
Abstract
Psychoactive drug use is a common behavior in many societies worldwide, frequently associated with drug instrumentalization. Regular use may develop into drug addiction, which is a severe psychiatric disorder with multiple pathological effects to virtually all organ systems. Treatment strategies for addiction are often insufficient with no broadly working pharmaco-treatment available. Recently, lipids, and particularly sphingolipids, have been considered as new mediators in the pathogenic pathways and as possible therapeutic targets for the treatment of addictive states. In our review, we discuss the contribution of sphingolipids in the development of addictive states including alcohol consumption, nicotine, amphetamine, morphine, and cocaine dependencies. Recent data show that the involvement of various classes of sphingolipids, such as sphingomyelins, ceramides, globosides, sulfatides, and cerebrosides, might explain the development of some specific features of addictive states, for example, apoptotic neurodegeneration induced by psychoactive substances. On the other hand, protective effects of sphingolipids are discussed. Sphingolipids might be a key mechanism in the development of beneficial effects of moderate alcohol consumption. Therefore, sphingolipid systems emerge as possible new pathways involved in the development of addiction and its pathophysiological consequences. However, further analysis is still needed to investigate the exact mechanisms of sphingolipid contribution and possibility of using of sphingolipids as new therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipids are a large family of chemically distinct molecules containing combinations of fatty acids with various backbone structures. Mammalian cells include thousands of lipid species (Wenk 2005; Kimura et al. 2016). A diverse group of hydrophobic or amphipathic lipid molecules possess a variety of functions such as the maintenance of membrane structure, energy storage, signal transduction, regulation of gene expression, and others (Hyötyläinen and Orešič 2014). In accordance with one of the most used classification system LIPID MAPS Lipid, lipids might be classified into eight main groups including fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, prenol lipids, saccharolipids, and polyketides (Fahy et al. 2005). Lipid metabolism is particularly important for the central nervous system due to the high concentration of lipids in the brain, second only to adipose tissue. Dry human brain contains approximately 60% lipids (Tamiji and Crawford 2010).
Sphingolipids (SLs), a complex family of compounds with a common long-chain sphingoid base backbone, are particularly abundant in the brain as they constitute 10–20% of the membrane lipids (Holthuis et al. 2001). They comprise ceramides, phosphosphingolipids, glycosphingolipids, and other species, including protein adducts (Fahy et al. 2005). SLs are essential for the development and maintenance of the functional integrity of the nervous system (van Echten-Deckert and Herget 2006; Piccinini et al. 2010; Olsen and Færgeman 2017). Gangliosides are the most abundant group of lipids in the gray matter and in neurons. Sphingomyelin (SM), galactosylceramide (GalCer), and sulfatides are enriched in oligodendrocytes and myelin (Aureli et al. 2015; Olsen and Færgeman 2017). Ceramide serves as the key molecule of the SL metabolism and is involved in the maintenance of vital physiological processes such as cell apoptosis, inflammatory responses and others (Kornhuber et al. 2014).
SL synthesis starts in the endoplasmic reticulum (ER) from the condensation of l-serine and palmitoyl-CoA to 3-ketosphinganine (Fig. 1). This is a rate-limiting reaction catalyzed by serine palmitoyltransferase (SPT) (Merrill et al. 1985; Deevska and Nikolova-Karakashian 2017). Then, 3-ketosphinganine reductase transforms 3-ketosphinganine to sphinganine in a nicotinamide adenine dinucleotide phosphate (NADPH)-dependent manner. Sphinganine is acetylated to dihydroceramide by ceramide synthases (CerS). CerS differ in their specificity to fatty acids of particular acyl chain length and saturation and tissue distribution. CerS1 and CerS4 mainly produce C18-dihydroceramide and are expressed in the brain, skin, muscles, heart, and liver. CerS5 and CerS6 synthesize C16-dihydroceramide and are expressed in the brain, adipose tissue, intestine, and kidneys. CerS2 is expressed in practically all organs and generates long-chain dihydroceramide such as C20–C26. Similarly, CerS3 produces ceramide (Cer) 24–Cer26. After acetylation, a 4,5-trans-double bond is generated in the sphingoid base to form ceramide (Fei et al. 2011; Brodowicz et al. 2017). This pathway of ceramide biosynthesis is known as de novo pathway. Ceramide is then transferred from the ER to the Golgi apparatus by either vesicular transport or the ceramide transfer protein, CERT1, to reach glucosylceramide synthase or sphingomyelin synthase (SMS) (van Meer and Sprong 2004; Deevska and Nikolova-Karakashian 2017). Then, it is converted to complex sphingolipids through the addition of a phosphocholine group (for SM), glucose (for glucosylceramide), or a phosphate group (for ceramide-1-phosphate). SMS produces SM by transferring the phosphocholine group of phosphatidylcholine to ceramide with the production of diacylglycerol (Deevska and Nikolova-Karakashian 2017).

Sphingolipids are a class of lipids containing a sphingoid base as a backbone, most commonly the unsaturated amino alcohol sphingosine. Sphingolipids can be synthesized de novo by the condensation of palmitate and serine, which is catalyzed by serine palmitoyl transferase (SPT). 3-Ketodihydrosphingosine is then reduced to dihydrosphingosine by 3-ketosphinganine reductase (KDSR) and then to dihydroceramide by ceramidases (CerS). Ceramide, the key molecule of sphingolipid metabolism, is generated from dihydroceramide by the action of dihydroceramide desaturase. Alternatively, ceramide might be generated from sphingosine by ceramide synthases (CerS) in a salvage pathway of synthesis or from ceramide-1-phosphate by phosphatases (PT). Sphingomyelin is transformed to ceramide by sphingomyelinases (SMase). The only exit pathway from the sphingolipid pathways is mediated by sphingosine-1-phosphate lyase (S1P lyase), which metabolises sphingosine-1-phosphate to ethanolamine and hexadecenal. Ceramide might serve as a basis for the generation of complex sphingolipids such as sulfatides, globosides, cerebrosides, and gangliosides via glucosylceramide (Hannun and Obeid 2008; Kornhuber et al. 2014). SPT serine palmitoyl transferase, KDSR 3-ketosphinganine reductase, CerS ceramide synthase, CDase ceramidase, SMase sphingomyelinase, SMS sphingomyelin synthase, CK ceramide kinase, PT phosphatase, GCS glucosylceramide synthase, GCase glucosyl ceramidase, SPPase sphingosine phosphate phosphatase, SK sphingosine kinase, S1P lyase sphingosine-1-phosphate lyase
SM turnover is mediated by sphingomyelinases, a family of enzymes removing the phosphocholine group of SM to produce ceramide. Sphingomyelinases are classified depending on their pH optimum and subcellular localization as either neutral sphingomyelinase (NSM), acid sphingomyelinase (ASM), or alkaline sphingomyelinase (alkSM). ASM has a pH optimum of 5 and is located in acidic lysosomes. In mammals, the expression of a single gene, SMPD1, results in two forms of ASM: the Zn2+-dependent lysosomal and Zn2+-independent secretory ASM (Kornhuber et al. 2015). Several NSM genes have been described: NSM1 (SMPD2), NSM2 (SMPD3), NSM3 (SMPD4), and mitochondrial-associated NSM (SMPD5). NSM-1 is localized in the ER and Golgi apparatus, but its functions are not well understood. NSM-2 catalyzes SM hydrolysis at the cytosolic face of the plasma membrane, in the multi-lamellar bodies in the cytosol, and at the nuclear envelop (Airola and Hannun 2013; Deevska and Nikolova-Karakashian 2017). Mitochondrial-associated NSM has been discovered in 2010, and has comparable domain architecture and biochemical properties as NSM2 (Wu et al. 2010; Airola and Hannun 2013). Human NSM3 was identified in 2006, but the functional role of it still has to be determined (Krut et al. 2006). This pathway of ceramide synthesis is known as sphingomyelinase pathway.
The last known ceramide synthesis pathway refers to SM recycling called the salvage pathway (Kornhuber et al. 2015). Glycosphingolipids are hydrolyzed to ceramide only in the lysosomes by the combined activity of specific acid hydrolases. Ceramide is further degraded to sphingosine by the ceramidases ACER1, ACER2, and ACER3. Ceramidases are expressed in various cellular compartments and have specific pH optima (Coant et al. 2017; Deevska and Nikolova-Karakashian 2017). Sphingosine kinases 1 and 2 (SK1 and SK2) catalyze the phosphorylation of sphingosine to sphingosine-1-phosphate (S1P). Consequently, S1P is either dephosphorylated by the sphingosine-1-phosphate phosphatase (SPP) back to sphingosine, or degraded by the sphingosine-1-phosphate lyase (SPL) to alcoholamine phosphate and trans‑2-hexadecenal, two products that cannot re-enter the sphingolipid metabolic pathway (Deevska and Nikolova-Karakashian 2017).
Once formed, ceramide can be converted into more complex sphingolipids through different pathways. Ceramide can be glycosylated by glucosylceramide synthase to glucosylceramide (GlcCer), the main cerebroside, in the Golgi apparatus. More than 300 complex glycosphingolipids can be produced from GlcCer (Messner and Cabot 2010; Deevska and Nikolova-Karakashian 2017). In the ER lumen, ceramide can be glycosylated to GalCer (Deevska et al. 2009), a precursor of sulfatides. Together with GalCer, sulfatides serve as important components of neuronal myelin in the central nervous system. The addition of galactose transforms GlcCer to lactosylceramide (LacCer) by an intermediate product in the synthesis of more complex glycosphingolipids (Nikolova-Karakashian and Rozenova 2010; Deevska and Nikolova-Karakashian 2017). The biosynthesis of gangliosides also takes place in the Golgi apparatus and starts with the transfer of sialic acid residues to LacCer by specific sialyltransferases—the GM3 synthase, the GD3 synthase, and the GT3 synthase (Groux-Degroote et al. 2017). When the synthesis is complete, SM and glycosphingolipids (GSL) are relocated to the plasma membrane, where they are known to participate in micro-domain formation (Olsen and Færgeman 2017).
Due to the variety of SL species and the complexity of SL families, many classifications of them exist. For example, SLs can be divided into several major classes: the sphingoid bases and their simple derivatives (e.g., sphingoside-1-phosphate), the sphingoid bases with an amide-linked fatty acid (e.g., ceramides), and more complex sphingolipids with head groups that are attached via phosphodiester linkages (e.g., phosphosphingolipids), via glycosidic bonds (e.g., cerebrosides and gangliosides), and other groups (e.g., phosphono- and arseno-sphingolipids) (Fahy et al. 2005). Slightly simpler classification suggests that SL can be subdivided into SMs/ceramides and GSLs including cerebrosides, sulfatides, globosides, and gangliosides (Dong et al. 2017). In our review, we will use this classification.
Structural diversity of SL determines their multiple functions at the cellular level. SLs modulate the chemical and mechanical properties of all membranes, mediate protein trafficking, ion channel functioning, and cell-to-cell communication. They regulate membrane assembly, vesicle synthesis and trafficking, neurotransmitter release, synaptogenesis, rapid nerve impulse conduction, and signal propagation (Adibhatla et al. 2006; Piomelli et al. 2007; Veloso et al. 2011; Miranda and Oliveira 2015). In the plasma membrane, lipids regulate the membrane’s function and generate a barrier between the intracellular and extracellular spaces. Membrane SLs also determine the localization and function of proteins within the membrane and regulate functions of synapses. Moreover, SLs may function within the membrane as second messengers (Müller et al. 2015; Schneider et al. 2017). Moreover, the generation of lipid rafts might serve as one of these mechanisms mediating the effects of SLs. Thereby, SLs and cholesterol form distinct domains in the cell membrane (Brown and London 1998). Hydrophilic carbohydrate head groups of sphingolipids mediate an association between these lipids. Hydrophobic Van der Waals interactions associate the saturated side chains. Cholesterol fills the spaces between the glycerosphingolipids to form an ordered structure and to stabilize the domain by tight interactions between its sterol ring system and the sphingosine residue, and hydrogen bonding between the hydroxyl group and the hydrophilic head group of SM. This interaction between SLs and cholesterol produces lipid rafts—distinct membrane domains containing or recruiting proteins under the influence of an appropriate stimulus (Simons and Ikonen 1997; Gulbins et al. 2004). The generation of ceramide in the plasma membrane results in the formation of ceramide-enriched micro-domains that exhibit altered membrane fluidity, change membrane shape, and induce trans-bilayer flip-flop transport (Rebillard et al. 2007; Jenkins et al. 2009). Membrane domains trap membrane-related proteins and enable the oligomerization of G-protein-coupled receptors (Gulbins et al. 2004; Kornhuber et al. 2014). Thus, changes in the composition of the lipid rafts may affect receptor affinity, signaling, and subsequent internalization. Lipid rafts regulate receptor density and contribute to the regulation of signal transmission (Fantini and Barrantes 2009; Müller et al. 2015). For instance, ceramide-rich platforms mediate a variety of signaling cascades in cells, including activation of B cells, bacterial pathogen infection and cytokine release during infections, and also induction of apoptosis (Maceyka and Spiegel 2014). Lipid rafts are the specific sites for ASM activation and subsequent generation of ceramide, which can explain the involvement of these molecules in the regulation of neurotransmitter homeostasis and development of neurological and psychiatric disorders (Kornhuber et al. 2014; Müller et al. 2015; Schneider et al. 2017; Brodowicz et al. 2017; Müller and Kornhuber 2017). As the brain contains huge amounts of lipids, lipid disturbances in the central nervous system are involved in the pathophysiology of different neurological disorders, including depression, bipolar disorder, schizophrenia, Alzheimer, Hungtington’s, and Parkinson’s diseases, amyotrophic lateral sclerosis, cerebral ischemic injury, and substance use disorder (Wenk 2005; Hillard 2005; Adibhatla et al. 2006; Ciarlo et al. 2012; Blasco et al. 2017; Brodowicz et al. 2017).
