
Abstract
Sulfatides are sphingolipids commonly found at the surface of most of eukaryotic cells. Sulfatides are not just structural components of the plasma membrane but also participate in a wide range of cellular processes including protein trafficking, cell adhesion and aggregation, axon-myelin interactions, neural plasticity, and immune responses, among others. The intriguing question is how can sulfatides trigger such cellular processes? Their dynamic presence and specific localization at plasma membrane sites may explain their multitasking role. Crystal and NMR structural studies have provided the basis for understanding the mechanism of binding by sulfatide-interacting proteins. These proteins generally exhibit a hydrophobic cavity that is responsible for the interaction with the sulfatide acyl chain, whereas the hydrophilic, negatively charged moiety can be found either buried in the hydrophobic cavity of the protein or exposed for additional intermolecular associations. Since sulfatides vary in their acyl chain composition, which are tissue-dependent, more emphasis on understanding acyl chain specificity by sulfatide-binding proteins is warranted. Importantly, changes in cellular sulfatide levels as well as circulating sulfatides in serum directly impact cardiovascular and cancer disease development and progress. Therefore, sulfatides might prove useful as novel biomarkers. The scope of this review is to overview cell functions and mechanisms of sulfatide recognition to better understand the role of these lipids in health and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sulfatides
- Ceramide
- Plasma membrane
- Sulfatide-binding proteins
- Platelet aggregation
- Disabled-2
- Cluster of differentiation 1
3.1 Introduction to Sulfatides
Sulfatides (also known as 3-O-sulfogalactosylce-ramides, sulfated galactocerebrosides, or SM4) are sphingolipids found at the extracellular leaflet of the plasma membrane of most eukaryotic cells. They were first isolated from human brain tissue by Thudichum in 1884 [105]. Sulfatides are not only membrane components but they are also involved in protein trafficking, cell adhesion and aggregation, axon-myelin interactions, modulation of sodium and potassium channels, learning and memory, and neural plasticity [9, 13, 16, 66, 109, 113]. Sulfatides are expressed in a variety of cells, predominantly in the myelin sheath of the nervous system, representing ∼4 % of the total myelin lipids [47]. Also, these sphingolipids are largely found at the surface of blood cells such as erythrocytes [57], neutrophils [93], and platelets [85] and they are major component of lipoproteins in blood serum [100]. Sulfatides are esters of sulfuric acid with galactosylceramides at C3 of the galactose moiety, which is connected to the primary hydroxyl group of the N-acylated D-erythro-sphingosine base via a β-glycosidic bond (Fig. 3.1). The fatty acid chain length of sulfatides varies, with the majority being composed of C16 to C26, including 2-hydroxy fatty acids [47]. Sulfatides containing nervonic acid (C24:1) are the most abundant in myelin, whereas high levels of the lipid with stearic acid (C18:0) are present in the cortical grey matter [46]. Other structural variants of sulfatides (C22:0) are found in kidney tissue [47], with shorter acyl chains (C16:0) being predominant form in pancreas [23]. Sulfatides are also modified by hydroxylation at the α-2 carbon of the fatty acid by the fatty acid 2-hydroxylase [4] and both hydroxylated and nonhydroxylated forms of the lipid are found distinctly distributed in the cerebral cortex [119].

