
Abstract
Duck enteritis virus (DEV) is classified to the family Herpesviridae, but has not been grouped into any genus so far. Four overlapped fragments were amplified from the DEV genome with polymerase chain reaction (PCR). The assembled length of the four fragments was 6202 bp, which contained the genes encoding unique long (UL) 24, thymidine kinase (TK) and glycoprotein H (gH) proteins. The UL24 overlapped with TK by 64 nucleotides (nt), in a head-to-head transcription orientation, and the TK and gH had the same transcription orientation. The comparison of amino acid sequences of these 3 deduced DEV proteins with other 12 alphaherpesviruses displayed 5 highly conserved sites in the UL24, as well as another 5 consensus regions in the TK and 4 consensus regions in the gH. The RNA polymerase II transcriptional control elements were identified in all the UL24, TK and gH of DEV. These elements included core promoters, TATA motifs and polyadenylation sites. Phylogenetic analysis for the genetic classification of DEV in the Alphaherpesvirinae subfamily with other 12 alphaherpesviruses was computed. The result showed that DEV was more closely related to avian herpesviruses, except infectious laryngotracheitis virus (ILTV), than to other alphaherpesviruses. Conclusively, according to the phylogenesis-based analysis and the homology comparison of functional domains of UL24, TK and gH, DEV should be classified to a separate genus of the Alphaherpesvirinae subfamily in the family Herpesviridae.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Duck viral enteritis (DVE), also known as duck plague, is an acute, contagious and lethal disease of waterfowl, which can cause substantial mortalities in both farmed and wild waterfowl. DVE is caused by duck enteritis virus (DEV). This disease appears in waterfowl of all ages. Several studies indicate that survivors of DVE may become carriers of the virus for up to 4 years [1]. DVE was first reported in China in 1957 [2]. This disease is difficult to be monitored and controlled, because DEV establishes an asymptomatic carrier state in waterfowl that is detectable only during the intermittent shedding period of the virus [1].
Based on the report of Eighth International Committee on Taxonomy of Viruses (ICTV) [3], DEV is currently classified to the family Herpesviridae, but has not been grouped into any genus yet. Only limited sequences of DEV were published and the DEV genome has not been sequenced so far. The genome of DEV is a linear, double stranded DNA and the G + C content is 64.3%, which tops any reported avian herpesvirus in Alphaherpesvirinae subfamily [4]. However, three genes were considered relatively conserved among herpesviruses. UL24 is evolutionarily conserved and is considered to be a core herpesvirus gene [5]. The gH product is the second most highly conserved group of the herpesviral glycoproteins. TK is well suited for detailed genetic analysis [6], and the encoded kinase has a highly conserved ATP-binding site that provides the basis for evolution analysis [7]. Many properties of gH are identical among the members of herpesvirus family. In addition, gH forms heterodimers with glycoprotein L (gL). This form of dimers plays an essential role in mediating membrane fusion [8, 9]. More important, UL24, TK and gH forms a cluster, and their relative positions in the cluster are highly conserved among herpesviruses.
DEV was grouped into the subfamily of Alphaherpesvirinae previously [10, 11]. However, DEV has been assigned as an unclassified virus within the family Herpesviridae since the seventh ICTV [12]. The proper classification of DEV helps the better understanding of the characters and behaviors of this virus, which provides a platform for further study on the diseases caused by DEV. In the present study, we sequenced and compared three genes, UL24, TK, and gH of DEV Clone-03, and suggest DEV being classified to a separate genus of subfamily of Alphaherpesvirinae in the family Herpesviridae according to the molecular characterization of the three genes.
Materials and methods
Virus strain and propagation
DEV Clone-03 was a strain of Chinese commercial DEV vaccine and was purified in chicken embryo fibroblasts (CEF) using plaque assay. Virus was propagated in CEF growing in Dulbecco’s Minimum Essential Medium (D-MEM) for two passages. Viral particles were harvested when the cytopathic effect (CPE) reached 80%. Cell lysate containing DEV was subjected to three freezing-and-thaw cycles and stored at −70°C till use.