Drug addiction is a chronically relapsing disorder that has been characterized by compulsion to seek and take the drug, loss of control in limiting intake, and the emergence of a negative emotional state reflecting a motivational withdrawal syndrome when access to the drug is prevented (Koob and Volkow 2010). Numerous epidemiological surveys show the world-wide importance of drug dependence and abuse (Whiteford et al. 2013; Spagnolo and Goldman 2017). For example, in 2014, 240 million people worldwide suffered from alcohol use disorder, 15 million people consume injectable substances, and 1 billion people smoked tobacco products (Gowing et al. 2015; Brodowicz et al. 2017). However, the majority of people regularly consuming psychoactive drugs are not addicts and will never become such (Abel 1980; Waldorf et al. 1991 Heath 2000; O’Malley and Johnston 2002). For example, in the United States, only 14.9% of the current alcohol drinkers are alcohol dependent. In the European Union, this level is even lower—only 7.1% (Anderson and Baumberg 2006). In most cases, psychoactive drugs are consumed for instrumentalization goals such as the facilitation of social connections, the facilitation of sexual and mating behavior, coping with stress and psychiatric disorders, or cognitive enhancement (Müller and Schumann 2011a, b; McCreary et al. 2015; Müller 2017a, b). The main biological mechanism of controlled drug consumption is related to learning and memory processes. Psychoactive drugs drive learning mechanisms as natural reinforcers. Positive effects of the drugs and altered mood induce positive memories and maintain the regular consumption (White 1996; Robbins et al. 2008; Müller and Schumann 2011a, b; Müller 2013). A small regular consumption of, e.g., alcohol, was associated with better mental as well as physical health compared to complete abstinence (Skogen et al. 2009). At the same time, negative toxic effects of drugs are associated with negative memories, which determine the controlled use of drugs. However, drug instrumentalization works only in a narrow dose and frequency window. Controlled drug use behavior can be a transitory state for the development of addiction (Müller 2013). Thus, negative life events might lead to an over-instrumentalization and an escalation of drug consumption and drug addiction (Müller and Schumann 2011b; Müller 2017b). These states are characterized by compulsive drug consumption when free choice is not possible anymore. The dosage and frequency of drug intake are substantially increasing. This leads to the development of negative consequences of drug consumption due to chronic poisoning by these substances in toxic doses, and might result in lethal outcome (Hassan et al. 2017).
Although drug instrumentalization is a relatively safe state of drug consumption not requiring pharmacological treatment, drug addiction is a severe medical and social problem, which arises due to the limited number of effective treatments. It might be related to the insufficient understanding of the pathogenic pathways of drug addiction and a lack of effective pharmaco-therapies (McCreary et al. 2015). Recently, it has been shown that various types of SL contribute to the pathogenesis of drug dependence and are proposed as a possible new target for preventive treatment of drug abuse (Schneider et al. 2017). Here, we will discuss the involvement of main classes of SL such as ceramides, gangliosides, sulfatides, and cerebrosides as the most studied SL in the development of addictive states and their health impact induced by various psychoactive substances.
Sphingomyelins and addictive disorders
Alcohol
Peripheral ceramides
Human studies show that patients with acute alcohol intoxication are characterized by a significant increase in lysosomal ASM activity in the peripheral blood cells. ASM is an enzyme catalyzing the breakdown of sphingomyelin to ceramide. Thus, an increase in ASM activity might indicate an elevated ceramide production. This increase is followed by a significant decline within 24 h after beginning of withdrawal. However, withdrawal for 7–10 days is accompanied by a secondary increase in ASM activity and an enhancement of Cer18:0 levels (Reichel et al. 2010, 2011). In contrast, patients display low ASM activity in the peripheral blood cells in the early phase of withdrawal and increasing activity towards the end of the qualified withdrawal (Reichel et al. 2010). Analysis of secretory ASM activity showed a significant increase in blood serum of alcohol-dependent patients, which was reduced during the abstinence period. Moreover, serum ASM activity reaches normal values already after 1 week of withdrawal (Reichel et al. 2011). Another study has confirmed these findings and showed a gradual decrease of ASM activity during abstinence in patients with acute alcohol intoxication upon admission for treatment. Analysis of gender effect has shown that this trend is similar in females and males, although the initial enzyme activities in females were significantly lower (Mühle et al. 2014).
Animal studies are largely in line with the human data. Clugston et al. (2011) observed a significant increase in the levels of total ceramide, sphingosine, and sphinganine in the liver of alcohol-fed mice. Taking into account that sphingosine and sphinganine are the precursors of ceramide, this increase correlates with the increased ceramide level. However, among the genes controlling ceramide metabolism, only the expression of Kdsr, a gene associated with sphinganine synthesis, is changed in alcohol-fed mice. On the contrary, the ceramide plasma concentration in alcohol-fed mice did not differ under these conditions from controls. The authors also showed that alcohol exposure was accompanied by a significant decrease in serum levels of sphingosine and sphinganine in wild-type mice, although the phosphorylated forms of these SLs were unchanged (Clugston et al. 2011). Similarly, a significant decrease in the amount of SMs is found in the blood serum of rats fed with liquid alcohol-containing diet for 8 weeks as well as in alcohol-dependent patients with or without liver disease (Marmillot et al. 2007).
It has been shown that alcohol treatment of rodents re-establishes the composition of SL with various chain lengths and their saturation in the tissue. The levels of SM16:0, SM18:1, and SM18:0 were decreased in the serum, but increased in the heart of mice fed with the Lieber–DeCarli liquid alcohol diet for 4 weeks. Concentrations of Cer18:0 and Cer16:0 were reduced in the liver of these rodents. In the kidneys, the amounts of these ceramides as well as those of Cer18:1, Cer20:0, and Cer22:0 were enhanced after alcohol treatment. Levels of Cer24:1 and Cer24:0 were reduced in the kidneys (Zhao et al. 2011). Another study revealed higher levels of Cer14:0 and Cer18:0 and relatively lower levels of Cer16:0 and Cer20:0 in the livers of chronic alcohol-fed rats. Increased levels of Cer16:0 ceramide could promote cell death (Osawa et al. 2005; Seumois et al. 2007), while Cer18:0 ceramide may inhibit cell growth (Koybasi et al. 2004; Yang et al. 2016). Carr et al. (2014) found an increase in Cer22:0 and Cer24:0 levels in the liver of mice fed with alcohol for 6 weeks, but no changes in concentrations of Cer16:0, Cer16:1, or Cer24:1 species. The authors reported that the onset of hepatic steatosis and insulin resistance in experimental alcoholic liver disease correlates with an increase in long-chain hepatic ceramides. Liangpunsakul et al. (2010) observed a significant rise in Cer16:0 and Cer18:0 species and a relatively minor increase in Cer24:0 in the liver of alcohol-fed animals (6 weeks protocol), while the concentration of these ceramides was decreased in alcohol-fed ASM knockout (KO) mice.
Altogether, these data indicate that a significant shift in ceramide pattern occurs in peripheral tissues during ethanol treatment. It should be emphasized that several studies reveal a correlation between ceramide accumulation and negative consequences of alcohol consumption. However, as the changes in SL concentrations are multidirectional and differ in various tissues, further investigations are needed to systematize the observed alterations.
Several investigations are devoted to the genetic mechanisms mediating the described changes in ceramide profile induced by toxic effects of alcohol. Longato et al. (2012) have shown that chronic alcohol-related liver disease in humans is associated with an increased expression of multiple genes and proteins promoting ceramide production. These patients are characterized by a significantly increased expression of CerS1, CerS5, CerS6, and SPT1, reflecting an increased ceramide biosynthesis. An mRNA expression of sphingomyelin phosphodiesterase 1 (SMPD1) and SMPD3 genes, which code for ASM and NSM, indicates a high level of SM hydrolysis. A decreased expression of ceramidase 2 (CerD2), an enzyme responsible for deacetylation of ceramide to sphingosine and fatty acids, is also registered during this disorder. Chronic alcohol-related liver disease is also accompanied by significantly higher activities of GM3 synthase and UDP glucose ceramide glycosyltransferase, which mediate the formation of complex SLs from ceramides. Activities of ASM, NSM, and alkSM were also enhanced in the liver of patients with chronic alcohol-related liver disease. These alterations in enzyme activity were associated with higher hepatic levels of the Cer14:0, Cer16:0, Cer18:0, and Cer20:0 species, but not Cer24:0 (Longato et al. 2012). The authors speculate that the cell death associated with chronic alcohol-related liver disease in humans could be mediated in part by increased levels of Cer16:0, as it was shown to promote cell death (Osawa et al. 2005; Seumois et al. 2007), while Cer18:0 inhibits cell growth (Koybasi et al. 2004). Similar results have been found by Ramirez et al. (2013), showing that rats chronically fed with alcohol develop steatohepatitis associated with increased hepatic mRNA expression of CerS4, CerD3, SMPD1, and long-chain subunit 2 of SPT, and significantly reduced expression of SMPD3 and GM3 synthase. Chronic alcohol feeding significantly increases levels of Cer14:0 and Cer18, and reduces the levels of Cer16:0 in the liver of rats receiving alcohol.
It is not clear yet which exact pathway of ceramide biosynthesis is responsible for alcohol-induced ceramide accumulation. A study of Tong et al. (2014) showed that an inhibitor of de novo ceramide synthesis, myriocin, reduces the concentrations of ceramide in the liver of rats treated with alcohol for 8 weeks and the severity of alcohol-related steatohepatitis including the abundance and sizes of lipid droplets and mitochondria, inflammation, and architectural disruption of the endoplasmic reticulum. These findings suggest that excessive ceramide accumulation is a critical mediator of steatohepatitis in alcohol-related liver disease. Myriocin-mediated reductions in hepatic lipid and ceramide levels are associated with constitutive enhancement of insulin signaling through the insulin receptor and IRS-2, reduced hepatic oxidative stress and modulation of endoplasmic reticulum stress signaling mechanisms. However, focal necrosis and apoptosis persists after myriocin treatment suggesting that the de novo pathway of ceramide synthesis may not play the key role in alcohol-induced cell degeneration in the liver (Ramirez et al. 2013; Correnti et al. 2014). It has also been shown that ASM contributes to the ceramide elevation in alcohol-fed animals (Supakul and Liangpunsakul 2011). An increase in hepatic ceramide after 6-weeks alcohol exposure was associated with enhanced ASM but not NSM activity in the liver (Liangpunsakul et al. 2010). Moreover, ASM KO mice are resistant to the steatosis and apoptotic effects of tumor necrosis factor-α (TNFα) in the liver (Iimuro et al. 1997; Fernandez-Checa et al. 2005). Liangpunsakul et al. (2010, 2012) observed that the functional inhibitor of ASM (FIASMA; Kornhuber et al. 2010), imipramine, significantly reduces the alcohol-induced increase in the levels of total ceramide as well as Cer16:0 and Cer24:0 species. These changes were associated with an improvement of hepatic steatosis. Presented data confirm the hypothesis that the alcohol-induced ceramide increase is mediated by ASM. However, treatment of alcohol-fed animals with imipramine does not completely inhibit the alcohol-mediated increase in liver ceramide levels (Liangpunsakul et al. 2012), suggesting that alcohol contributes to the elevation of ceramide levels via both de novo and sphingomyelinase pathways. Another ASM inhibitor amitriptyline limits the formation of ceramide in the liver of Long–Evans Cinnamon rats, a genetic model for Wilson disease. It reduces the manifestations of hepatic steatosis, fibrosis and nuclear pyknosis, and improved animal survival (Lang et al. 2007). In ASM KO mice, amitriptyline treatment could also decrease alcohol-induced macrosteatosis and the accumulation of triglycerides and free fatty acids, without causing an SM accumulation (Supakul and Liangpunsakul 2011). Another mechanism for the alcohol-induced increase in ceramide level is the induction of stearoyl-CoA desaturase-1 (Scd-1) (You and Crabb 2004). Ceramide concentration and SPT mRNA expression are diminishes in Scd-1 KO mice (You and Crabb 2004; Supakul and Liangpunsakul 2011).
In vitro studies are in line with in vivo investigations of alcohol effects. A recent study showed that alcohol treatment of isolated hepatocytes reduced the levels of SM and sphingosine and increased ceramide content (Viktorov and Yurkiv 2008). Therefore, it is clear that at least SM pathway is involved in alcohol-induced changes in ceramide metabolism in the liver. However, it cannot be excluded that other mechanisms, particularly the de novo pathway and changes in Scd-1 activity, might also contribute to the observed alterations. Further analysis of possible pathways is needed to understand detailed mechanisms of alcohol action.
It is well known that toxic effects of alcohol result in a variety of degenerative processes in the brain, liver, pancreas, and other organs. It was shown that alcohol-related conditions, such as alcoholic liver disease, alcohol-induced neurotoxicity, hepatic steatosis, or fetal alcohol syndrome are associated with an enhanced production of ceramide and activation of enzymes involved in ceramide metabolism (de la Monte et al. 2009; Wang and Bieberich 2010; Liangpunsakul et al. 2010, 2012; Carr et al. 2014; Yang et al. 2016). Hepatic and serum ceramide levels are shown to correlate with the severity of alcohol-induced steatohepatitis in rats (de la Monte et al. 2009). Alcohol-fed animals are characterized by the augmentation of liver apoptosis. This might be related to the changes in hepatic ceramide level, which are associated with a similar pattern of alterations of caspase-3 activity. These degenerative processes are tightly bound to cell apoptosis. The ceramide/sphingosine rheostat was shown to significantly contribute to the initiation and progression of cell apoptosis in a variety of cells (Cuvillier et al. 1996). Ceramide may participate in hepatocyte apoptosis (Hoekstra 1999; Tsugane et al. 1999) via mitochondria-dependent mechanisms including an increase in reactive oxygen species production and cytochrome c release (Garcia-Ruiz et al. 1997; Deaciuc et al. 2000). However, the mechanism of alcohol-induced cell apoptosis mediated by ceramide is still insufficiently studied.
It was also suggested that ceramide could inhibit AMP-activated protein kinase (AMPK) and promote local hepatocellular injury via intensification of the ER stress and apoptosis (Anderson and Borlak 2008; Liangpunsakul et al. 2010; Kuznetsov et al. 2011; Yang et al. 2016). ER stress promotes oxidative injury and inflammation, which might contribute to the progression of alcoholic liver disease. Ceramide accelerates oxidative stress and pro-apoptotic signaling resulting in reactive oxygen species formation, an increase in lipid peroxidation and in cell death. In turn, reactive oxygen species and inflammatory mediators could increase ceramide generation (Lim et al. 2011). It was hypothesized that ASM could induce ER stress reflected by an up-regulation of ER stress markers in both HepG2 cells and primary hepatocytes. Currently, it is not clear whether NSM contributes to this pathway. Cultivation of HepG2 cells with exogenous ASM or NSM is followed by an increase in ceramide levels and, in case of ASM, but not NSM, a rise in the levels of ER stress markers. Another study has shown that exogenous ASM, but not NSM increases the expression of ER stress markers (Garcia-Ruiz et al. 2000). Furthermore, inhibition of ASM with amitriptyline prevents alcohol-induced ER stress. Interestingly, ASM KO mice are also resistant to alcohol-induced ER stress and alcohol-mediated sensitization of the liver to lipopolysaccharide effects (Fernandez et al. 2013; Yang et al. 2016).
In HepG2 cells, ASM decreases thapsigargin-induced ER Ca2+ release, indicating lower ER Ca2+ storage. The authors suggest that alcohol-induced ASM activation and ER stress, at least in part, by disrupting ER Ca2+ homeostasis (Yang et al. 2016). The ER structure was severely disrupted in alcohol-exposed liver cells, accompanied by increased mRNA levels of ER stress-related genes (Longato et al. 2012; Ramirez et al. 2013). ER stress may play a critical role in alcoholic liver disease, as it regulates hepatic steatosis and liver injury (Ji and Kaplowitz 2006; Fernandez et al. 2013; Yang et al. 2016). Thus, the ceramide system significantly contributes to alcohol-induced injuries and apoptosis (Yang et al. 2016).