The synthesis and degradation pathway of sulfatides. Ceramide is converted to galactocerebroside by addition of a galactose group from UDP-galactose, a reaction catalyzed by UDP-galactose:ceramide galactosyltransferase (CGT). Galactocerebroside is a substrate of 3′-phosphoadenosine-5′-phosphosulfate:cerebroside sulfotransferase (CST), which adds a sulfate group to the galactose moiety, using 3′phosphoadenosine-5′-phospho-sulfate (PAPS), to generate sulfatide. Sulfatide turnover is mediated by arylsulfatase A (ASA), an enzyme that removes the sulfate group and generates galactocerebroside. ASA requires saposin B activity, a cysteine-rich protein that extracts sulfatides from membranes and allows ASA to catalyze the reaction on diffusible protein-lipid complexes. The chemical structure of ceramides is characterized by the presence of a sphingosine group and an additional fatty chain, which usually varies with different lengths and, therefore, depicted with an R group. Commonly found R-groups in ceramides and sulfatides are depicted at the bottom
3.2 Sulfatide Synthesis and Degradation
Synthesis of sulfatides occurs in the endoplasmic reticulum and the Golgi apparatus. Initially, a galactose residue is transferred from UDP-galactose to 2-hydroxylated or nonhydroxylated ceramide at the luminal membrane leaflet of the endoplasmic reticulum, a reaction catalyzed by the UDP-galactose:ceramide galactosyltransferase (CGT; C 2.4.1.45) (Fig. 3.1). The product of this reaction, galactocerebroside, is delivered to the Golgi apparatus where it is modified by sulfation at position 3 of the galactose moiety through the action of a 3′-phosphoadenosine-5′-phosphosulfate:cerebroside sulfotransferase (CST; EC 2.8.2.11) [117]. Tissue-dependent expression of sulfatides correlates with the expression of both CGT and CST genes [35, 39, 123]. Recently, Aoyama and colleagues determined that the CST gene is transcriptionally stimulated by the activated peroxisome proliferator-activated receptor α and this effect directly enhances sulfatide levels in mice [75]. Mice lacking CST or CGT cannot produce sulfatides [38, 89]. Absence of the CST gene leads to disorganized paranodes and a lack of septate-like junctions, defects that promote a reduction of the nerve conduction velocity due to the lack of sulfatides [15, 19, 38]. Degradation of sulfatides is mediated by lysosomal arylsulfatase A (ASA; EC 3.1.6.8), which hydrolyzes the sulfate group from the galactose moiety leading to the formation of galactocerebroside. Sulfatide accumulation by the lack of ASA is associated with demyelination and metachromatic leukodystrophy (MLD), a lethal neurological disease [21, 84]. Overall, accumulated evidences indicate that alteration of sulfatide synthesis has a major impact on the generation of neuronal defects.
The reaction catalyzed by ASA depends upon the presence of saposin B, a sphingolipid activator protein that removes sulfatides from membranes and, thus, allows sulfatides to interact with ASA [56]. The crystal structure of saposin B shows a shell-like dimer of a helical bundle that encloses a hydrophobic cavity [2], a structural organization that is observed in many sulfatide-binding proteins [91]. Saposin B adopts a V-shaped conformation with five amphipathic α-helices, which associates to another saposin B molecule to build a large hydrophobic cavity in the dimer. The structure also reveals a region of elongated electron density that could be a potential lipid-binding site, an association that may require a conformational change of saposin B to expose its inner hydrophobic cavity to membranes [2].
Sulfatides can be intracellularly distributed by action of the glycolipid transfer protein (GLTP), a cytosolic peripheral protein that transfers glycolipids from the cytosolic leaflet of the plasma membrane or the endoplasmic reticulum and acts as a sensor of glycolipid levels [68]. GLTP employs a helical two-layer sandwich motif to transfer glycolipids and is able to recognize the sugar head group using hydrogen bonds and a hydrophobic pocket that associates with most of the nonpolar hydrocarbon chains of the ceramide region of the glycolipid [65]. There are two modes of glycosphingolipid binding by GLTPs [64]: (i) “Sphingosine in” mode, in which both the acyl and sphingosine chains are located in the same hydrophobic pocket of GLTP and (ii) “Sphingosine out” mode, in which the acyl chain of the sphingolipid remains in the hydrophobic pocket of GLTP, where the sphingosine backbone becomes exposed to the protein surface and allows interaction with another GLTP, forming a dimer. Recently, studies using the crystal structures of the wild-type human GLTP and a mutant (Asp48Val; D48V) version of the protein in complex with sulfatides reveal that the D48V mutation favors the transfer selectivity to sulfatides by switching GLTP to the “sphingosine in” mode [91]. The D48V GLTP exhibits a cavity that allows the sulfate group to efficiently accommodate the sulfatide molecule in the protein, enhancing sulfatide binding over other neutral glycosphingolipids, such as galactoceramides. Consequently, sulfatides favor dimerization of GLTP, whose dimerization interface resembles the membrane-binding domains of the protein [54, 64].