A pair of primers, P1(+) 5′-GTGCATGAGGCATTTAGAAC-3′ and P2(-) 5′-TGCAACGAGGAGAGTTATTG-3′, was used to verify that the Clone-03 was derived from DEV. Nucleotide sequence analysis of the 516-bp amplified fragment showed an identity of 99.8% compared with a published sequence of the 516-bp fragment of UL6 (AF043730) in DEV. Therefore, it was clear that the virus clone purified from the commercial vaccine belonged to DEV.
Preparation of viral genomic DNA
Cell lysate containing DEV was clarified by centrifugation at 10,000 rpm for 10 min using the F2402H rotor of Beckman. SDS (10%, Sigma) and proteinase K (20 mg/ml, TaKaRa, Dalian) were added into the supernatant to the final concentration of 1% and 500 μg/m1, respectively. After incubation at 56°C for 1 h, the mixture was extracted twice with equal volume of phenol–chloroform (1:1), and was then centrifuged at 12,000 rpm for 10 min. The viral DNA was precipitated with 2.5-fold volume of ethanol at −20°C for 30 min. The preparation was centrifuged at 13,000 rpm for 15 min, and the pellet was washed once with 70% ethanol. The DNA pellet was dissolved in deionized water and stored at −20°C. Extracts of noninfected CEF lysate were prepared as the negative control.
PCR amplification of UL24, TK and gH
After comparing the nucleotide sequences of UL24, TK and gH of 12 alphaherpesviruses [Herpes simplex virus 1 (HSV-1, X14112); Herpes simplex virus 2 (HSV-2, NC001798); Varicella-zoster virus (VZV, NC001348); Bovine herpesvirus 1 (BHV-1, NC001847); Bovine herpesvirus 2 (BHV-2, D00537); Equine herpesvirus 1 (EHV-1, AY464052); Equine herpesvirus 4 (EHV-4, NC001844); Pseudorabies virus (PRV, NC006151); Marek’s disease virus 1 (MDV-1, NC002229); Marek’s disease virus 2 (MDV-2, NC002577); Herpesvirus of turkeys (HVT, AF291866); Infectious laryngotracheitis virus (ILTV, NC006623)], we noticed that the UL24 and TK among these alphaherpesviruses were overlapped and transcribed in a head-to-head orientation, and there was an internal sequence between the TK and gH. Therefore, we amplified a 1302-bp of TK fragment from the DEV Clone-03 by using primers T1 at UL24 and T2 at the internal sequence. The determined sequence of this 1302-bp fragment was then used as the start-point to walk the DEV gene fragment in both the directions using two pairs of primers, T3 and T4, as well as T5 and T6. Consequently, two fragments were amplified. The sequences of these two fragments were used as the second start-points of PCR walking. Based on the sequence of the fragment amplified by T3 and T4, a pair of primers, T9 and T10, was synthesized and applied to amplify the 3′ fragment of gH. Primers used in the above experiments were listed in Fig. 1.

Schematic representation of the relative positions of the UL24, TK and gH gene in DEV genome. Three ORFs were showed in different shaded box. The black arrows show the transcription orientation of the three genes, UL24 and TK are in a head-to-head orientation, TK and gH are in the same orientation. The position of the oligonucleotide primers used in PCR was labeled in the diagram and the sequences were listed as below: T1(+) 5′-GACGTGTTGGCATCGGTTC-3′; T2(−) 5′-AAACAAATAGGGAGTAGCGAAGG-3′; T3(+) 5′-AGCTCTGATGGGCGGTTCTG-3′; T4(−) 5′-CCATGTGCAGCATTACGATGTC-3′; T5(+) 5′-AGCGCAATGGTGTATCGTTC-3′; T6(+)5′-GCCTTGTAGACGCGTAGCAG-3′ T9(+) 5′-CTAGATAAGCTCTCAACATCAC-3′; T10(−) 5′-TTCAATTACAACGTATAGCG-3′
PCR was carried out in a 25 μl reaction mixture containing 2.5 μl of 10× reaction buffer, 2.0 μ1 dNTPs (2.5 mM for each of the four dNTPs), 1.0 μl of each primer (10 pmol each), 5.0 μl DNA template, 0.25 μl Taq DNA polymerase (5 U/μl), and 13.25 μl water (all the reagents were purchased from TaKaRa). Reactions were performed at 95°C for 5 min, followed by 35 cycles of 94°C for 50 s, 50°C for 50 s and 72°C for 2 min, followed by 72°C for 10 min.