There may be a functional relationship between ASM and cathepsins, molecules triggering apoptosis (Chwieralski et al. 2006). Cathepsin D is a lysosomal target of ceramide (Heinrich et al. 1999). The differentiation of primary mouse hematopoietic stem cells (HSCs) into myofibroblast-like cells is associated with selective ASM stimulation and a cathepsin B and D increase. Inhibition of ASM is followed by the disruption of proliferation of mouse and human hepatic stellate cells (Moles et al. 2010; Yang et al. 2016). Thus, cathepsin-induced apoptosis might serve as another pathway of alcohol-induced cell loss mediated by ceramide.
Taken together, ceramides are shown to contribute to the peripheral effects of alcohol. Ceramides might serve as a key mechanism for alcohol-induced apoptosis in cells of the liver, pancreas, and other peripheral organs. Recent investigations have unraveled a few possible molecular cascades involved in the initiation of alcohol-induced and ceramide-mediated apoptosis. Besides the enhancement of oxidative stress, which is tightly bound to the molecular effects of ceramide (Park et al. 2011), other molecular targets such as cathepsins and Ca2+ have been observed. However, further investigations are required to understand the mechanisms of ceramide-mediated apoptotic effects of alcohol.
Ceramides in the central nervous system
A study of Roux et al. (2016) compared alcohol effects on SL levels in the whole brain of juvenile and adult rodents. It was found that ceramide levels, especially Cer16:0, were significantly increased after long-term alcohol drinking in adult animals, but were either decreased or unchanged in the juvenile animals. Alcohol consumption decreases Cer18:0 level in adults, but increases it in juvenile animals (Roux et al. 2016). Thus, a shift in ceramide level is also typical for the central nervous system of animals exposed to alcohol treatment.
Similar results were observed in newborn mice after acute alcohol administration. Time course studies indicate that the levels of ceramide increase significantly in the cortex, hippocampus, and inferior colliculus of 7-day-old mice between 4 and 8 h after alcohol exposure. A robust apoptotic neurodegeneration was induced by acute administration of alcohol associated with ceramide accumulation. An increase in ceramide concentration was coincident with caspase-3 activation in the forebrain. It should be noticed that an increase in the amount of other lipids occurs at later time points mostly 24 h after alcohol exposure. Thus, ceramide may trigger apoptosis in the developing murine brain after alcohol treatment and be a possible missing point between alcohol consumption and development of neurodegenerative disorders (Jana et al. 2009; Saito et al. 2007, 2010).
Several investigations show that ceramide contributes to the development of apoptosis during fetal alcohol disorder. A single dose of alcohol injected to pregnant C57BL/6J mice at gestational day 15–16 resulted in enhanced ceramide and sphingosine amounts in the brain of the offspring. These SL might be a potential mediator of fetal alcohol-induced neuronal loss (Dasgupta et al. 2007). Dasgupta and Hogan (2001) proposed that alcohol-mediated sphingosine toxicity is one possible mechanism of neuronal apoptosis in the brain of progeny mice(Dasgupta et al. 2007). Sphingosine was shown to be cytotoxic in cultured cells (Hannun and Bell 1989), although recent studies did not detect toxicity of even high sphingosine concentrations in epithelial cells (Grassmé et al. 2017; Martin et al. 2017). Acute alcohol administration to mice on postnatal day 7, equivalent to the late third trimester in humans (Godfrey et al. 2015), elevated levels of ceramide in several brain areas such as the cortex, inferior colliculus, and hippocampus. As in adult mice, in pups, these changes were accompanied by an increase in the level of caspase 3, an enzymatic marker of apoptosis. Since alcohol in these animals directly inhibits SPT, it was suggested that the de novo pathway significantly contributes to the development of alcohol-induced apoptosis, particularly in neurons (Olney et al. 2002; Godfrey et al. 2015). These data are confirmed by in vitro studies. The exposure of rat cerebral granule neurons to alcohol results in a ceramide accumulation. Myriocin inhibits the alcohol-induced ceramide elevation, thus, confirming that the effects of alcohol on SL content in these cells are mediated by the de novo pathway (Saito et al. 2005). In vitro experiments using SPT inhibitors indicate the involvement of de novo ceramide synthesis in various apoptotic pathways such as retinoic acid-induced apoptosis in PCC7-Mz1 cells, Aβ-induced toxicity in hippocampal neurons, oxidative and excitotoxic insults in motor neurons, and hypoxia-induced apoptosis in SHSY5Y cells (Herget et al. 2000; Cutler et al. 2002, 2004; Kang et al. 2009; Saito et al. 2010). However, the activation of sphingomyelinase—and the salvage pathway of ceramide synthesis cannot be excluded yet, as SPT inhibitors do not suppress alcohol-induced caspase-3 activation completely (Saito et al. 2005).
The CerS inhibitor fumonosin B1 did not affect ceramide levels in astrocytes and also not the alcohol-induced increase in ceramide level. Alcohol-induced apoptosis in astrocytes also did not change after fumonosin B1 treatment. However, palmitate, a biosynthetic ceramide precursor, increased ceramide and ceramide-mediated apoptosis in control and alcohol-treated cells. The FIASMA desipramine attenuated the alcohol-induced enhancement in ceramide concentration and in apoptosis (Pascual et al. 2003). Thus, it might be suggested that both, de novo and sphingomyelinase pathways of ceramide synthesis, are implicated in alcohol-dependent accumulation of ceramide and subsequent apoptosis.
Thus, under physiological conditions, SLs such as ceramide are important physiological modulators of normal neuronal development, differentiation, and apoptosis. However, increased levels of these lipids induced by a variety of intra- and extracellular stimuli are implicated in pathological apoptosis of neurons and oligodendrocytes in neuroinflammatory and neurodegenerative disorders such as Alzheimer’s disease, HIV-associated dementia, multiple sclerosis, amyotrophic lateral sclerosis, stroke, and particularly, alcohol-induced central nervous system damage (de la Monte et al. 2009; Saito et al. 2010; Chakraborty et al. 2012; Godfrey et al. 2015). Several studies suggest that the ceramide system is a mediator of alcohol-induced apoptosis. Neurodegeneration, white matter atrophy, and increased levels of oxidative stress in human alcoholics are associated with increased expression of genes promoting ceramide production (de la Monte et al. 2008).
Protective properties of alcohol are mediated by ceramides
Even though ceramide seems to be tightly involved in the development of alcohol-induced neuronal loss and following neurodegeneration, some studies indicate also protective effects of ceramide after alcohol treatment. Experiments of Bae et al. (2014) showed that acute alcohol-induced reduction in ceramides is consistent with a neuroprotective phenotype. However, an increase in ceramide and a decrease in sphingomyelin levels during withdrawal are associated with a neurotoxic phenotype. Binge alcohol drinking is accompanied by a decrease in the levels of Cer26:0, Cer16:1, Cer18:1, Cer20:1, and Cer22:1 species in the frontal cortex of rodents. An increase in the expression of SMS1 and SMS2 and ACER suggests that ceramide is probably converted to SM and sphingosine/S1P. As an inhibition of ceramide production and an increase in S1P were shown to be protective in a variety of model systems (Culmsee et al. 2002; Haughey et al. 2004; Yung et al. 2012), these data suggest that a shift in ceramide metabolism toward S1P may protect the central nervous system during alcohol intoxication. However, acute withdrawal after binge drinking is followed by a significant increase in the concentrations of Cer16:0, Cer18:0, and Cer20:0 species, which is mostly driven by an enhanced expression of CerS2 and CerS4 (Bae et al. 2014). Although the authors have not identified the neural cell types affected by alcohol, they suggest that astroglia and neurons of the brain may be particularly vulnerable to the toxic effects of alcohol via ASM and NSM-mediated pathways or the de novo pathway of ceramide synthesis (De Vito et al. 2000; Pascual et al. 2003; Schatter et al. 2005; Bae et al. 2014). It was proposed that alcohol withdrawal might induce inflammatory and stress pathways that promote the catabolic formation of ceramide in astrocytes, while de novo ceramide production may be the preferred metabolic pathway for ceramide production in neurons during withdrawal (Bae et al. 2014).
Wang et al. (2013) have shown that prenatal exposure to alcohol induces compensatory neural proliferation and the formation of newborn neurons. Daily intragastrical administration of 25% alcohol to dams promotes an increase in the neural proliferation and the number of newborn neurons in the dental gyrus of pups. Authors speculate that neural proliferation might be the result of a compensatory mechanism for neuroapoptosis induced by alcohol (Jiang et al. 2007; Wang et al. 2013). These data are confirmed by the discovery that the neural proliferation in SMS2 KO pups was enhanced after prenatal alcohol exposure. Thus, the accumulation of intracellular ceramide following the loss of the SMS2 gene results in subsequent compensatory cell proliferation. Neural proliferation is mainly achieved through its phosphorylated metabolite, ceramide-1-phosphate (C1P). The authors suggested that low expression level of protein kinase C (Huang et al. 1999) can be associated with the slowed-down ceramide/C1P cascade. In contrast, alcohol could induce the expression of protein kinase Cα. Thus, alcohol-induced accumulation of ceramide could upregulate neural proliferation. Protein kinase Cα phosphorylation is a key step for the activation of the ceramide pathway (Wang et al. 2013).
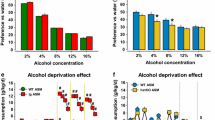
Similar findings on the involvement of ceramide in the protective properties of moderate voluntary alcohol consumption were observed by Müller et al. (2017). It is well known that moderate alcohol consumption reduces the risk for the development of mental disorders such as anxiety and depression (Peele and Brodsky 2000; Skogen et al. 2009). Moreover, moderate drinking, which can be maintained with a high degree of stability, is associated with better health, more close friendships, and more family support than total abstinence (Peele and Brodsky 2000; Skogen et al. 2009). The authors show that alcohol drinking has paradoxical antidepressant effects in initially depressed rodents with genetically enhanced ASM activity (Müller et al. 2017). Mice in which ASM hyperactivity (tgASM) causes a depressive phenotype (Gulbins et al. 2013; Jernigan et al. 2015), drink significantly more alcohol than the wild-type controls. Alcohol drinking is also more susceptible to withdrawal effects in tgASM animals. Interestingly, free-choice alcohol consumption, which allows for self-titration, selectively reduces depression-like behavior in initially depressed tgASM mice. Forced alcohol exposure, in contrast, enhances depression-like behavior in tgASM and wild-type mice alike. The paradoxical antidepressant effects of free-choice alcohol consumption in tgASM mice might be related to the changes in sphingolipid balance of the brain. In wild-type mice, free-choice alcohol drinking reduces the tissue levels of the most abundant SM species, SM18:0, SM18:1, and SM20:0, in the nucleus accumbens and dorsal hippocampus suggesting an increase in corresponding ceramide species. In tgASM mice which showed attenuated basal levels of these SM species, alcohol drinking had the opposite effect and partially reversed the SM decline, thus, at least partly, re-establishing SM homeostasis in the nucleus accumbens, but not in the dorsal hippocampus. In wild-type animals, alcohol drinking had no effect on ASM activity in the brain. In tgASM mice, ASM activity was reduced by free-choice drinking further supporting a drop in ceramide level in this brain structure. Another possible pathway for the paradox antidepressant properties of alcohol in tgASM mice might be mediated by brain monoamines, which may, however, be down-stream of SL homeostasis. Depression induced by ASM hyperactivity coincided with reduced tissue levels of serotonin and dopamine, but not norepinephrine, in several brain structures including the ventral striatum, dorsal hippocampus, and prefrontal cortex. A permanent decline of monoamine tissue levels is associated with depression, and may also render an organisms more prone to addiction (Krishnan and Nestler 2008; Carey et al. 2008; Carr and Lucki 2010; Müller and Homberg 2015). Serotonin and dopamine deficits were partially reversed by alcohol drinking in depressed tgASM mice. In wild-type mice, alcohol drinking by itself reduced dopamine and serotonin tissue levels. These findings suggest that re-establishing SL homeostasis may down-stream normalize also the serotonin- and dopamine deficits in the brain. The authors speculate that the nucleus accumbens may appear to be a brain region that is particularly sensitive to an SL-dopamine/serotonin interaction, and thus the alcohol effects on SL allostasis (Müller et al. 2017; Müller and Kornhuber 2017).
Experiments by Godfrey et al. (2015) have also shown that ceramides contribute to the beneficial cardioprotective and neuroprotective effects of moderate alcohol consumption. Selectively bred alcohol-preferring rats exposed chronically to alcohol intermittent access drinking showed significantly reduced level of total ceramide in the forebrain and in the heart. These data indicate that alcohol intake during chronic voluntary exposure may have favorable effects on lipid profiles, which may contribute to the beneficial cardioprotective and neuroprotective effects of moderate alcohol consumption (Standridge et al. 2004; Collins et al. 2009; Godfrey et al. 2015). The reduction in the activity of sphingolipid desaturase, Degs2, catalyzing the conversion of dihydroceramide to ceramide, and ACER1 might mediate alcohol-induced decrease in ceramide levels. However, the activity and expression rate of other enzymes, such as CerS or ceramidases, are not changed under these conditions. Therefore, it is proposed that the beneficial effects of voluntary drinking of moderate amounts of alcohol are possibly mediated by a reduction in ceramide synthesis (Godfrey et al. 2015).
Altogether, several investigations reflect the involvement of SLs in the development of alcohol use disorder and related to neurodegeneration and neuronal loss. SLs mediate the toxic effects of alcohol and may contribute to the mechanisms of alcohol addiction. However, another line of studies indicates the protective effects of ceramides against alcohol-induced pathological changes, mostly in the central nervous system. SLs are suggested to mediate beneficial effects of moderate alcohol consumption and might be involved in the mechanisms determining controlled alcohol consumption. These mechanisms might be based on the contribution of SLs to the development of memory traces associated with alcohol consumption, as it is known that ASM/ceramide system is tightly involved in the mechanisms of learning and particularly behavioral extinction (Huston et al. 2016).
Nicotine
Nicotine smoking is associated with the development of a variety of pulmonary diseases, which are shown to affect SL systems (Tibboel et al. 2013; Seitz et al. 2015). An accumulation of 168 ceramide and SM species with different chain lengths (28 ceramides, 11 dihydroceramides, 19 phytoceramides, 36 sphingomyelins, and 74 GSLs) was observed in the sputum of smokers with and without chronic obstructive pulmonary disease (Telenga et al. 2014). Similar findings were reported for the lung of cigarette smoke-exposed animals (Petrache et al. 2005; Zulueta et al. 2017). The association between enhanced ceramide levels and lung endothelial and epithelial cell apoptosis was shown in cigarette-smoking-exposed mice (Bodas et al. 2011; Filosto et al. 2011; Zulueta et al. 2017).