3.3 Cellular Mechanisms Mediated by Sulfatides
3.3.1 Nervous System
Sulfatides are present in high levels in the myelin sheath, in both the central and peripheral nervous systems [108]. Myelin contains 70–75 % lipid, 4–7 % of which are sulfatides [77]. Sulfatides are also found in other glial cells, astrocytes, and neurons [11, 46, 81] and are myelin-associated inhibitors of central nervous system axon regeneration [114]. Increased cellular concentration of sulfatides is associated with MLD, in which patients exhibit accumulation of the lipid in lysosomes of oligodendrocytes, Schwann cells, macrophages, astrocytes, and neurons [79]; elevated levels of sulfatides are also associated with epileptic and audiogenic seizures [107]. Although unusual, deficiency in saposin B has also been observed in MLD [124]. Nonetheless, MLD leads to a progressive loss of myelin, in which the individual ultimately dies in a decerebrated state. Patients with Multiple Sclerosis or Parkinson’s disease exhibit elevated levels of anti-sulfatide antibodies in serum and cerebrospinal fluid compared to healthy individuals [55]. Indeed, sulfatides act as autoantigens in Multiple Sclerosis patients [33]. Overall, these findings indicate that release of sulfatides from myelin is associated with the development of central nervous system diseases. Changes in sulfatide levels have been observed in other neuronal diseases, including epilepsy with mental retardation and Alzheimer’s disease (for more details, see [20]).
3.3.2 Platelet Adhesion and Aggregation
Platelets represent an important linkage between thrombus formation and inflammatory processes. First, they prevent post-traumatic blood loss by forming fibrin-containing thrombi at the site of vascular injury, followed by the release of a battery of potent inflammatory and mitogenic molecules within the microenvironment that alters the chemotactic and adhesive properties of endothelial cells. These events facilitate the tethering and rolling of leukocytes over an inflamed vessel wall (activated endothelium [24, 25]), which then either firmly adhere and transmigrate into the arterial intima or simply detach [26, 27]. Among the various glycoproteins involved in these events, selectins are crucial for the initial contact between platelets and the vascular endothelium, and remarkably, mediate rosetting of platelets with monocytes and neutrophils to form platelet-leukocyte aggregates [60, 104]. Despite some contradictory results reviewed by Kyogashima [59], accumulated recent evidence suggests that sulfatides promote platelet adhesion and aggregation [18, 31, 70, 113].
One of the key cell surface receptors mediating leukocyte recruitment and exhibiting pro-aggregatory activity is P-selectin (for a review, see [12]). Most of the P-selectin ligands contain post-translational modifications needed for receptor binding and signal transduction in which sulfate moieties are frequently present [83, 88]. Sulfatides modulate P-selectin activity at the platelet surface [72] leading to further degranulation and increased surface P-selectin expression, which reinforces platelet aggregation [70, 113]. Moreover, P-selectin-sulfatide interaction leads to the formation of stable platelet aggregates and surface sulfatides enhance the formation of platelet-leukocyte aggregates [70]. Platelet P-selectin expression is decreased by fibrinogen deficiency [118].
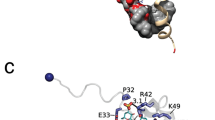
Frequently, soluble platelet aggregation agonists bind to and induce conformational changes in the extracellular domains of the αIIbβ3 integrin receptor, triggering the inside-out integrin-signaling pathway [49]. Simultaneously, fibrinogen activates the outside-in signaling pathway by association with the αIIbβ3 integrin receptor via two Arg-Gly-Asp (RGD) motifs located in its α-chain [82]. In addition to fibrinogen, other integrin receptor agonists include the von Willebrand factor (vWF) and fibronectin and these associations stimulate platelet spreading and aggregation on vascular surfaces [49]. The adaptor protein Disabled-2 (Dab2) negatively regulates fibrinogen-αIIbβ3 integrin receptor association and, consequently, inhibits cell adhesion and cell signaling [18, 41]. The inhibitory function of the cytosolic pool of Dab2 is mediated by phosphorylation in its Ser24 residue, a post-translational modification that triggers the association of Dab2 to the cytoplasmic tail of the β3 subunit of the integrin receptor [41]. Binding of Dab2 to the integrin receptor is likely to be enhanced by phosphatidylinositol 4,5-bisphosphate-mediated membrane anchoring (Fig. 3.2). Consequently, Dab2 acts as a negative regulator of integrin receptor inside-out signaling.