Cloning and sequencing of PCR products
PCR products were purified using the DNA Gel Extraction Kit UNIQ-l0 (Sangon, Shanghai) and cloned into the pMDl8-T vector (TaKaRa) according to the manufacturer’s instructions. Three clones of each fragment were sequenced with a CEQTM DTCS-Quick Stare Kit and a CEQTM 8800 Genetic Analysis System (Beckman Coulter).
ORF determination
To search for ORFs encoding UL24, TK and gH, the full-length assembled sequence was analyzed with the program of GENERUNR (version 3.00) in both strands. In order to verify the deduced genes, the predicted amino acid sequences of these ORFs were compared with counterparts in other alphaherpesvirus using the program of DNASTAR (version 5.01).
Promoter, motif and Poly(A) search
The sequence of the DEV gene fragment containing UL24, TK and gH was analyzed by the program of Berkeley Drosophila Genome project’s Neural Network Promoter Prediction, a eukaryotic (human) core promoter search engine (http://www.fruitfly.org/seq_tools/promoter.html). The initial search was performed at a high stringency (cutoff score of 0.85 out of 1.00). The core promoters found in this search were examined for the presence of TATA box consensuses using the TRANSFACFind search engine (http://motif.genome.ad.jp/). POLYADQ, a eukaryotic (human) polyadenylation [poly(A)] signal search engine (Cold Spring Harbor Laboratory ([http://argon.cshl.org/tabaska/polyadq_form.html]), was used to search poly(A) signals for the prediction of transcription terminal signals. All cutoff parameters were initially set at zero to return the location of all AATAAA and ATTAAA consensus signals, along with an associated score between 0 and 1.
Phylogenetic analysis
Phylogenetic analysis was performed by using the program of MEGALIGN in LASERGENE (DNASTAR) with CLUSTAL V multiple alignment and weight matrix PAM250 of 13 alphaherpesviruses (HSV-1, HSV-2, VZV, BHV-1, BHV-2, EHV-1, EHV-4, PRV, MDV-1, MDV-2, HVT, ILTV and DEV).
Results
ORF determination
Based on the published partial sequence of DEV genome, we cloned and sequenced four overlapped DNA fragments that were expected containing the genes of UL24, TK and gH from the DEV Clone-03. Three ORFs were identified in the assembled sequence that formed a length of 6202 bp. We designated these three ORFs as UL24, TK and gH homolog of HSV-1, which encoded 409, 358 and 834 amino acids, respectively. The sequences determined in this study are available in the GenBank under accession numbers: UL24, DQ227739; TK, AY963569 and gH, DQ227740. While TK and gH had the same transcription orientation, UL24 was in a head-to-head orientation respecting to TK, with an overlap of 64 nt. The relative positions of the three genes in DEV genome are shown in Fig. 1.
The search for promoters, motifs and poly(A) signals were performed by using online programs as described in the ‘Materials and methods’. These predicted functional regions were shown in the Tables 1 and 2. The canonical polyadenylation signal AATAAA was found downstream of gH and TK termination codons. In addition, a TG rich sequence, which may play a role in improving the efficient termination of transcript, was located downstream of the TK poly(A) signal. However, there was no typical poly(A) signal sequence downstream of the UL24. A Kozak’s ribosome-binding sequence was found at the translation start sequence of UL24, but was absent in TK and gH.