These data are in line with the changes observed in the central nervous system of rodents. The administration of one of the main tobacco-specific nitrosamines, nicotine-derived nitrosamine ketone (NNK), to rats was followed by an increase in PS36:1 phosphatidylserine in the cortex and hypothalamus and PS40:6 phosphatidylserine in medial temporal structures, in the cortex and hypothalamus. The mechanisms of smoking-induced changes in the ceramide system are currently unclear. Ceramide accumulation in patients with chronic obstructive pulmonary disease and in cigarette smoke-exposed mice is associated with increased activity of NSM and ASM, thus, reflecting an enhanced SM hydrolysis and ceramide production (Filosto et al. 2011; Lea et al. 2016). On the other hand, inhibition of ceramide synthesis by myriocin and XM462, acting on two different steps of de novo pathway, significantly reduced the outcome of smoking-induced inflammatory cascades, responsible for the induction of chronic damage in the lungs (Zulueta et al. 2017). Thus, ceramide might act as a major mediator of cigarette-smoking-induced damage the peripheral tissues and central nervous system.
Cocaine
In vitro studies showed that cocaine treatment of cultured Rat-1 fibroblasts induces an increase in total cell phospholipid content, including phosphatidylcholine, phosphatidylserine, and phosphatidylalcoholamine, but not phosphatidylinositol, lysophosphatidylcholine, and cholesterol. A dose-dependent decrease in lysosomal ASM activity can also be induced by cocaine. Cocaine withdrawal was accompanied by a full restoration of ASM activity within 16 h. The authors suggest that the proteolytic degradation of ASM by cocaine might be associated with structural alteration of the enzyme, unknown signal transduction processes (e.g., phosphorylation events) or modification of the lysosomal membrane permeability, resulting in a more pronounced susceptibility to degradation. It was speculated that cocaine and cocaine-induced lipidosis in lysosomes could induce membrane rupture followed by the activation of apoptosis-inducer caspase-3 (Nassogne et al. 2004). Thus, cocaine-induced neurodegeneration might be explained by membrane damage mediated by ceramide as one of the main compounds of biological membranes (Goñi et al. 2014). These data might unravel interesting new therapeutic approaches for the cocaine-induced neurodegenerative disorders by targeting the SL systems.
Methamphetamine
Self-administration of d-methamphetamine increases ceramide content in the frontal cortex, dorsal, and ventral striatum and, to a greater extent, in the heart, liver, and skeletal muscles of rats. The main effects were observed in the levels of Cer16:0, Cer18:0, Cer24:0, and Cer24:1. A d-methamphetamine-induced increase in the levels of SM16:0, the obligatory biosynthetic precursor of Cer16:0 through the de novo pathway (Gault et al. 2010), and no changes in the levels of SM16:0 or dihydrosphingomyelin 16:0, which generate ceramide through sphingomyelinase-mediated hydrolysis (Gault et al. 2010), reflect the main contribution of de novo pathway to ceramide accumulation. Consistent with this suggestion, d-methamphetamine self-administration was associated with an increase in the transcription of SPT1 and SPT2, and CerS2, CerS4, and CerS6 (Astarita et al. 2015).
Enhanced d-methamphetamine-induced ceramide production was associated with the enhanced expression of genes involved in cell-cycle control (e.g., p21 and p16) and chronic inflammation (e.g., IL-6 and TNF-α). The authors suggested that d-methamphetamine promotes the generation of free oxygen radicals via cytochrome P450 and nuclear factor-κB, which induce the expression of enzymes involved in the de novo pathway of ceramide biosynthesis, particularly SPT and CerS. In turn, ceramide may induce apoptosis, thus, possibly contributing to d-methamphetamine-induced neurotoxicity (Ellison 1992; Cherner et al. 2010). In line with this suggestion, l-cycloserine, myriocin, and fumonisin B1, inhibitors of SPT and CerS, can prevent d-methamphetamine-induced ceramide accumulation in primary mouse embryonic fibroblast cultures (Astarita et al. 2015). These findings suggest a contribution of SLs and especially ceramides to the neurotoxic effects of d-methamphetamine. Interestingly, as distinct from all previously described addictive drugs, methamphetamine-induced changes in SL content and associated cell injury are mostly driven by the de novo pathway. Further analysis of this feature is needed focusing in particular on SL effects in the brain and their contribution to addiction-related behaviors (Müller 2017a).
Morphine
Little is known about the contribution of ceramide to the development of opioid dependence. Studies on morphine-induced hyperalgesia and antinociceptive tolerance show that morphine treatment induces an up-regulation of ceramide in the periaqueductal gray and dorsal horn (Bryant et al. 2009; Ritter et al. 2012). This increase was associated with an enhanced activity of ASM, SPT, and CerS, indicating that both de novo and SM pathways are involved in these effects (Ndengele et al. 2009; Ritter et al. 2012). Moreover, the morphine-induced accumulation of ceramide results in the development of peroxynitrite-derived nitroxidative stress and neuroimmune activation including the activation of glial cells and increased production of TNFα, IL-1β, and IL-6. Inhibition of ceramide biosynthesis with pharmacological inhibitors (myriocin, D609, and FB1) significantly attenuated the increase in spinal ceramide production, nitroxidative stress, and neuroimmune activation (Ndengele et al. 2009). It was also shown that ceramide contributes to the development of morphine-induced apoptosis, which can be fully blocked by FB1 (Bryant et al. 2009). However, even though ceramide contributes to morphine effects, the increased ceramide generation and enzyme expression in the periaqueductal gray region and dorsal horn tissues is most likely involved in the analgesic action of morphine, which might differ from its role in addiction-related behaviors. Thus, further investigation of ceramide involvement in the development of morphine abuse is needed, as this SL seems to interact with the important pathways of drug dependence such as oxidative stress and activation of immune system. Altogether, accumulating evidence shows that various addictive substances increase ceramide activity in peripheral tissues and the brain. So far, this was mostly related to the aversive side effects and/or neurodegenerative effects of those drugs in the brain. Effects in the brain reward system that could contribute to neuroplasticity associated with controlled drug use and addiction are so far only shown for alcohol and await further characterization for other drugs of abuse.
Glycosphingolipids and addictive disorders
Gangliosides
Gangliosides are major the components of the neuronal membranes as they account for 10–12% of the lipid content (Posse de Chaves and Sipione 2010). They are located on the external leaflet of the plasma membrane and contribute to the maintenance of neuronal functions such as neuronal development and myelin stability (Palmano et al. 2015; Posse de Chaves and Sipione 2010). The four major brain gangliosides in the adult mammalian brain are GM1, GD1a, GD1b, and GT1b (Tettamanti 2004; Olsen and Færgeman 2017). It is well known that the ganglioside profile changes remarkably during early development of the nervous system as well as throughout life. These changes are region specific (Olsen and Færgeman 2017). Gangliosides have many biological functions as antigens, mediators of cell adhesion, and modulators of signal transduction (Ledeen and Wu 2002). They also play an important role in the development of apoptosis. Specifically, the involvement of GD3 in CD95/Fas-mediated apoptosis in lymphocytes has been extensively studied (Malorni et al. 2007; Saito et al. 2012). Although the majority of gangliosides are found in glycosphingolipid-enriched micro-domains (lipid rafts) of the plasma membrane (Hakomori 2003), GD3 accumulates within mitochondria of cells undergoing apoptosis (Rippo et al. 2000). The direct interaction of GD3 with mitochondria induces cytochrome c release and caspase-3 activation (García-Ruiz et al. 2002; Saito et al. 2012). Recent investigations reveal the involvement of gangliosides in the mechanisms of drug dependence.
Alcohol
It is known that alcohol treatment alters levels and profiles of gangliosides in several organ systems including the liver and blood plasma (Klemm and Foster 1986; Ruano et al. 1994; Senn et al. 1990; Hannuksela et al. 2002). Long-term alcohol ingestion for 35–56 days reduced the content of gangliosides in the small intestine of rats. The main changes are typical for GM3, a ganglioside accounting for nearly 70–80% of total intestinal gangliosides (Grewal and Mahmood 2010).
In line with these data, the alcohol-preferring line of mice C57BL/6ByJis characterized by a significantly lower concentration of another ganglioside GM1 in the serum, blood cells, and liver, but not in the cerebellum compared to a non-preferring BALB/cJ line. Interestingly, both acute alcohol treatment and subchronic oral self-administration of alcohol in an alcohol preference test paradigm reduced serum GM1 level in alcohol-preferring animals. However, acute alcohol treatment did not affect hepatic GM1 levels probably due to a higher turnover rate of serum GM1 compared to the liver plasma membranes (Dumontet et al. 1992). Chronic oral alcohol self-administration also reduced the GM1 content in the liver probably mediated by the inhibition of GM1 synthesis or the augmentation of GM1 degradation (Saito et al. 2004). On the other hand, long-term administration of GM1 for 21 days attenuated behavioral sensitization to alcohol measured as locomotion frequency of mice in the open-field test, but did not affect alcohol blood levels (Bellot et al. 1996). Thus, it can be suggested that peripheral GM1 might play an important role in the regulation of controlled alcohol use. However, further analysis of changes in GM1 level during voluntary drinking is needed to understand the role of central structures in this process.
As distinct from the peripheral tissues, an increase in GM3 levels in the frontal cortex was observed after the subchronic treatment with alcohol for 20 days. However, levels of GM1, GM2, GQ1b, GD1a, or GT1b were not affected by this type of treatment (Haselhorst et al. 1991).
Prenatal treatment with alcohol was accompanied by a decrease in whole brain GM1 levels. GD3 was decreased at postnatal day 5, then increased by day 15, and again decreased at day 21. Level of GM1 increased at postnatal days 15 and 21. A reduction in GM1 concentration indicates abnormal myelinogenesis, as GM1 is considered as a marker of myelin. GM1 was shown to promote in vitro neuritogenesis and in vivo re-myelination (Farooqui et al. 1997; Fang et al. 2000; Dasgupta et al. 2007).
In vitro studies show that alcohol treatment induces a decrease in ceramide long-chain base moiety in the rat cerebral granular cells, but does not affect the rate of de novo biosynthesis of gangliosides labeled in the oligosaccharide moiety. These data indicate that in the presence of alcohol, de novo biosynthesis of ganglioside long-chain base moiety strongly decreases, while the biosynthesis of the ganglioside oligosaccharide portion is almost unaffected. An alcohol-induced increase in the levels of radioactive GD1b and GT1b suggests that alcohol increases the sphingosine salvage pathway for ganglioside biosynthesis. The authors suggest that the alcohol-induced increase of sphingosine metabolism constitutes a mechanism of cell defense during alcoholic stress. Moreover, a decrease in ganglioside concentration in the cells induced by alcohol might also have important consequences on cell events such as growth, signal transduction, apoptosis, and proliferation (Ravasi et al. 2002). However, gangliosides do not react uniformly to alcohol administration. Both main pathways of ganglioside synthesis seem to contribute to alcohol-induced changes in ganglioside pattern of the brain and peripheral tissues.
Gangliosides may also contribute to alcohol-induced apoptosis. It was observed that prenatal alcohol treatment induces a wide-spread apoptotic neurodegeneration within 24 h associated with a significant increase in GM2 level in the brain of progeny mice. This increase was most pronounced in the cingulum and the cingulate/retrosplenial cortex. However, the levels of other major gangliosides, such as GD1a, GD1b, and GM1, were unchanged after prenatal alcohol exposure. The elevation of GM2 is observed initially in apoptotic neurons and later in activated microglia. The authors suggest that GM2 is present in both mitochondria and lysosomes/phagolysosomes of neurons and activated microglia in the alcohol-exposed brains, although GM2 expression is stronger in activated microglia than in neurons. Thus, GM2 elevation in mitochondria and GM2-induced cytochrome c release from mitochondria reflect the involvement of GM2 in alcohol-induced mitochondrial apoptosis (Saito et al. 2012). It can be suggested that as well as ceramides, an increase in the levels of some gangliosides in the brain after alcohol treatment might mediate the neurodegenerative effects of this substance.
In contrast, another ganglioside, GM1, appears to have protective properties against alcohol-induced adverse effects in the central nervous system. The addition of GM1 to culture media or in vivo administration to rodents reduces the teratogenic/toxic effects of alcohol (Hungund and Mahadik 1993; Hungund et al. 1994). GM1 reverses alcohol-induced growth retardation and alteration of morphological properties in cortical cells (Laev et al. 1995). GM1 provided protection against alcohol neurotoxicity in hippocampal neurons of fetal rats and in dorsal-root ganglion neurons from embryonic chicks (Heaton et al. 1994). Pretreatment with gangliosides and LIGA20, a semisynthetic derivative of GM1 ganglioside, attenuated alcohol-induced apoptosis and the following apoptotic processes, such as cell death, DNA fragmentation, and caspase-3 activation, in cultured rat cerebellar granule neurons. The effects of GD1b and GT1b under these conditions were more pronounced than the effects of GM1 (Saito et al. 1999).
Similar findings were obtained by in vivo models. LIGA20 and GM1 partially reduced alcohol-induced wide-spread neurodegeneration and activation of caspase-3 in the cingulate and retrosplenial cortices of mice. The attenuation of alcohol-induced apoptotic neurodegeneration by these substances might be related to the regulation of growth factors and neurotrophic factors by gangliosides (Hakomori et al. 1998; Ledeen et al. 1998; Saito et al. 2007). It was shown that GM1 activates Trk-type receptors in cultured neurons (Bachis et al. 2002; Duchemin et al. 2002). Alcohol induces impairments in the functions of neurotrophic factors including brain-derived neurotrophic factor (BDNF) via the blockade of NMDA receptors (Spanagel 2009). This leads to a Bax-dependent activation of caspase-3. The activation of Trk receptors by GM1 stimulates survival pathways such as the PI3K/Akt pathway (Yao and Cooper 1995) and the extracellular signal-regulated kinase pathway (Climent et al. 2002). Thereby, GM1 protects against alcohol-induced apoptotic neurodegeneration. Authors speculate that GM1 and LIGA20 also regulate intracellular Ca2+ levels (Ledeen and Wu 2006; Saito et al. 2007), which are known to have a critical role in apoptosis and are disrupted by alcohol through the blockade of NMDA receptors (Saito et al. 2007). This, under certain conditions of alcohol treatment gangliosides might have neuroprotective and anti-apoptotic effects at least partly mediated by growth and neurotrophic factors. These data might provide an input in the hypothesis on SL contribution to positive effects of alcohol instrumentalization described previously.
Altogether, accumulating evidence suggests that gangliosides are involved in the regulation of both, controlled alcohol consumption and toxic effects of alcohol. Opposite effects of various gangliosides, particularly neuroprotective effects and pro-apoptotic properties, might determine the contribution of SLs to those effects. It might be suggested that the transition from controlled alcohol use to alcohol addiction is at least partly mediated by the changes in the synthesis pattern of gangliosides.
Cocaine
An accumulation of complex gangliosides such as GM1, GD1a, GD1b, GT1b, and GQ1b, and a reduction of precursors GM3, GM2, GD3, and GD2 in the rat liver were observed after repeated cocaine administration. The authors suggest that the observed changes in ganglioside synthesis are mediated by an alteration of vesicular transport from cis- to trans-Golgi cisternae, induced by either cocaine or its metabolites (Cabello et al. 1994).
