseudorabies virus UL51 gene product is a 30-kilodalton virion component. J Virol 71:5635–5638
Li BW, Rush AC, Tan J, Weil GJ (2004) Quantitative analysis of gender-regulated transcripts in the filarial nematode Brugia malayi by real-time RT-PCR. Mol Biochem Parasitol 137:329–337
Li HX, Liu SW, Kong XG (2006) Characterization of the genes encoding UL24, TK and gH proteins from duck enteritis virus (DEV): a proof for the classification of DEV. Virus genes 33:221–227
Liu FY, Ma B, Zhao Y, Zhang Y, Wu YH, Liu XM, Wang JW (2008) Characterization of the gene encoding glycoprotein C of duck enteritis virus. Virus Genes 37:328–332
Liu SW, Chen SH, Li HX, Kong XG (2007) Molecular characterization of the herpes simplex virus 1 (HSV-1) homologues, UL25 to UL30, in duck enteritis virus (DEV). Gene 401:88–96
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25:402–408
Loomis JS, Bowzard JB, Courtney RJ, Wills JW (2001) Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J Virol 75:12209–12219
McGuire JM, Douglas M, Smith KD (1996) The resolution of the neutral N-linked oligosaccharides of IgG by high pH anion-exchange chromatography. Carbohydr Res 292:1–9
Mettenleiter TC (2002) Herpesvirus assembly and egress. J Virol 76:1537–1547
Miller WJ, Skinner JA, Foss GS, Davies KE (2000) Localization of the fragile X mental retardation 2 (FMR2) protein in mammalian brain. Eur J Neurosci 12:381–384
Mukhopadhyay A, Lee GE, Wilson DW (2006) The amino terminus of the herpes simplex virus 1 protein Vhs mediates membrane association and tegument incorporation. J Virol 80:10117–10127
Nozawa N, Daikoku T, Koshizuka T, Yamauchi Y, Yoshikawa T, Nishiyama Y (2003) Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J Virol 77:3204–3216
Nozawa N, Kawaguchi Y, Tanaka M, Kato A, Kato A, Kimura H, Nishiyama Y (2005) Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J Virol 79:6947–6956
Pan H, Cao R, Liu L, Niu M, Zhou B, Chen P, Hu J (2008) Molecular cloning and sequence analysis of the duck enteritis virus UL5 gene. Virus Res 136:152–156
Plummer PJ, Alefantis T, Kaplan S, O’Connell P, Shawky S, Schat KA (1998) Detection of duck enteritis virus by polymerase chain reaction. Avian Dis 42:554–564
Qi XF, Yang XY, Cheng AC, Wang MS, Zhu DK, Jia RY (2008) Quantitative analysis of virulent duck enteritis virus loads in experimentally infected ducklings. Avian Dis 52:338–344
Sandhu TS, Shawky SA (2003) Duck virus enteritis. In: Saif YM (ed) Diseases of poultry, 11th edn. Wiley, New York, pp 354–363
Telford EA, Watson MS, McBride K, Davison AJ (1992) The DNA sequence of equine herpesvirus-1. Virology 189:304–316
Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ (2005) Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79:9566–9571
Wada K, Goshima F, Takakuwa H, Yamada H, Daikoku T, Nishiyama Y (1999) Identification and characterization of the UL14 gene product of herpes simplex virus type 2. J Gen Virol 80:2423–2431
Wolf K, Burke CN, Quimby MC (1976) Duck viral enteritis: a comparison of replication by CCL-141 and primary cultures of duck embryo fibroblasts. Avian Dis 20:447–454
Xie W, Cheng AC, Wang MS, Chang H, Zhu DK, Luo QH, Jia RY, Chen XY (2009) Expression and characterization of the UL31 protein from Duck enteritis virus. Virol J 6:19
Yamauchi Y, Kiriyama K, Kubota N, Kimura H, Usukura J, Nishiyama Y (2008) The UL14 tegument protein of herpes simplex virus type 1 is required for efficient nuclear transport of the alpha transinducing factor VP16 and viral capsids. J Virol 82:1094–1106
Yuan GP, Cheng AC, Wang MS, Liu F, Han XY, Liao YH, Xu C (2005) Electron microscopic studies of the morphogenesis of duck enteritis virus. Avian Dis 49:50–55
Zhao LC, Cheng AC, Wang MS, Yuan GP, Jia RY, Zhou DC, Qi XF, Ge H, Sun T (2008) Identification and characterization of duck enteritis virus dUTPase gene. Avian Dis 52:324–331
Zhao Y, Wang JW, Liu F, Ma B (2009) Molecular analysis of US10, S3, and US2 in duck enteritis virus. Virus Genes doi:10.1007/s11262-008-0315-0
Zhao Y, Wang JW, Ma B, Liu F (2009) Molecular analysis of duck enteritis virus US3, US4, and US5 gene. Virus Genes 38:289–294
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (No. 30771598), Changjiang Scholars and Innovative Research Team in University (No. PCSIRT0848), the earmarked fund for Modern Agro-industry Technology Research System (No. nycytx-45-12), the Cultivation Fund of the Key Scientific and Technical Innovation Project, the Ministry of Education of China (No. 706050), the Cultivation Fund of the Key Scientific and Technical Innovation Project, Department of Education of Sichuan Province (No. 07ZZ028), the Sichuan Province Outstanding Youths Fund (No. 07ZQ026-132) and the Sichuan Province Basic Research Program (No. 07JY029-016/07JY029-017).
Author information
Authors and Affiliations
Corresponding author
Additional information
Chan-Juan Shen, An-Chun Cheng, Ming-Shu Wang and Yu-Fei Guo were contributed equally to this work.
Rights and permissions
About this article
Cite this article
Shen, CJ., Cheng, AC., Wang, MS. et al. Identification and characterization of the duck enteritis virus UL51 gene. Arch Virol 154, 1061–1069 (2009). https://doi.org/10.1007/s00705-009-0407-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-009-0407-8