Comparison of DNA sequence of UL24, TK and gH in DEV with the corresponding gene in other Alphaherpesviruses
The results of homology comparison revealed that DEV was closer to the subfamily of Alphaherpesvirinae than to Betaherpesvirinae and Gammaherpesvirinae (data not shown). This result was consistent with the finding of Plummer et al. [13]. The predicted amino acid sequences of the viral protein UL24 from 13 alphaherpesviruses (including DEV) were compared by using the program of DNASTAR. The alignment revealed five high consensus regions, which were listed in Fig. 2A. Although UL24 is not essential for the propagation of viruses, it is expressed during HSV-1 infection and is important for the growth of these viruses in cultured cells. And the small plague phenotype of HSV-1 is closely correlated with mutations in UL24 that are strongly conserved among the herpesviruses [14].

Conserved sites in the deduced amino acid sequences of UL24 (A), TK (B) and gH (C) among 13 alphaherpesviruses. Highly conserved sites were shaded, and the four conserved glycosylation sites in gH were blackened and named as a, b, c and d, respectively. Site 1 and site 3 in B are active centers of TK, which represents ATP-binding and nucleoside-binding domain, respectively
Five consensus regions of amino acids in TK were identified (Sites 1–5, Fig. 2B). The consensus sequence in the Site 1 contained a predicted ATP-binding domain. The function of Site 2 was unknown. The Site 3 was located in a region implicated in the binding nucleosides. The Sites 4 and 5 were in regions that did not correlate to any known function but were conserved among the TKs of herpesviruses.
All the four conserved sites of amino acids in the gH of DEV did not contain the known functional motifs. These sequences were shown in Fig. 2C. In addition, eight predicted glycosylation sites were found in the deduced amino acid sequence of gH. Four of these glycosylation sites were conserved among most of the 13 alphaherpesviruses. Highly conserved amino acid residues were shaded.
Phylogenetic analysis
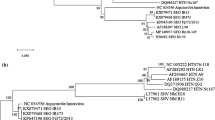
Phylogenetic trees of viral proteins UL24, TK and gH of the Alphaherpesvirinae subfamily were shown in Fig. 3. In all trees, the genera Simplexvirus, Varicellovirus, Mardivirus and Iltovirus [15] were separated into distinct clusters. The gH and TK of DEV were more closely related to the genus Mardivirus, while the UL24 of DEV formed a single cluster. Therefore, the overall result of phylogenetic analysis indicates that DEV forms a unique, separated cluster within the Alphaherpesvirinae subfamily of Herpesviridae.

Phylogenetic analysis of UL24, TK and gH in 13 viral strains of the Alphaherpesvirinae subfamily. Phylogenetic trees of these three proteins in these alphaherpesviruses were generated by using the program of MEGALIGN (DNASTAR). Sequence distances indicated by the scale were calculated by using the PAM250 matrix in LASERGENE
Discussion
Phylogenetic trees showed that DEV was more closely related to avian herpesviruses, including MDV-1, MDV-2 and HVT, than to other herpesviruses. The distance between DEV and ILTV is the farthest among these 13 alphaherpesviruses. Amino acid homology of the ATP-binding domain among herpesviruses represents the relationship of phylogenesis [6]. Therefore, the phylogenetic tree of TK may be used as a basis for DEV classification. Among avian herpesviruses, DEV was classified to the Group II and the Serotype 2 according to Günther’s report [16]. However, the genus of DEV is undetermined. The limited knowledge on the genome structure of DEV is one of the major reasons that prevent the determination. The partial DNA sequence of UL6 (Accession no. AF043730) was initially reported in 1998 [13], which provided the first sequence information for DEV that classified this virus into the group of alphaherpesvirus. Our result is consistent with this classification. The sequences of the three DEV ORFs we reported have provided solid evidence for DEV classification. Based on the phylogenetic trees of the deduced amino acid sequences of these ORFs, we suggest that DEV belongs to an individual genus within the Alphaherpesvirinae subfamily of Herpesviridae.