Prenatal treatment with cocaine was followed by a significant elevation of whole brain content of neutral glycosphingolipids and total gangliosides in 1-day-old mice (Leskawa et al. 1994). Elevated levels returned to control value already at postnatal day 11 (Leskawa et al. 1994).
Experiments by Valdomero et al. (2010) showed that GM1 pretreatment 2 h before cocaine injection, which did not have any effect by itself, increased the rewarding effect of cocaine in the conditioned place preference test (Huston et al. 2013). This enhancement correlated with a significant increase in the brain’s cocaine level. However, pharmacokinetic parameters, such as plasma bound/free cocaine ratio, permeability of the blood–brain barrier for cocaine, and activity of brain and serum acetylcholinesterase and butyrylcholinesterase, were not changed by GM1. It also did not influence the inhibitory effect of cocaine on the dopamine transporter, a major mediator of cocaine addictive properties in the brain. A follow-up study showed that GM1 administration induces a significant increase in BDNF protein levels in the nucleus accumbens, which might explain the increased rewarding effects of cocaine in the conditioned place preference test (Valdomero et al. 2015). Similar findings have been observed by Lim et al. (2011), demonstrating an enhanced release of mature BDNFin hippocampal neurons and both mature BDNF and pro-BDNF in human neuroblastoma cells after GM1 treatment. Moreover, GM-1 also partially reversed the anti-proliferative effect of cocaine in PC12 cells. It was shown that GM1 can act as a neurotrophic factor supporting the development of some midbrain dopaminergic neurons (Tosk et al. 1996). Thus, it can be suggested that an increase in GM1 level observed in the brain after repeated cocaine administration might serve as a mechanism for the development of cocaine addiction as it enhances the rewarding properties of this drug. Interestingly, this effect of GM1 is mediated by BDNF, which is considered as a brain-protective molecule. Further analysis of GM1-induced involvement of BDNF in cocaine dependence is warranted.
Amphetamine
Subchronic treatment with amphetamine for 20 days resulted in an increase in GQ1b, but not GD1a, GM2, or GM3 concentrations in the brain of rats (Haselhorst et al. 1991). Bellot et al. (1997) showed that repeated administration of the exogenous ganglioside GM1 for 7 days does not affect the locomotor activity behavior of mice, but decreases amphetamine-induced hyperlocomotion. However, acutely administered GM1 does not affect amphetamine-induced locomotion. The authors proposed that ganglioside treatment may affect synaptic plasticity, thus, modifying the induction of the adaptive changes following drug treatment. The GM1-induced attenuation of the development of behavioral sensitization to the locomotor-stimulant effect of amphetamine might be mediated by the influence of this ganglioside on the dopaminergic system. This view is supported by the observation that GM1 has neuroprotective effects in different lesion models of the dopaminergic nigrostriatal pathway (Jackson et al. 1989; Schneider and DiStefano 1995). Supersensitivity of dopaminergic neurons developed after the treatment combination haloperidol/GM1, measured in the open-field test and by apomorphine-induced stereotypy (Vital et al. 1995). GM1 administered after haloperidol withdrawal enhanced the increases in both, general activity in the open field and apomorphine-induced stereotypies (Bellot et al. 1997). Thus, it can be suggested that GM1 affects synaptic plasticity in the brain of rodents, facilitate the induction of the adaptive changes in receptor function, and, thus, exerts protective effects at various stages in the development of amphetamine dependence.
Interestingly, as practically all types of drug addiction are mediated by dopaminergic system (Volkow et al. 2017), the effects of gangliosides on this neurotransmitter might be critical for understanding of the role of these lipids in the mechanisms of drug dependence. Further detailed analysis of molecular mechanisms of ganglioside effects during dependence states is needed. These data might explain the contribution of gangliosides not only to amphetamine dependence, but also to all other types of drug abuse.
Morphine
It is now agreed that gangliosides contribute to the development of morphine tolerance and antinociception (Crain and Shen 2000, 2007; Mayer et al. 1995; Anghelescu et al. 2015). Moreover, some authors suggested that these effects might also play an important role in morphine dependence (Crain and Shen 1998). Treatment of dorsal-root ganglion neurons with GM1 in low doses induced an opioid excitatory supersensitivity, manifested by a decrease in the threshold concentration of a κ-opioid peptide dynorphin (1–13) and morphine required to prolong action-potential duration. Other gangliosides, such as GD1a, GD1b, GQ1b, GM2, and GM3, did not have these effects. The efficiency of opioid excitatory receptor functions after GM1 treatment increased in a way that even a weak partial agonist, naloxone, became effective in prolonging action potentials in these neurons. However, antagonist properties of naloxone at opioid receptors were not altered by GM1. The authors suggested that the effects of GM1 are related to the enhanced binding activity of naloxone to Gs-coupled opioid receptors. GM1 binds to opioid receptors and induces a conformational change in the receptor–G-protein association, thus resulting in switching of receptor-coupling from normal inhibitory Gi-coupled signaling mode to an excitatory Gs-coupled signaling mode. These data indicate that GM1 is required for the development of cellular manifestations of tolerance and physical dependence (Crain and Shen 1998, 2000, 2007). Systemic administration of the CTX-B subunit of cholera toxin, which selectively binds GM1 at the plasma membrane, also enhances morphine’s analgesic potency and blocks opioid tolerance (Crain and Shen 2004).
However, it should be noticed that these effects are typical only for GM1 in low doses. High doses of GM1 attenuated the development of morphine tolerance and dependence (Shen and Crain 1990). GM1 in high doses blocked the translocation of protein kinase C from the cytosol to the neuronal surface membranes (Shen and Crain 1990; Crain and Shen 2004).
Thus, GM1 in various concentrations is shown to have different effects on cell response to morphine. Further analysis of thin mechanisms of these differences might not only unravel the pathways of morphine dependence mediated by SLs, but also propose new possible treatment strategies for this disorder.
Sulfatides
Sulfatides (ST) are glycosphingolipids composed of ceramide, galactose, and sulfate. They are expressed in many organs such as the brain, kidney, liver, heart, intestine, muscle, and pancreas (Kyogashima 2004; Hara et al. 1993). Together with galactosylceramides, STs are crucial myelin lipids required for the stability and maintenance of the myelin sheet (Schmitt et al. 2015). They are also expressed in neurons and astrocytes of the gray matter of the brain (Pernber et al. 2002; Isaac et al. 2006; Blomqvist et al. 2017). Sulfatides contribute to a variety of cellular processes including protein trafficking, cell adhesion, aggregation, and immune responses (Xiao and Finkielstein 2013). Sulfatide release from myelin is associated with the development of neurological diseases such as early Alzheimer’s disease (Cheng et al. 2013), as the presence of ST in the cerebrospinal fluid might serve as a marker of demyelination, myelin damage, and myelin turnover (Cheng et al. 2013; Blomqvist et al. 2017). It is known that chronic drug consumption, e.g., alcoholism, causes neurodegeneration with major impairment in the structural integrity of myelin (Yalcin et al. 2017). Thus, the investigation of sulfatide involvement in the development of drug dependence is recently of specific interest.
Alcohol
A study by Yalcin et al. (2015) revealed a decrease in the content of ST18:0, ST24:0, ST24:1 and PI36:4, PI36:1; PI38:4, PI38:3 phosphatidylinositols in the brain of rats receiving alcohol p.o. and i.p. in an 8-week protocol (Yalcin et al. 2015). Middle and high doses, but not low-dose alcohol treatment significantly reduced serum and liver ST levels via down-regulation of cerebroside sulfotransferase (CST) activity in the liver. CST is a key enzyme of ST synthesis catalyzing the final step in the synthesis by transferring the sulfate from 3′-phosphoadenosine 5′-phosphosulfate to galactocerebroside. CST mRNA levels were reduced by the middle- and high-dose alcohol, but not by low-dose alcohol treatment. However, mRNA of arylsulfatase A, an enzyme catalyzing ST degradation, was not affected by alcohol in any of the study doses. The authors suggest that the pronounced decrease in the expression of catalase and superoxide dismutases and increase in the intensity of lipid peroxidation in the liver of alcohol-treated mice are associated with CST suppression in the liver (Kanbe et al. 2014). These data might suggest an interaction between various pathways of alcohol use disorder, such as oxidative stress and alterations in the SL systems. Altogether, a decrease in ST levels in the brain of alcohol-treated mice might reflect neurodegenerative processes. However, these changes are not in line with a reduction in ST concentration in the peripheral tissues, as demyelinisation processes is often associated with an increase in peripheral ST. Further investigation of these changes is needed.
Nicotine
Administration of nicotine-derived nitrosamine ketone induced a decrease in the expression of ST24:0 and ST24:1(OH) in the corpus callosum, cortex, and hypothalamus of rats. This decrease correlated with white matter demyelination or hypo-myelination. The authors suggested that nicotine-derived nitrosamine ketone-induced axonal loss is mediated by the reduction in ST concentration in those brain structures (Yalcin et al. 2015). These data provide evidence for a contribution of STs to the toxic effects of cigarette smoking, particularly smoking-induced neuroapoptosis. Altogether, these data indicate that ST is particularly important for neurodegenerative processes induced by drug consumption. Further investigations of this pathway of neuron demyelinisation should be performed to understand if modulation of ST concentration might be used for the prevention of drug-induced neurodegeneration associated with cognitive impairment.
Cerebrosides
Cerebrosides constitute a family of neutral glycosphingolipids containing a 3-O-acetyl-sphingosine galactosylceramide consisting of two main subgroups—galactosylceramides and glucosylceramides. They are the most abundant glycolipid components of myelin enriched in both central and peripheral nervous system myelin. The average GalCer concentration is around 20% for the central and around 30% for the peripheral nervous system (Dasgupta et al. 2002; Podbielska et al. 2011).
Alcohol
An increase in the levels of cerebrosides, glucosylceramide, lactosylceramide, and globotriaosylceramide was found in microvilli membranes of the intestine of rats chronically fed with alcohol for 35 or 56 days (Grewal and Mahmood 2010). The authors suggested that an increase in the concentrations of these SLs in the intestine of rodents might be related to the dystrophic changes in the intestinal epithelium typical for alcohol consumers. Long-term exposure to alcohol resulted in extensive mucosal damage, including thinning of brush borders, shortening of villi, and reduced mucosal surface in the intestine (Bode and Bode 2003; Peres et al. 2004). Moreover, alcohol consumption was associated with a compromised immune status of the intestine and an increased incidence of general infection by facilitation of bacterial passage through the intestinal epithelium (Bode and Bode 2003). These changes in the immune status of epithelial cells of the intestine are considered to be at least partly mediated by increased levels of ceramides, which form an activating loop with pro-inflammatory cytokines such as IL-6 and TNF-α (Józefowski et al. 2010; Hamada et al. 2014; Gomez-Muñoz et al. 2016). These data suggest an involvement of cerebrosides in the peripheral toxic effects of alcohol.
Conclusions
In summary, numerous studies in recent years revealed new pathogenesis pathways involved in the action of addictive drugs. It was shown that SLs are tightly involved in the pathogenesis of addictive disorders. As such, they might be interesting as novel targets for the treatment of this disease. One of the most important findings is related to the apoptotic neurodegeneration typical for many addictive drugs. Especially, for alcohol use disorder, this seems to be determined by SLs. Particularly, ceramides and gangliosides mediate the imbalance of neuronal death and proliferation induced by psychoactive substances. Single investigations pointed out a role of SLs in plastic processes in the brain that are required for the normal development of drug-related behavior. Interestingly, an increasing number of studies showed dual properties of SLs during moderate alcohol consumption and revealed beneficial effects of these lipids in the central nervous system. These data allow proposing that SLs contribute to the specific type of controlled consumption of psychoactive substances named drug instrumentalization, in which beneficial and adverse effects of a drug can still be balanced (Müller and Schumann 2011a, b). However, during long-term intake of addictive drugs, these classes of lipids might also play a role in the transition to addiction, where adverse drug effects dominate.