An updated model of sulfatide- and Dab2-mediated modulation of platelet aggregation. Resting platelets are enriched in α-granules, which contain pro-coagulant (i.e., P-selectin, αIIbβ3 integrin receptor) and anti-coagulant proteins (i.e., Dab2). Another pool of platelet Dab2 is distributed cytosolically. Also, platelets contain signaling lipids including sulfatides (found at the outer leaflet of the plasma membrane) and PtdIns(4,5)P2 (found at the inner leaflet of the plasma membrane). Upon activation, platelets change shape and release the α-granular content. Released Dab2 is partitioned in two pools: one associates with the αIIb subunit of the integrin receptor through its RGD motif, and therefore, competes with fibrinogen for integrin receptor binding. Consequently, Dab2 negatively controls clot formation by modulating platelet aggregation. The second pool of Dab2 associates with cell surface sulfatides, whose levels are increased upon platelet activation. Upon platelet activation, cytosolic Dab2 is recruited to the plasma membrane in a phosphorylated state where interacts and inhibits the β3 subunit of the integrin receptor. Membrane recruitment of Dab2 is likely enhanced by its association to PtdIns(4,5)P2. The fate of phosphorylated Dab2 after membrane recruitment is unknown. The function of extracellular Dab2 is modulated by the agonist thrombin, which cleaves Dab2 making it inactive (Dab2(i)). Both P-selectin and L-selectin bind to cell surface sulfatides mediating platelet-platelet platelet-leukocyte interactions, respectively. Furthermore, platelet-leukocyte interactions are enhanced by the association of P-selectin with PSGL-1. Both homotypic and heterotypic interactions are negatively modulated by Dab2. The presence of Dab2 at the cell surface is transient since it has been shown to be internalized back to α-granules (dotted arrows)
Dab2 is also localized in α-granules of both megakaryocytes [41] and resting platelets [18, 42]. Upon activation, Dab2 is secreted to the megakaryocyte and platelet surface via the α-granule secretory pathway where it binds to the αIIbβ3 integrin receptor, blocking fibrinogen-platelet interactions [42]. Integrin-binding takes place because of the presence of an RDG motif in Dab2, an association that can be inhibited by the fibrinogen-derived Arg-Gly-Asp-Ser (RGDS) peptide [42]. Dab2 targets platelet surface membranes, as a result of platelet activation, via its N-terminal region containing the phosphotyrosine-binding (N-PTB) domain [18]. N-PTB is necessary and sufficient to inhibit platelet adhesion and aggregation by competing with fibrinogen for binding to the αIIbβ3 integrin receptor through its RGD motif [18]. In addition, Dab2 binds membrane sulfatides, an association that redistributes the protein at the platelet surface [18]. Dab2 recognizes sulfatides through the residues Lys25, Lys49, Lys51, and Lys53, which are located within the XBBXBX (B, basic residue; X, any residue) and BXBXBX motifs in its N-PTB region [18]. This class of basic clusters also mediates sulfatide binding of other cell adhesive proteins, including thrombospondins, laminins, and selectins [47]. The sulfatide-binding site of Dab2 overlaps with that of the phosphoinositide PtdIns(4,5)P2 binding site [3], but competition likely does not occur in a physiological context since sulfatides are predominantly found at the plasma membrane surface, presumably in lipid rafts [96], whereas the phosphoinositide is predominantly found at the cytosolic leaflet of the plasma membrane [58]. Whereas sulfatides contribute to Dab2 membrane insertion, which is likely accompanied by a conformational change of the protein, phosphoinositide recognition occurs by electrostatic