In this study, we adopted the strategy of targeted gene walking PCR [17, 18] for the amplification of partial unique long fragment from the DEV genome, which consisted of 6202 nt. ORF search revealed three complete ORFs that were identified as homologs to the UL24, TK and gH gene of HSV-l, respectively. Two potential promoters, both of which had high prediction scores, were found in the UL24 of DEV. It is remained to be determined that which one predominates the transcription. This result of promoter prediction was similar to the UL24 of HSV-1, in which two 5′-ends of UL24 transcripts in vivo were mapped [19]. Therefore, it is possible that both of the potential promoters are functional and regulated differently by means of transcription regulatory factors, DNA accessibilities during different stages of infection or in different types of cells.
Recent database analysis of Drosophila and human core promoters have revealed that only 30–40% of these promoters contain TATAAA consensus regions that have less than one mismatch [20]. It is possible that some TATA-less promoters exist in DEV. We have identified TATA-like consensus sequences in all potential core promoters in the three ORFs of DEV by using relaxed search parameters. These TATA-like elements were located 34–28 nt upstream of the transcription start sites (TSS). These predicted TATA-like elements are unlikely being picked up by the preprogrammed bias of the promoter prediction program we used, because this program had been trained with a set of naturally occurring core promoter sequences of 51-bp long (−40 to +11 relative to the TSS).
In the ORF of TK, there are two potential AUG start codons in-frame with a distance of 54 nt. Both of these codons lie in translation initiation contexts of low score. The first AUG context is GAUCAAAUGU and the second one is CCUUCGAUGC. It is reported that a few kinds of eukaryotic mRNAs initiate translation independent of the Cap sequence. In these cases, regulatory elements known as internal ribosome entry sites (IRESs) are required. The IRESs directly recruit and bind ribosomes to an initiation codon without requiring the assembly of factors at the 5′-end of the transcript [21]. We did not find the IRESs between the two AUG codons. However, the cap sequence we found in TK is spaced by 157 nt from the first AUG codon, by using the program of GENERUNR. For the majority of eukaryotic mRNAs, the most 5′ proximal AUG is used as the translation start site [22]. It was reported that a purine at the −6 of AUG (A as +1) was able to enhance the initiation of translation, although it functioned less effectively than a purine at the position of −3 [22, 23]. The first AUG context of TK matches this rule. Since the amino acid sequence of the TK in DEV has not been determined yet, the identification of actual translation initiation codon of TK depends on further experimental data. The translation initiation context of gH is UUUAGAAUGU. Since there is an A at the position of −3, we presume that this start codon initiates the translation of gH, although the sequence of this context does not meet the parameter of a favorable translation context.
The poly(A) signal was not found in the sequence of UL24. We reviewed the genome structure of HSV-1 and PRV [24, 25], and found that the genes of UL24, UL25 and UL26 (and UL26.5 in PRV) shared the same termination sequence of transcription in these two viruses. In addition, the transcription of UL24 in HSV-1 is quite complex, with six UL24 transcripts detected in cells infected by HSV-1 [5]. Therefore, we presume that the UL24 of DEV share the same poly(A) signal with downstream genes (UL25 and UL26), which have not been identified yet.
The identity comparisons of deduced amino acid sequence of UL24, TK and gH from DEV with the corresponding proteins of other alphaherpesviruses have revealed five conserved sites in the UL24 of DEV. This result is consistent with the report of Jacobson et al. for the alphaherpevirus subfamily [14]. In addition, five conserved sites have been identified in TK. Two domains, ATP-binding domain and the nucleoside-binding domain, are active centers of TK. This structure is similar with the TKs of other alphaherpesviruses reported by Nunberg et al. [26]. Furthermore, four conserved sites without known function and eight potential glycosylation sites (N-X-S/T) exist in the gH of DEV. This feature of gH has not been reported before. Among the eight potential glycosylation sites, four are conserved in alphaherpesvirus. The gH protein mediates viral entry and direct cell–cell spread of the virus, both process constituting membrane fusion events. These four conserved glycosylation sites may play important role for this function.