Abbreviations
- alkSM:
-
Alkaline sphingomyelinase
- AMPK:
-
AMP-activated protein kinase
- ASM:
-
Acid sphingomyelinase
- ASM KO:
-
ASM knockout mice
- BDNF:
-
Brain-derived neurotrophic factor
- C1P:
-
Ceramide-1-phosphate
- cAMP:
-
Cyclic adenosine monophosphate
- Cer:
-
Ceramide
- CerD:
-
Ceramidase
- CerS:
-
Ceramide synthases
- CST:
-
Cerebroside sulfotransferase
- ER:
-
Endoplasmic reticulum
- FIASMA:
-
Functional inhibitor of ASM
- GluCer:
-
Glucosylceramide
- GSL:
-
Glycosphingolipids
- IL:
-
Interleukin
- LacCer:
-
Lactosylceramide
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NMDA:
-
N-methyl-d-aspartate
- NNK:
-
Nicotine-derived nitrosamine ketone
- NSM:
-
Neutral sphingomyelinase
- S1P:
-
Sphingosine-1-phosphate
- Scd-1:
-
Stearoyl-CoA desaturase-1
- SK:
-
Sphingosine kinase
- SL:
-
Sphingolipids
- SM:
-
Sphingomyelin
- SMPD:
-
Sphingomyelin phosphodiesterase
- SMS:
-
Sphingomyelin synthase
- SPL:
-
Sphingosine-1-phosphate lyase
- SPP:
-
Sphingosine-1-phosphate phosphatase
- SPT:
-
Serine palmitoyltransferase
- ST:
-
Sulfatides
- tgASM:
-
Transgenic mice with ASM hyperactivity
- TNFα:
-
Tumor necrosis factor α
References
Abel EL (1980) Marihuana: the first twelve thousand years. Plenum Press, New York
Adibhatla RM, Hatcher JF, Dempsey RJ (2006) Lipids and lipidomics in brain injury and diseases. AAPS J 8(2):E314–E321
Airola MV, Hannun YA (2013) Sphingolipid metabolism and neutral sphingomyelinases. Handb Exp Pharmacol 215:57–76
Anderson P, Baumberg B (2006) Alcohol in Europe. Institute of Alcohol Studies, Luxembourg
Anderson N, Borlak J (2008) Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev 60(3):311–357
Anghelescu DL, Goldberg JL, Faughnan LG, Wu J, Mao S, Furman WL, Santana VM, Navid F (2015) Comparison of pain outcomes between two anti-GD2 antibodies in patients with neuroblastoma. Pediatr Blood Cancer 62(2):224–228
Astarita G, Avanesian A, Grimaldi B, Realini N, Justinova Z, Panlilio LV, Basit A, Goldberg SR, Piomelli D (2015) Methamphetamine accelerates cellular senescence through stimulation of de novo ceramide biosynthesis. PLoS One 10(2):e0116961
Aureli M, Grassi S, Prioni S, Sonnino S, Prinetti A (2015) Lipid membrane domains in the brain. Biochim Biophys Acta 1851:1006–1016
Bachis A, Rabin SJ, Del Fiacco M, Mocchetti I (2002) Gangliosides prevent excitotoxicity through activation of TrkB receptor. Neurotox Res 4:225–234
Bae M, Bandaru VV, Patel N, Haughey NJ (2014) Ceramide metabolism analysis in a model of binge drinking reveals both neuroprotective and toxic effects of alcohol. J Neurochem 131(5):645–654
Bellot RG, Camarini R, Vital MABF, Palermo-Neto J, Leyton V, Frussa-Filho R (1996) Monosialoganglioside attenuates the excitatory and behavioural sensitization effects of alcohol. EurJ Pharmacol 313:175–179
Bellot RG, Vital MA, Palermo-Neto J, Frussa-Filho R (1997) Repeated monosialoganglioside administration attenuates behavioral sensitization to amphetamine. Brain Res 747(1):169–172
Blasco H, Veyrat-Durebex C, Bocca C, Patin F, Vourc’h P, Kouassi Nzoughet J, Lenaers G, Andres CR, Simard G, Corcia P, Reynier P (2017) Lipidomics reveals cerebrospinal-fluid signatures of ALS. Sci Rep. 7(1):17652
Blomqvist M, Borén J, Zetterberg H, Blennow K, Månsson JE, Ståhlman M (2017) High-throughput analysis of sulfatides in cerebrospinal fluid using automated extraction and UPLC-MS/MS(S). J Lipid Res 58:1482–1489
Bodas M, Min T, Vij N (2011) Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. Am J Physiol Lung Cell Mol Physiol 300:L811–L820
Bode C, Bode JC (2003) Effect of alcohol consumption on the gut. Best Pract Res Clin Gastroenterol 17(4):575–592
Brodowicz J, Przegaliński E, Müller CP, Filip M (2017) Ceramide and its related neurochemical networks as targets for some brain disorder therapies. Neurotox Res. https://doi.org/10.1007/s12640-017-9798-6
Brown DA, London E (1998) Structure and origin of ordered lipid domains in biological membranes. J Membr Biol 164:103–114
Bryant L, Doyle T, Chen Z, Cuzzocrea S, Masini E, Vinci C, Esposito E, Mazzon E, Petrusca DN, Petrache I, Salvemini D (2009) Spinal ceramide and neuronal apoptosis in morphine antinociceptive tolerance. Neurosci Lett 463(1):49–53
Cabello J, Ruano MJ, Cabezas JA, Hueso P (1994) Cocaine exposure induces changes in the ganglioside content of rat liver. Biol Chem 375(12):817–819
Carey RJ, Huston JP, Müller CP (2008) Pharmacological inhibition of dopamine- and serotonin activity blocks spontaneous and cocaine-activated behavior. Prog Brain Res 172:347–360
Carr GV, Lucki I (2010) The role of serotonin in depression. In: Müller CP, Jacobs BL (eds) Handbook of the behavioral neurobiology of serotonin. Academic Press, London, pp 493–506
Carr RM, Peralta G, Yin X, Ahima RS (2014) Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol-fed mice. PLoS One 9(5):e97118
Chakraborty G, Saito M, Shah R, Mao RF, Vadasz C, Saito M (2012) Alcohol triggers sphingosine 1-phosphate elevation along with neuroapoptosis in the developing mouse brain. J Neurochem 121:806–817
Cheng H, Wang M, Li JL, Cairns NJ, Han X (2013) Specific changes of sulfatide levels in individuals with pre-clinical Alzheimer’s disease: an early event in disease pathogenesis. J Neurochem 127:733–738
Cherner M, Bousman C, Everall I, Barron D, Letendre S et al (2010) Cytochrome P450–2D6 extensive metabolizers are more vulnerable to methamphetamine-associated neurocognitive impairment: preliminary findings. J Int Neuropsychol Soc 16:890–901
Chwieralski CE, Welte T, Bühling F (2006) Cathepsin-regulated apoptosis. Apoptosis 11(2):143–149
Ciarlo L, Manganelli V, Matarrese P, Garofalo T, Tinari A, Gambardella L, Marconi M, Grasso M, Misasi R, Sorice M, Malorni W (2012) Raft-like microdomains play a key role in mitochondrial impairment in lymphoid cells from patients with Huntington’s disease. J Lipid Res 53(10):2057–2068
Climent E, Pascual M, Renau-Piqueras J, Guerri C (2002) Alcohol exposure enhances cell death in the developing cerebral cortex: role of brain-derived neurotrophic factor and its signaling pathways. J Neurosci Res 68:213–225
Clugston RD, Jiang H, Lee MX, Piantedosi R, Yuen JJ, Ramakrishnan R, Lewis MJ, Gottesman ME, Huang LS, Goldberg IJ, Berk PD, Blaner WS (2011) Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: a targeted lipidomic and gene expression study. J Lipid Res 52(11):2021–2031
Coant N, Sakamoto W, Mao C, Hannun YA (2017) Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul 63:122–131
Collins MA, Neafsey EJ, Mukamal KJ, Gray MO, Parks DA, Das DK, Korthius RJ (2009) Alcohol in moderation, cardioprotection, and neuroprotection: epidemiological considerations and mechanistic studies. Alcohol Clin Exp Res 33:206–219
Correnti JM, Juskeviciute E, Swarup A, Hoek JB (2014) Pharmacological ceramide reduction alleviates alcohol-induced steatosis and hepatomegaly in adiponectin knockout mice. Am J Physiol Gastrointest Liver Physiol 306(11):G959–G973
Crain SM, Shen KF (1998) Modulation of opioid analgesia, tolerance and dependence by Gs-coupled, GM1 ganglioside-regulated opioid receptor functions. Trends Pharmacol Sci 19(9):358–365
Crain SM, Shen KF (2000) Enhanced analgesic potency and reduced tolerance of morphine in 129/SvEv mice: evidence for a deficiency in GM1 ganglioside-regulated excitatory opioid receptor functions. Brain Res 856(1–2):227–235
Crain SM, Shen KF (2004) Neuraminidase inhibitor, oseltamivir blocks GM1 ganglioside-regulated excitatory opioid receptor-mediated hyperalgesia, enhances opioid analgesia and attenuates tolerance in mice. Brain Res 995(2):260–266
Crain SM, Shen KF (2007) Naloxone rapidly evokes endogenous kappa opioid receptor-mediated hyperalgesia in naïve mice pretreated briefly with GM1 ganglioside or in chronic morphine-dependent mice. Brain Res 1167:31–41
Culmsee C, Gerling N, Lehmann M, Nikolova-Karakashian M, Prehn JH, Mattson MP, Krieglstein J (2002) Nerve growth factor survival signaling in cultured hippocampal neurons is mediated through TrkA and requires the common neurotrophin receptor P75. Neuroscience 115:1089–1108
Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP (2002) Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol 52:448–457
Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA 101:2070–2075
Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381:800–803
Dasgupta S, Hogan EL (2001) Chromatographic resolution and quantitative assay of CNS tissue sphingoids and sphingolipids. J Lipid Res 42:301–308
Dasgupta S, Levery SB, Hogan EL (2002) 3-O-acetyl-sphingosine-series myelin glycolipids: characterization of novel 3-O-acetyl-sphingosine galactosylceramide. J Lipid Res 43(5):751–761
Dasgupta S, Adams JA, Hogan EL (2007) Maternal alcohol consumption increases sphingosine levels in the brains of progeny mice. Neurochem Res 32(12):2217–2224
de la Monte SM, Yeon JE, Tong M, Longato L, Chaudhry R, Pang MY, Duan K, Wands JR (2008) Insulin resistance in experimental alcohol-induced liver disease. J Gastroenterol Hepatol 23(8 Pt 2):e477–e486
de la Monte SM, Longato L, Tong M, DeNucci S, Wands JR (2009) The liver–brain axis of alcohol-mediated neurodegeneration: role of toxic lipids. Int J Environ Res Public Health 6(7):2055–2075
De Vito WJ, Xhaja K, Stone S (2000) Prenatal alcohol exposure increases TNFalpha-induced cytotoxicity in primary astrocytes. Alcohol 21:63–71
Deaciuc IV, Nikolova-Karakashian M, Fortunato F, Lee EY, Hill DB, McClain CJ (2000) Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol Clin Exp Res 24(10):1557–1565
Deevska GM, Nikolova-Karakashian MN (2017) The expanding role of sphingolipids in lipid droplet biogenesis. Biochim Biophys Acta 1862:1155–1165
Deevska GM, Rozenova KA, Giltiay NV, Chambers MA, White J, Boyanovsky BB, Wei J, Daugherty A, Smart EJ, Reid MB, Merrill AH, Nikolova-Karakashian M (2009) Acid sphingomyelinase deficiency prevents diet-induced hepatic triacylglycerol accumulation and hyperglycemia in mice. J Biol Chem 284:8359–8368
Dong S, Zhang R, Liang Y, Shi J, Li J, Shang F, Mao X, Sun J (2017) Changes of myocardial lipidomics profiling in a rat model of diabetic cardiomyopathy using UPLC/Q-TOF/MS analysis. Diabetol Metab Syndr 9:56
Duchemin AM, Ren Q, Mo L, Neff NH, Hadjiconstantinou M (2002) GM1 ganglioside induces phosphorylation and activation of Trk and Erk in brain. J Neurochem 81:696–707
Dumontet C, Rebbaa A, Portoukalian J (1992) Kinetics and organ distribution of [14C]-sialic acid-GM3 and [3H]-sphingosine-GM1 after intravenous injection in rats. Biochem Biophys Res Commun 189(3):1410–1416
Ellison G (1992) Continuous amphetamine and cocaine have similar neurotoxic effects in lateral habenular nucleus and fasciculus retroflexus. Brain Res 598(1–2):353–356
Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Murphy RC, Raetz CRH, Russell DV, Seyama Y, Shaw W, Shimizu T, Spener F, van Meer G, Van Nieuwenhze MS, White SH, Witztum JL, Dennis EA (2005) A comprehensive classification system for lipids. J Lipid Res 46:839–861
Fang Y, Wu G, Xie X, Lu ZH, Ledeen RW (2000) Endogenous GM1 ganglioside of the plasma membrane promotes neuritogenesis by two mechanisms. Neurochem Res 25:931–940
Fantini J, Barrantes FJ (2009) Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim Biophys Acta 1788:2345–2361
Farooqui T, Franklin T, Pearl DK et al (1997) Ganglioside GM1 enhances induction by nerve growth factor of a putative dimer of TrkA. J Neurochem 68:2348–2355
Fei W, Shui G, Zhang Y, Krahmer N, Ferguson C, Kapterian TS, Lin RC, Dawes IW, Brown AJ, Li P, Huang X, Parton RG, Wenk MR, Walther TC, Yang H (2011) A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet 7:e1002201
Fernandez A, Matias N, Fucho R, Ribas V, Von Montfort C, Nuño N, Baulies A, Martinez L, Tarrats N, Mari M, Colell A, Morales A, Dubuquoy L, Mathurin P, Bataller R, Caballeria J, Elena M, Balsinde J, Kaplowitz N, Garcia-Ruiz C, Fernandez-Checa JC (2013) ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J Hepatol 59:805–813
Fernandez-Checa JC, Colell A, Mari M, García-Ruiz C (2005) Ceramide, tumor necrosis factor and alcohol-induced liver disease. Alcohol Clin Exp Res 29(11):151S–157S
Filosto S, Castillo S, Danielson A, Franzi L, Khan E, Kenyon N, Last J, Pinkerton K, Tuder R, Goldkorn T (2011) Neutral sphingomyelinase 2: a novel target in cigarette smoke-induced apoptosis and lung injury. Am J Respir Cell Mol Biol 44:350–360
Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernkndez-Checa JC (1997) Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem 272:11369–11377
Garcia-Ruiz C, Mari M, Morales A, Colell A, Ardite E, Fernández-Checa JC (2000) Human placenta sphingomyelinase, an exogenous acidic pH-optimum sphingomyelinase, induces oxidative stress, glutathione depletion, and apoptosis in rat hepatocytes. Hepatology 32:56–65
García-Ruiz C, Colell A, Morales A, Calvo M, Enrich C, Fernández-Checa JC (2002) Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J Biol Chem 277(39):36443–36448
Gault CR, Obeid LM, Hannun YA (2010) An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688:1–23
Godfrey J, Jeanguenin L, Castro N, Olney JJ, Dudley J, Pipkin J, Walls SM, Wang W, Herr DR, Harris GL, Brasser SM (2015) Chronic voluntary alcohol consumption induces favorable ceramide profiles in selectively bred alcohol-preferring (P) rats. PLoS One 10(9):e0139012
Gomez-Muñoz A, Presa N, Gomez-Larrauri A, Rivera IG, Trueba M, Ordoñez M (2016) Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res 61:51–62
Goñi FM, Sot J, Alonso A (2014) Biophysical properties of sphingosine, ceramides and other simple sphingolipids. Biochem Soc Trans 42:1401–1408
Gowing LR, Ali L, Allsop S, Marsden J, Turf EE, Wes R, Witton J (2015) Global statistics on addictive behaviours: 2014 status report. Addiction 110:904–919
Grassmé H, Henry B, Ziobro R, Becker KA, Riethmüller J, Gardner A, Seitz AP, Steinmann J, Lang S, Ward C, Schuchman EH, Caldwell CC, Kamler M, Edwards MJ, Brodlie M, Gulbins E (2017) β1-Integrin accumulates in cystic fibrosis luminal airway epithelial membranes and decreases sphingosine, promoting bacterial infections. Cell Host Microbe 21:707–718