interactions associated with minor local structural changes in Dab2 [3]. Sulfatide recognition by Dab2 impairs cleavage by thrombin, a strong platelet agonist [18]. Consequently, a pool of Dab2 remains intact at the platelet surface upon activation, and is eventually internalized back to α-granules by an actin cytoskeleton-dependent mechanism [18]. Also, sulfatides modulate the availability of Dab2 for binding to the integrin receptor [18]. Taken together, Dab2 may be distributed in two pools at the platelet surface (Fig. 3.2). One pool of Dab2 competes with fibrinogen for binding to the integrin receptor, whereas a second pool binds sulfatides at the platelet surface. The second pool of Dab2 also exerts an additional layer of modulation of platelet aggregation since sulfatide binding by Dab2 blocks P-selectin-sulfatide interactions (Fig. 3.2) [113], which are required to sustain platelet aggregation [71]. Indeed, sulfatides promote surface expression of P-selectin in activated platelets [70, 113]. The N-PTB region of Dab2 not only blocks platelet-platelet interactions, but also controls the extent of heterotypic cell interactions, such as those with leukocytes via its recognition to cell surface sulfatides [113].
We have recently generated a Dab2-derived peptide that contains the two sulfatide-binding motifs (SBMs) of the protein [116]. The Dab2 SBM peptide adopts a helical and amphipathic structure when embedded in dodecylphosphocholine (DPC) micelles. The majority of the sulfatide-interacting residues map to the second sulfatide-binding motif with the basic residues Lys49, Lys51, and Lys53 as well as the nonpolar residues Ala52, Leu54 and Ile55 playing a major role in the interaction with the sphingolipid [116]. Using a combination of paramagnetic probes, we established that the peptide lies in a parallel orientation below the sulfatide-enriched DPC micellar surface but does not cross the hydrophobic core of the micelle. Using mic-rofluidic devices that readily mimic vasculature, we showed that Dab2 SBM displays anti-aggregatory platelet activity, comparable to that described for the fibrinogen-derived peptide, Arg-Gly-Asp-Ser (RGDS) [116]. Thus, by binding to cell surface sulfatides, Dab2 SBM provides the basis for rational design, promising anti-aggregatory low-molecular mass molecules for therapeutic applications.
Sulfatides also interact with homeostatic cell adhesion proteins, such as vWF [86], chemokines [92], laminin [86], and thrombospondin [85]. Sulfatides inhibit vWF’s platelet adhesion in flowing blood and under physiological shear stress conditions [9]. The sulfatide-binding site in vWF overlaps with that of the glycoprotein Ib and, consequently, the lipid can inhibit glycoprotein Ib-mediated platelet adhesion [9]. vWF binds sulfatides by a region comprising residues 1,391–1,409 within the A1 domain of the protein [5]. Further site-directed mutagenesis studies demonstrated that the residues Arg1392, Arg1395, Arg1399, and Lys1423 are critical for sulfatide recognition as shown using ELISA-based plates coated with sulfatides [76]. The residues Arg1392 and Arg1395 within the A1 domain of vWF are also relevant for glycoprotein Ib binding [67], confirming that sulfatides and glycoprotein Ib compete with each other for vWF binding.
Chemokines are cytokines that bind to cell surface sulfated glycosaminoglycans, modulating the activity of chemokine receptors. In addition to glycosaminoglycan binding, chemokines bind sulfatides [92], although the role of sulfatide recognition by these proteins is not clear. Whereas chemokine production is reduced by sulfatides when tested in peripheral leukocytes and fat cells [10, 87], it is stimulated in brain immune cells [52].