References
E.C. Burgess, J. Ossa, M. Yuill, Avian Dis. 23, 940–949 (1979)
Y.X. Huang, J. South China Agric. Univ. 1, 1–12 (1959)
C.M. Fauquet, M.A. Mayo, J. Maniloff, U. Desselberger, L.A. Ball, Virus Taxonomy: Eighth Report of the International Committee on Taxonomy of Viruses (Elsevier Academic Press, California, 2005), p. 208
R. Gardner, J. Wilkerson, J.C. Johnson, Intervirology 36, 99–112 (1993)
A. Pearson, A. Coen, J. Virol. 76, 10821–10828 (2002)
S.L. McKnight, Nucleic Acids Res. 8, 5949–5964 (1980)
G.R. Robertson, J.M. Whalley, Nucleic Acids Res. 16, 11303–11317 (1988)
B.G. Klupp, N. Visser, T.C. Mettenleiter, J. Virol. 66, 3048–3055 (1992)
T.J. Pasieka, L. Maresova, C. Grose, J. Virol. 77, 4191–4202 (2003)
E.F. Kaleta, Avian pathol. 19, 193–211 (1990)
S. Shawky, K. Schat, Avian Dis. 46, 308–313 (2002)
M.H.V. Regenmortel, C.M. Fauquet, D.H.L. Bishop, E.B. Carstens, M.K. Estes, S.M. Lemon, J. Maniloff, M.A. Mayo, D.J. McGeoch, C.R. Pringle, R.B. Wickner, Virus Taxonomy: Seventh Report of the International Committee on Taxonomy of Viruses. (Academic Press, California, 2000), p. 221
P.J. Plummer, T. Alefantis, S. Kaplan, P. O’Connell, S. Shawky, K.A. Schat, Avian Dis. 42, 554–564 (1998)
J.G. Jacobson, S.L. Martin, D.M. Coen, J. Virol. 63, 1839–1843 (1989)
M.A. Mayo, Arch. Virol. 150, 189–198 (2005)
B.M.F. Günther, B.G. Klupp, M. Gravendyck, J.E. Lohr, T.C. Mettenleiter, E.F. Kaleta, Avian Pathol. 26, 305–316 (1997)
P.-C. Chang, M.-L. Hsieh, J.-H. Shien, D.A. Graham, M.-S. Lee, H.K. Shien, J. Gen. Virol. 82, 2157–2168 (2001)
J.D. Parker, P.S. Rabinovitch, G.C. Burmer, Nucleic Acids Res. 19, 3055–3060 (1991)
G.S. Read, J.A. Sharp, W.S. Summers, J. Virol. 138, 368–372 (1984)
S.T. Smale, J.T. Kadonaga, Annu. Rev. Biochem. 72, 449–479, (2003)
C.U. Hellen, P. Sarnow, Genes Dev. 15, 1593–1612 (2001)
M. Kozak, J. Mol. Biol. 196, 947–950 (1987)
M. Kozak, Cell 44, 283–292 (1986)
D.J. Mcgeoch, M.A. Dalrymple, A.J. Davison, A. Kolan, M.C. Frame, D. Mcnab, L.J. Perry, J.E. Scott, P. Taylor, J. Gen. Virol. 69, 1531–1574, (1988)
B.G. Klupp, C.J. Hengartner, T.C. Mettenleiter, L.W. Enquist, J. Virol. 78, 424–440 (2004)
J.H. Nunberg, D.K. Wright, G.E. Cole, E.A. Petrovskis, L.E. Post, T. Compton, J.H. Gilvert, J. Virol. 63, 3240–3249 (1989)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, H., Liu, S. & Kong, X. Characterization of the genes encoding UL24, TK and gH proteins from duck enteritis virus (DEV): a proof for the classification of DEV. Virus Genes 33, 221–227 (2006). https://doi.org/10.1007/s11262-005-0060-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-005-0060-6