Grewal RK, Mahmood A (2010) The effects of alcohol administration on brush border membrane glycolipids in rat intestine. Alcohol 44(6):515–522
Groux-Degroote S, Guérardel Y, Delannoy P (2017) Gangliosides: structures, biosynthesis, analysis, and roles in cancer. ChemBioChem 18(13):1146–1154
Gulbins E, Dreschers S, Wilker B, Grassmé H (2004) Ceramide, membrane rafts and infections. J Mol Med (Berl) 82(6):357–363
Gulbins E, Palmada M, Reichel M, Lüth A, Böhmer C, Amato D, Müller CP, Tischbirek CH, Groemer TW, Tabatabai G, Becker KA, Tripal P, Staedtler S, Ackermann TF, van Brederode J, Alzheimer C, Weller M, Lang UE, Kleuser B, Grassmé H, Kornhuber J (2013) Acid sphingomyelinase/ceramide system mediates effects of antidepressant drugs. Nat Med 19:934–938
Hakomori S (2003) Structure, organization, and function of glycosphingolipids in membrane. Curr Opin Hematol 10(1):16–24
Hakomori S, Yamamura S, Handa AK (1998) Signal transduction through glyco(sphingo)lipids. Introduction and recent studies on glyco(sphingo)lipid-enriched microdomains. Ann N Y Acad Sci 845:1–10
Hamada Y, Nagasaki H, Fujiya A, Seino Y, Shang QL, Suzuki T, Hashimoto H, Oiso Y (2014) Involvement of de novo ceramide synthesis in pro-inflammatory adipokine secretion and adipocyte–macrophage interaction. J Nutr Biochem 25(12):1309–1316
Hannuksela ML, Liisanantti MK, Savolainen MJ (2002) Effect of alcohol on lipids and lipoproteins in relation to atherosclerosis. Crit Rev Clin Lab Sci 39(3):225–283
Hannun YA, Bell RM (1989) Function of sphingolipid and sphingolipid products in cellular regulation. Science 243:500–507
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signaling: lessons from sphingolipids. Nat Rev 9:139–150
Hara A, Kutsukake Y, Uemura KI, Taketomi T (1993) Anticoagulant activity of sulfatide and its anti-thrombotic effect in rabbit. J Biochem 113(6):781–785
Haselhorst U, Ghidoni R, Schenk H (1991) Changes of brain gangliosides in the frontal cortex of rats chronically treated with amphetamine, clozapine, haloperidol and alcohol. Biomed Biochim Acta 50(7):931–935
Hassan Z, Bosch OG, Singh D, Narayanan S, Kasinather BV, Seifritz E, Kornhuber J, Quednow BB, Müller CP (2017) Novel psychoactive substances—an update on use, abuse, behavioural effects and mechanisms of action. Front Psychiatry 8:152
Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J, Nath A, Mattson MP (2004) Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol 55:257–267
Heath DB (2000) Drinking occasions: comparative perspectives on alcohol and culture, Brunner/Mazel
Heaton MB, Paiva M, Swanson DJ, Walker DW (1994) Ethanol neurotoxicity in vitro: effects of GM1 ganglioside and protein synthesis inhibition. Brain Res 654(2):336–342
Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J, Krönke M, Schütze S (1999) Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J 18(19):5252–5263
Herget T, Esdar C, Oehrlein SA, Heinrich M, Schutze S, Maelicke A, van Echten-Deckert G (2000) Production of ceramides causes apoptosis during early neural differentiation in vitro. J Biol Chem 275:30344–30354
Hillard CJ (2005) Lipids and drugs of abuse. Life Sci 77(14):1531–1542
Hoekstra D (1999) Ceramide-mediated apoptosis of hepatocytes in vivo: a matter of nucleus? J Hepatol 31:161–164
Holthuis JC, Pomorski T, Raggers RJ, Sprong H, Van Meer G (2001) The organizing potential of sphingolipids in intracellular membrane transport. Physiol Rev 81(4):1689–1723
Huang HW, Goldberg EM, Zidovetzki R (1999) Ceramides modulate protein kinase C activity and perturb the structure of phosphatidylcholine/phosphatidylserine bilayers. Biophys J 77(3):1489–1497
Hungund BL, Mahadik SP (1993) Role of gangliosides in behavioral and biochemical actions of alcohol: cell membrane structure and function. Alcohol Clin Exp Res 17(2):329–339
Hungund BL, Ross DC, Gokhale VS (1994) Ganglioside GM1 reduces fetal alcohol effects in rat pups exposed to alcohol in utero. Alcohol Clin Exp Res 18(5):1248–1251
Huston JP, de Souza Silva MA, Topic B, Müller CP (2013) What’s conditioned in conditioned place preference? Trends Pharmacol Sci 34(3):162–166
Huston JP, Kornhuber J, Mühle C, Japtok L, Komorowski M, Mattern C, Reichel M, Gulbins E, Kleuser B, Topic B, De Souza Silva MA, Müller CP (2016) A sphingolipid mechanism for behavioral extinction. J Neurochem 137(4):589–603
Hyötyläinen T, Orešič M (2014) Systems biology strategies to study lipidomes in health and disease. Prog Lipid Res 55:43–60
Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG (1997) Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to alcohol in the rat. Hepatology 26:1530–1537
Isaac G, Pernber Z, Gieselmann V, Hansson E, Bergquist J, Mansson JE (2006) Sulfatide with short fatty acid dominates in astrocytes and neurons. FEBS J 273:1782–1790
Jackson EA, Jenner P, Marsden CD (1989) Behavioural and morphological changes following treatment with GM1 ganglioside of rats with an electrolytic lesion of the substantia nigra. Neuropharmacology 28:543–555
Jana A, Hogan EL, Pahan K (2009) Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci 278:5–15
Jenkins RW, Canals D, Hannun YA (2009) Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal 21(6):836–846
Jernigan P, Hoehn R, Grassmé H, Edwards MJ, Müller CP, Kornhuber J, Gulbins E (2015) Sphingolipids in major depression. Neurosignals 23:49–58
Ji C, Kaplowitz N (2006) ER stress: can the liver cope? J Hepatol 45:321–333
Jiang Q, Hu Y, Wu P, Cheng X, Li M, Yu D, Deng J (2007) Prenatal alcohol exposure and the neuroapoptosis with long-term effect in visual cortex of mice. Alcohol Alcohol 42(4):285–290
Józefowski S, Czerkies M, Łukasik A, Bielawska A, Bielawski J, Kwiatkowska K, Sobota A (2010) Ceramide and ceramide-1-phosphate are negative regulators of TNF-α production induced by lipopolysaccharide. J Immunol 185(11):6960–6973
Kanbe H, Kamijo Y, Nakajima T, Tanaka N, Sugiyama E, Wang L, Fang ZZ, Hara A, Gonzalez FJ, Aoyama T (2014) Chronic alcohol consumption decreases serum sulfatide levels by suppressing hepatic cerebroside sulfotransferase expression in mice. Arch Toxicol 88(2):367–379
Kang MS, Ahn KH, Kim SK, Jeon HJ, Ji JE, Choi JM, Jung KM, Jung SY, Kim DK (2009) Hypoxia-induced neuronal apoptosis is mediated by de novo synthesis of ceramide through activation of serine palmitoyltransferase. Cell Signal 22:610–618
Kimura T, Jennings W, Epand RM (2016) Roles of specific lipid species in the cell and their molecular mechanism. Prog Lipid Res 62:75–92
Klemm WR, Foster DM (1986) Alcohol, in a single pharmacological dose, decreases brain gangliosides. Life Sci 39(10):897–902
Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35(1):217–238
Kornhuber J, Tripal P, Reichel M, Mühle C, Rhein C, Muehlbacher M, Groemer TW, Gulbins E (2010) Functional inhibitors of acid sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem 26(1):9–20
Kornhuber J, Müller CP, Becker KA, Reichel M, Gulbins E (2014) The ceramide system as a novel antidepressant target. Trends Pharmacol Sci 35(6):293–304
Kornhuber J, Rhein C, Müller CP, Mühle C (2015) Secretory sphingomyelinase in health and disease. Biol Chem 396(6–7):707–736
Koybasi S, Senkal CE, Sundararaj K, Spassieva S, Bielawski J, Osta W, Day TA, Jiang JC, Jazwinski SM, Hannun YA, Obeid LM, Ogretmen B (2004) Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J Biol Chem 279:44311–44319
Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455:894–902
Krut O, Wiegmann K, Kashkar H, Yazdanpanah B, Kronke M (2006) Novel tumor necrosis factor-responsive mammalian neutral sphingomyelinase-3 is a C-tail-anchored protein. J Biol Chem 281(19):13784–13793
Kuznetsov JN, Leclerc GJ, Leclerc GM, Barredo JC (2011) AMPK and Akt determine apoptotic cell death following perturbations of one-carbon metabolism by regulating ER stress in acute lymphoblastic leukemia. Mol Cancer Ther 10:437–447
Kyogashima M (2004) The role of sulfatide in thrombogenesis and haemostasis. Arch Biochem Biophys 426(2):157–162
Laev H, Karpiak SE, Gokhale VS, Hungund BL (1995) In utero alcohol exposure retards growth and alters morphology of cortical cultures: GM1 reverses effects. Alcohol Clin Exp Res 19(5):1226–1233
Lang PA, Schenck M, Nicolay JP, Becker JU, Kempe DS, Lupescu A, Koka S, Eisele K, Klarl BA, Rübben H, Schmid KW, Mann K, Hildenbrand S, Hefter H, Huber SM, Wieder T, Erhardt A, Häussinger D, Gulbins E, Lang F (2007) Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat Med 13(2):164–170
Lea SR, Metcalfe HJ, Plumb J, Beerli C, Poll C, Singh D, Abbott-Banner KH (2016) Neutral sphingomyelinase-2, acid sphingomyelinase, and ceramide levels in COPD patients compared to controls. Int J Chron Obstr Pulmon Dis 11:2139–2147
Ledeen RW, Wu G (2002) Ganglioside function in calcium homeostasis and signaling. Neurochem Res 27(7–8):637–647
Ledeen RW, Wu G (2006) Gangliosides of the nuclear membrane: a crucial locus of cytoprotective modulation. J Cell Biochem 97:893–903
Ledeen RW, Wu G, Lu ZH, Kozireski-Chuback D, Fang Y (1998) The role of GM1 and other gangliosides in neuronal differentiation. Overview and new finding. Ann NY Acad Sci 845:161–175
Leskawa KC, Jackson GH, Moody CA, Spear LP (1994) Cocaine exposure during pregnancy affects rat neonate and maternal brain glycosphingolipids. Brain Res Bull 33(2):195–198
Liangpunsakul S, Sozio MS, Shin E, Zhao Z, Xu Y, Ross RA, Zeng Y, Crabb DW (2010) Inhibitory effect of alcohol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am J Physiol Gastrointest Liver Physiol 298(6):G1004–G1012
Liangpunsakul S, Rahmini Y, Ross RA, Zhao Z, Xu Y, Crabb DW (2012) Imipramine blocks alcohol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in alcohol-fed mice. Am J Physiol Gastrointest Liver Physiol 302(5):G515–G523
Lim ST, Esfahani K, Avdoshina V, Mocchetti I (2011) Exogenous gangliosides increase the release of brain-derived neurotrophic factor. Neuropharmacology 60:1160–1167
Longato L, Ripp K, Setshedi M, Dostalek M, Akhlaghi F, Branda M, Wands JR, de la Monte SM (2012) Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid Med Cell Longev 2012:479348
Maceyka M, Spiegel S (2014) Sphingolipid metabolites in inflammatory disease. Nature 510(7503):58–67
Malorni W, Giammarioli AM, Garofalo T, Sorice M (2007) Dynamics of lipid raft components during lymphocyte apoptosis: the paradigmatic role of GD3. Apoptosis 12(5):941–949
Marmillot P, Munoz J, Patel S, Garige M, Rosse RB, Lakshman MR (2007) Long-term alcohol consumption impairs reverse cholesterol transport function of high-density lipoproteins by depleting high-density lipoprotein sphingomyelin both in rats and in humans. Metabolism 56(7):947–953
Martin GE, Boudreau RM, Couch C, Becker KA, Edwards MJ, Caldwell CC, Gulbins E, Seitz A (2017) Sphingosine´s role in epithelial host defense: a natural antimicrobial and novel therapeutic. Biochimie. https://doi.org/10.1016/j.biochi.2017.03.014
Mayer DJ, Mao J, Price DD (1995) The development of morphine tolerance and dependence is associated with translocation of protein kinase C. Pain 61(3):365–374
McCreary AC, Müller CP, Filip M (2015) Psychostimulants: basic and clinical pharmacology. Int Rev Neurobiol 120:41–88
Merrill AH, Nixon DW, Williams RD (1985) Activities of serine palmitoyltransferase (3-ketosphinganine synthase) in microsomes from different rat tissues. J Lipid Res 26:617–622
Messner MC, Cabot MC (2010) Glucosylceramide in humans. Adv Exp Med Biol 688:156–164
Miranda AM, Oliveira TG (2015) Lipids under stress—a lipidomic approach for the study of mood disorders. BioEssays 37(11):1226–1235
Moles A, Tarrats N, Morales A, Domínguez M, Bataller R, Caballería J, García-Ruiz C, Fernández-Checa JC, Marí M (2010) Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. Am J Pathol 177:1214–1224
Mühle C, Amova V, Biermann T, Bayerlein K, Richter-Schmidinger T, Kraus T, Reichel M, Gulbins E, Kornhuber J (2014) Sex-dependent decrease of sphingomyelinase activity during alcohol withdrawal treatment. Cell Physiol Biochem 34(1):71–81
Müller CP (2013) Episodic memories and their relevance for psychoactive drug use and addiction. Front Behav Neurosci 7(34):1–13
Müller CP (2017a) Animal models of psychoactive drug use and addiction—present problems and future needs for translational approaches. Behav Brain Res. https://doi.org/10.1016/j.bbr.2017.06.028
Müller CP (2017b) Non addictive drug use: the way forward, chapter 42. In: Wolff K, White J, Karch S (eds) The SAGE handbook of drugs and alcohol studies—biological approaches, vol 2. SAGE, London, pp 411–434
Müller CP, Homberg J (2015) The role of serotonin in drug use and addiction. Behav Brain Res 277C:146–192
Müller CP, Kornhuber J (2017) Biological evidence for paradoxical improvement of psychiatric disorder symptoms by addictive drugs. Trends Pharmacol Sci 38(6):501–502
Müller CP, Schumann G (2011a) Drugs as an instrument: a new framework for non-addictive psychoactive drug use. Behav Brain Sci 34(6):293–310
Müller CP, Schumann G (2011b) To use or not to use: expanding the view on non-addictive psychoactive drug consumption and its implications. Behav Brain Sci 34(6):328–347
Müller CP, Reichel M, Mühle C, Rhein C, Gulbins E, Kornhuber J (2015) Brain membrane lipids in major depression and anxiety disorders. Biochim Biophys Acta 1851(8):1052–1065
Müller CP, Kalinichenko LS, Tiesel J, Witt M, Stöckl T, Sprenger E, Fuchser J, Beckmann J, Praetner M, Huber SE, Amato D, Mühle C, Büttner C, Ekici AB, Smaga I, Pomierny-Chamiolo L, Pomierny B, Filip M, Eulenburg V, Gulbins E, Lourdusamy A, Reichel M, Kornhuber J (2017) Paradoxical antidepressant effects of alcohol are related to acid sphingomyelinase and its control of sphingolipid homeostasis. Acta Neuropathol 133(3):463–483
Nassogne MC, Lizarraga C, N’Kuli F, Van Bambeke F, Van Binst R, Wallemacq P, Tulkens PM, Mingeot-Leclercq MP, Levade T, Courtoy PJ (2004) Cocaine induces a mixed lysosomal lipidosis in cultured fibroblasts, by inactivation of acid sphingomyelinase and inhibition of phospholipase A1. Toxicol Appl Pharmacol 194(2):101–110