Laminins contain a series of G-like modules of about 200 amino acids each that bind to sulfatides, an association that may facilitate the polymerization of the protein into networks [53]. Two XBBXBX and three BXBXBX sequences were initially suggested to be potential sulfatide-binding motifs for the protein [103]. Timpl and colleagues demonstrated that sulfatide binding is increased when laminin G-like modules are in tandem [102], indicating their cooperation in ligand recognition. Structural data indicate that residues K3027 and K3028 within the XBBXBX motif of laminin α2 G-like 4–5 domains are critical for sulfatide binding [37, 102]. Furthermore, residues K3088 and K3091 present in a basic cluster BXXBXXXB of the same protein contribute to sulfatide binding [37]. Likewise, the 2831RAR and 2766KGRTK residues of the related laminin α1 G-like 4–5 domains, which belong to potential BXBXBX motifs, are crucial for sulfatide binding [34]. However, other basic clusters involved in heparin recognition are dispensable for sulfatide binding [34], suggesting that the association of laminin to different ligands may trigger unique biological responses.
Thrombospondins are extracellular calcium-binding proteins that are involved in wound healing, angiogenesis, vessel wall biology, synaptogenesis, and connective tissue organization (for a review, see [1]). Thrombospondins are known to bind many partners [1]. Sulfatides and heparins show strong affinity to thrombospondin-derived peptides containing the WSXW (where X is any residue) sequence with no polybasic motif required for sulfatide binding [32]. Indeed, these peptides strongly inhibit sulfatide and heparin binding to the thrombospondin, blocking binding of this protein to melanoma cells [32].
3.3.3 Innate Immunity and Autoimmunity
T cells recognize antigens, such as foreign and self-lipids and peptides, leading to the production of cytokines and, therefore, contributing to immune responses [7]. T cells also use their cell surface receptor to recognize lipid antigen-bound cluster of differentiation 1 (CD1) molecules at the surface of professional antigen-presenting cells such as macrophages, dendritic cells, and a small group of B cells. There are three groups of CD1 surface proteins: (i) CD1a, CD1b, and CD1c (group 1), (ii) CD1d (group 2), and (iii) CD1e (group 3) [17]. CD1 proteins contain three extracellular domains (α1, α2, and α3), a transmembrane domain, and a cytoplasmic tail. The extracellular domains form a surface groove (named the lipid-binding groove) formed by two α-helices (α1 and α2) on top of a β-sheet [120, 122]. The lipid-binding groove, which is narrow and deep, contains hydrophobic residues that can interact with the acyl chains of the glycolipids [6], whereas the polar head group becomes exposed in the CD1-lipid complex, allowing recognition by T cell receptors [73]. In CD1a proteins, the lipid- binding groove contains two large hydrophobic regions termed A’ and F’ [120]. In the CD1a-sulfatide complex, the sulfatide adopts an S-shaped conformation in which the A’ pocket contributes to the C18 sphingosine backbone recognition, and the acyl chain of the lipid emerges from the A’ pocket and extends its association into the F’ pocket [120]. The galactose moiety forms hydrogen bonds with residues Arg76 and Ser77, whereas the sulfate group forms hydrogen bonds with residues Arg76 and Glu154 and with water that is in complex between residues Arg73 and Glu154 [120]. Consequently, the sulfated galactose residue becomes exposed at the surface of the complex for T cell receptor recognition. Sulfatides can be presented by all members of the CD1 group 1 and by CD1d [8, 94]. However, the sulfatide-binding affinity varies with each CD1 molecule, with the CD1a-sulfatide being the most stable complex [94].
Sulfatides have also been shown to be self-glycolipid antigens recognized by CD1d, assembling a complex that activates type II natural killer T (NKT) cells [50]. Sulfatides induce proliferation and expansion of memory, but not naïve, T cells [48]. The mechanism by which the T-cell receptor from type II NKT cells (XV19 hybridoma) interacts with the CD1d-sulfatide complex has been recently reported [78]. Whereas the type I NKT T-cell receptor exclusively contacts the F’ pocket of CD1d, the type II NKT T-cell receptor binds orthogonally above the A’ pocket of CD1d, emphasizing different CD1d points of contact. More importantly, T cells highly reactive to sulfatides are increased in number and CD1d is upregulated in the central nervous system of patients with experimental autoimmune encephalomyelitis [33, 50]. The presence of the sulfate group and the β-anomeric linkage are critical for CD1d activation-dependent T cells [94]. The dominant sulfatide species for CD1d-dependent immune responses is a C24:1 [121], which bears one unsaturation at the 8–9 position (Fig. 3.1). The crystal structure of the CD1d-C24:1 sulfatide complex shows the acyl chain in the A’ pocket, whereas the sphingosine chain associates with the F’ pocket, leaving the sulfated head group exposed at the protein surface [121].