Ndengele MM, Cuzzocrea S, Masini E, Vinci MC, Esposito E, Muscoli C, Petrusca DN, Mollace V, Mazzon E, Li D, Petrache I, Matuschak GM, Salvemini D (2009) Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther 329(1):64–75
Nikolova-Karakashian MN, Rozenova KA (2010) Ceramide in stress response. Adv Exp Med Biol 688:86–108
O’Malley PM, Johnston LD (2002) Epidemiology of alcohol and other drug use among American college students. J Stud Alcohol Suppl 14:23–39
Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C (2002) Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Dev Brain Res 133(2):115–126
Olsen ASB, Færgeman NJ (2017) Sphingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol 7(5):170069
Osawa Y, Uchinami H, Bielawski J, Schwabe RF, Hannun YA, Brenner DA (2005) Roles for C16-ceramide and sphingosine-1-phosphate in regulating hepatocyte apoptosis in response to tumor necrosis factor-α. J Biol Chem 280(30):27879–27887
Palmano K, Rowan A, Guillermo R, Guan J, McJarrow P (2015) The role of gangliosides in neurodevelopment. Nutrients 7:3891–3913
Park JY, Kim MJ, Kim YK, Woo JS (2011) Ceramide induces apoptosis via caspase-dependent and caspase-independent pathways in mesenchymal stem cells derived from human adipose tissue. Arch Toxicol 85:1057–1065
Pascual M, Valles SL, Renau-Piqueras J, Guerri C (2003) Ceramide pathways modulate alcohol-induced cell death in astrocytes. J Neurochem 87:1535–1545
Peele S, Brodsky A (2000) Exploring psychological benefits associated with moderate alcohol use: a necessary corrective to assessments of drinking outcomes? Drug Alcohol Depend 60:221–247
Peres WA, Carmo MG, Zucoloto S, Iglesias AC, Braulio VB (2004) Alcohol intake inhibits growth of the epithelium in the intestine of pregnant rats. Alcohol 33(2):83–89
Pernber Z, Molander-Melin M, Berthold CH, Hansson E, Fredman P (2002) Expression of the myelin and oligodendrocyte progenitor marker sulfatide in neurons and astrocytes of adult rat brain. J Neurosci Res 69:86–93
Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM (2005) Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 11:491–498
Piccinini M, Scandroglio F, Prioni S, Buccinnà B, Loberto N, Aureli M, Chigorno V, LupinoE DeMarco G, Lomartire A, Rinaudo MT, Sonnino S, Prinetti A (2010) Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol Neurobiol 41:314–340
Piomelli D, Astarita G, Rapaka R (2007) A neuroscientist’s guide to lipidomics. Nat Rev Neurosci 8(10):743–754
Podbielska M, Levery SB, Hogan EL (2011) The structural and functional role of myelin fast-migrating cerebrosides: pathological importance in multiple sclerosis. Clin Lipidol 6(2):159–179
Posse de Chaves E, Sipione S (2010) Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett 584:1748–1759
Ramirez T, Longato L, Dostalek M, Tong M, Wands JR, de la Monte SM (2013) Insulin resistance, ceramide accumulation and endoplasmic reticulum stress in experimental chronic alcohol-induced steatohepatitis. Alcohol Alcohol 48(1):39–52
Ravasi D, Ferraretto A, Omodeo-salè MF, Tettamanti G, Pitto M, Masserini M (2002) Alcohol-induced increase of sphingosine recycling for ganglioside biosynthesis: a study performed on cerebellar granule cells in culture. J Neurosci Res 69(1):80–85
Rebillard A, Tekpli X, Meurette O, Sergent O, LeMoigne-Muller G, Vernhet L, Gorria M, Chevanne M, Christmann M, Kaina B, Counillon L, Gulbins E, Lagadic-Gossmann D, Dimanche-Boitrel MT (2007) Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Cancer Res 67(16):7865–7874
Reichel M, Greiner E, Richter-Schmidinger T, Yedibela O, Tripal P, Jacobi A, Bleich S, Gulbins E, Kornhuber J (2010) Increased acid sphingomyelinase activity in peripheral blood cells of acutely intoxicated patients with alcohol dependence. Alcohol Clin Exp Res 34(1):46–50
Reichel M, Beck J, Mühle C, Rotter A, Bleich S, Gulbins E, Kornhuber J (2011) Activity of secretory sphingomyelinase is increased in plasma of alcohol-dependent patients. Alcohol Clin Exp Res 35(10):1852–1859
Rippo MR, Malisan F, Ravagnan L, Tomassini B, Condo I, Costantini P, Susin SA, Rufini A, Todaro M, Kroemer G, Testi R (2000) GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J 14(13):2047–2054
Ritter JK, Fang Y, Xia M, Li PL, Dewey WL (2012) Contribution of acid sphingomyelinase in the periaqueductal gray region to morphine-induced analgesia in mice. NeuroReport 23(13):780–785
Robbins TW, Ersche KD, Everitt BJ (2008) Drug addiction and the memory systems of the brain. Ann NY Acad Sci 1141:1–21
Roux A, Jackson SN, Muller L, Barbacci D, O’Rourke J, Thanos PK, Volkow ND, Balaban C, Schultz JA, Woods AS (2016) Alcohol induced brain lipid changes in mice assessed by mass spectrometry. ACS Chem Neurosci 7(8):1148–1156
Ruano MJ, Martínez-Zorzano VS, Cabezas JA, Hueso P (1994) Ganglioside content of rat liver after administration of alcohol and pentazocine or sucrose supplemented diets. Arch Toxicol 68(9):576–581
Saito M, Saito M, Berg MJ, Guidotti A, Marks N (1999) Gangliosides attenuate alcohol-induced apoptosis in rat cerebellar granule neurons. Neurochem Res 24(9):1107–1115
Saito M, Sait M, Cooper TB, Vadasz C (2004) Alcohol reduces GM1 ganglioside content in the serum of inbred mouse strains. Alcohol Clin Exp Res 28(7):1107–1113
Saito M, Saito M, Cooper TB, Vadasz C (2005) Alcohol-induced changes in the content of triglycerides, ceramides, and glucosylceramides in cultured neurons. Alcohol Clin Exp Res 29:1374–1383
Saito M, Mao RF, Wang R, Vadasz C, Saito M (2007) Effects of gangliosides on alcohol-induced neurodegeneration in the developing mouse brain. Alcohol Clin Exp Res 31(4):665–674
Saito M, Chakraborty G, Hegde M, Ohsie J, Paik SM, Vadasz C, Saito M (2010) Involvement of ceramide in alcohol-induced apoptotic neurodegeneration in the neonatal mouse brain. J Neurochem 115:168–177
Saito M, Chakraborty G, Shah R, Mao RF, Kumar A, Yang DS, Dobrenis K, Saito M (2012) Elevation of GM2 ganglioside during ethanol-induced apoptotic neurodegeneration in the developing mouse brain. J Neurochem 121(4):649–661
Schatter B, Jin S, Loffelholz K, Klein J (2005) Cross-talk between phosphatidic acid and ceramide during alcohol-induced apoptosis in astrocytes. BMC Pharmacol 5:3
Schmitt S, Castelvetri LC, Simons M (2015) Metabolism and functions of lipids in myelin. Biochim Biophys Acta 1851:999–1005
Schneider JS, DiStefano L (1995) Enhanced restoration of striatal dopamine concentrations by combined GM1 ganglioside and neurotrophic factor treatments. Brain Res 674:260–264
Schneider M, Levant B, Reichel M, Gulbins E, Kornhuber J, Müller CP (2017) Lipids in psychiatric disorders and preventive medicine. Neurosci Biobehav Rev 76(Pt B):336–362
Seitz AP, Grassmé H, Edwards MJ, Pewzner-Jung Y, Gulbins E (2015) Ceramide and sphingosine in pulmonary infections. Biol Chem 396(6–7):611–620
Senn HJ, Orth M, Fitzke E, Schölmerich J, Köster W, Wieland H, Gerok W (1990) Altered concentrations, patterns and distribution in lipoproteins of serum gangliosides in liver diseases of different etiologies. J Hepatol 11(3):290–296
Seumois G, Fillet M, Gillet L, Faccinetto C, Desmet C, François C, Dewals B, Oury C, Vanderplasschen A, Lekeux P, Bureau F (2007) De novo C16- and C24-ceramide generation contributes to spontaneous neutrophil apoptosis. J Leukoc Biol 81(6):1477–1486
Shen KF, Crain SM (1990) Cholera toxin-B subunit blocks opioid excitatory effects on sensory neuron action potentials indicating that GM1 ganglioside may regulate Gs-linked opioid receptor functions. Brain Res 531:1–7
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572
Skogen JC, Harvey SB, Henderson M, Stordal E, Mykletun A (2009) Anxiety and depression among abstainers and low-level alcohol consumers. The Nord-Trøndelag Health Study. Addiction 104(9):1519–1529
Spagnolo PA, Goldman D (2017) Neuromodulation interventions for addictive disorders: challenges, promise, and roadmap for future research. Brain 140(5):1183–1203
Spanagel R (2009) Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev 89(2):649–705
Standridge JB, Zylstra RG, Adams SM (2004) Alcohol consumption: an overview of benefits and risks. South Med J 97:664–672
Supakul R, Liangpunsakul S (2011) Alcoholic-induced hepatic steatosis—role of ceramide and protein phosphatase 2A. Transl Res 158:77–81
Tamiji J, Crawford DA (2010) The neurobiology of lipid metabolism in autism spectrum disorders. Neurosignals 18(2):98–112
Telenga ED, Hoffmann RF, t’Kindt R, Hoonhorst SJ, Willemse BW, van Oosterhout AJ, Heijink IH, van den Berge M, Jorge L, Sandra P, Postma DS, Sandra K, ten Hacken NH (2014) Untargeted lipidomic analysis in chronic obstructive pulmonary disease. Uncovering sphingolipids. Am J Respir Crit Care Med 190:155–164
Tettamanti G (2004) Ganglioside/glycosphingolipid turnover: new concepts. Glycoconjugate J20:301–317
Tibboel J, Reiss I, de Jongste JC, Post M (2013) Ceramides: a potential therapeutic target in pulmonary emphysema. Respir Res 14:96
Tong M, Longato L, Ramirez T, Zabala V, Wands JR, de la Monte SM (2014) Therapeutic reversal of chronic alcohol-related steatohepatitis with the ceramide inhibitor myriocin. Int J Exp Pathol 95:49–63
Tosk JM, Farag M, Ho JY, Lee CC, Maximos BB, Yung HH (1996) The effects of nerve growth factor and ganglioside GM1 on the anti-proliferative activity of cocaine in PC12 cells. Life Sci 59(20):1731–1737
Tsugane K, Tamiya-Koizumi K, Nagini M, Nimura Y, Yoshida S (1999) A possible role of nuclear ceramide and sphingosine in hepatocyte apoptosis in rat liver. J Hepatol 31:8–17
Valdomero A, Hansen C, de Burgos NG, Cuadra GR, Orsingher OA (2010) GM1 ganglioside enhances the rewarding properties of cocaine in rats. Eur J Pharmacol 630(1–3):79–83
Valdomero A, Perondi MC, Orsingher OA, Cuadra GR (2015) Exogenous GM1 ganglioside increases accumbal BDNF levels in rats. Behav Brain Res 278:303–306
van Echten-Deckert G, Herget T (2006) Sphingolipid metabolism in neural cells. Biochim Biophys Acta 1758:1978–1994
van Meer G, Sprong H (2004) Membrane lipids and vesicular traffic. Curr Opin Cell Biol 16:373–378
Veloso A, Fernández R, Astigarraga E, Barreda-Gómez G, Manuel I, Giralt MT, Ferrer I, Ochoa B, Rodríguez-Puertas R, Fernández JA (2011) Distribution of lipids in human brain. Anal Bioanal Chem 401(1):89–101
Viktorov AV, Yurkiv VA (2008) Effects of alcohol and lipopolysaccharide on the sphingomyelin cycle in rat hepatocytes. Bull Exp Biol Med 146:753–755
Vital MABF, Frussa-Filho R, Palermo-Neto J (1995) Effects of monosialoganglioside on dopaminergic supersensitivity. Life Sci 56:2299–2307
Volkow ND, Wise RA, Baler R (2017) The dopamine motive system: implications for drug and food addiction. Nat Rev Neurosci 18(12):741–752
Waldorf D, Reinarman C, Murphy S (1991) Cocaine changes: the experience of using and quitting. Temple University Press, Philadelphia
Wang G, Bieberich E (2010) Prenatal alcohol exposure triggers ceramide-induced apoptosis in neural crest-derived tissues concurrent with defective cranial development. Cell Death Dis 1:e46
Wang Z, Deng T, Deng J, Deng J, Gao X, Shi Y, Liu B, Ma Z, Jin H (2013) Ceramide is involved in alcohol-induced neural proliferation. Neural Regen Res 8(23):2178–2189
Wenk MR (2005) The emerging field of lipidomics. Nat Rev Drug Discov 4(7):594–610
White NM (1996) Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91:921–949
Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, Charlson FJ, Norman RE, Flaxman AD, Johns N, Burstein R, Murray CJ, Vos T (2013) Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 382(9904):1575–1586
Wu BX, Rajagopalan V, Roddy PL, Clarke CJ, Hannun YA (2010) Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J Biol Chem 285(23):17993–18002
Xiao S, FinkielsteinCV CapellutoDG (2013) The enigmatic role of sulfatides: new insights into cellular functions and mechanisms of protein recognition. Adv Exp Med Biol 991:27–40
Yalcin EB, Nunez K, Tong M, de la Monte SM (2015) Differential sphingolipid and phospholipid profiles in alcohol and nicotine-derived nitrosamine ketone-associated white matter degeneration. Alcohol Clin Exp Res 39(12):2324–2333
Yalcin EB, McLean T, Tong M, de la Monte SM (2017) Progressive white matter atrophy with altered lipid profiles is partially reversed by short-term abstinence in an experimental model of alcohol-related neurodegeneration. Alcohol 65:51–62
Yang L, Jin GH, Zhou JY (2016) The role of ceramide in the pathogenesis of alcoholic liver disease. Alcohol Alcohol 51(3):251–257
Yao R, Cooper GM (1995) Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 267:2003–2006
You M, Crabb DW (2004) Molecular mechanisms of alcoholic fatty liver: role of sterol regulatory element-binding proteins. Alcohol 34(1):39–43
Yung LM, Wei Y, Qin T, Wang Y, Smith CD, Waeber C (2012) Sphingosine kinase 2 mediates cerebral preconditioning and protects the mouse brain against ischemic injury. Stroke 43:199–204
Zhao Z, Yu M, Crabb D, Xu Y, Liangpunsakul S (2011) Alcohol-induced alterations in fatty acid-related lipids in serum and tissues in mice. Alcohol Clin Exp Res 35:229–234
Zulueta A, Caretti A, Campisi GM, Brizzolari A, Abad JL, Paroni R, Signorelli P, Ghidoni R (2017) Inhibitors of ceramide de novo biosynthesis rescue damages induced by cigarette smoke in airways epithelia. Naunyn Schmiedebergs Arch Pharmacol 390(7):753–759
Acknowledgements
This work was supported by funding from DFG Grants MU 2789/8-1 (C.P.M.), KO 947/15-1, KO 947/13-1 (J.K.), and GU 335/29-1 (E.G.) and by funding from the Interdisciplinary Center for Clinical Research (IZKF) Erlangen, Project E13 (L.S.K., C.P.M., J.K.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Kalinichenko, L.S., Gulbins, E., Kornhuber, J. et al. The role of sphingolipids in psychoactive drug use and addiction. J Neural Transm 125, 651–672 (2018). https://doi.org/10.1007/s00702-018-1840-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-018-1840-1