3.3.4 Host-Pathogen Interactions
The action of protein toxins from pathogenic organisms requires specific sphingolipids at the cell surface to mediate protein endocytosis and to enhance the virulence of the pathogen. Sulfatide recognition by pathogen proteins includes the coli surface antigen 6, the heat-stable toxin b, and the 987P-fimbriae from Escherichia coli [14, 30, 51], and heat shock proteins from Helicobacter pylori [43, 44]. The only structural data reported for this class of toxins is that for the Naja atra Taiwanese Cobra cardiotoxin A3 (CTX -A3) in complex with sulfatides using hexaethylene glycol monodecyl ether detergent as a membrane mimetic [110]. CTX-A3 acts as a toxin by a sulfatide-dependent internalization mechanism that leads to pore formation in the host cell membrane [115]. The crystal structure of CTX-A3 reveals a dimer of two β-sheet proteins, an oligomerization state that is induced upon sulfatide binding. In the CTX-A3-sulfatide complex, the sulfatide head group is buried so that the sulfate group forms a hydrogen bond with the amino group of the residue Lys35, whereas the galactose sugar forms hydrogen bonds with the amino groups of the residues Lys12 and Lys18 and the carbonyl oxygen group on Arg36 and Cys38 [110]. The side chain of Lys44 interacts with the amide region of the ceramide backbone through a single hydrogen bond. The remaining lipid tail becomes exposed to the detergent-enriched solvent that facilitates the dimerization of CTX-A3. Membrane insertion and pore formation by CTX-A3 requires both protein and sulfatide conformational changes [106] and the presence of sulfatide-containing lipid domains [115].
Glycosphingolipids are also employed as receptors for virus infection. Both galactocerebrosides and sulfatides facilitate HIV type 1 virus binding to the Cd4− cell surface via the viral envelope gp120 protein [22]. Similarly, sulfatides are thought to be alternative cell surface receptors for the Influenza A virus [99] and the vaccinia virus [80]. In addition, sulfatides have been shown to enhance the formation and release of the progeny of infectious Influenza A viruses as well as translocation of newly synthetized viral nucleoprotein to the cytoplasm [101]. Indeed, sulfatide administration prevents cell viral infection [22, 99, 112] as it has been demonstrated for the bovine immunodeficiency virus, in which its internalization is inhibited by the glycosphingolipid during syncytium formation [112]. More recently, Kumar and colleagues demonstrated that sulfatide administration in mice inhibits HIV type 1 replication more efficiently than treatment with the nucleoside analog reverse transcriptase inhibitor azidothymidine [98]. This is in agreement with the observation that antibodies that neutralize HIV-1 also recognize sulfatides [69]. Furthermore, the presence of sulfatides enhances mice hematopoiesis, which is usually lost during HIV-1 infection [98]. Overall, this evidence suggests that sulfatides represent novel tools to target viral infections.
3.4 Implications of Sulfatides in Disease Development and Progression
3.4.1 Cardiovascular Diseases
Sulfatides are known to play a critical role in the development of cardiovascular disease. Indeed, the measurement of serum sulfatide levels has been proposed to predict the incidence of cardiovascular disease in patients with end-stage renal disease (ESRD) [40]. The level of sulfatides in ESRD patients undergoing hemodialysis therapy and those with cardiovascular disease is consistently lower than in healthy individuals [40]. Patients with kidney transplantation show a significant increase of serum sulfatides in a time-dependent manner, which is correlated with an increment of platelet levels [111]. The recovery of sulfatide levels may be associated with the attenuation of the systemic oxidative stress triggered by the chronic kidney dysfunction in these patients [111]. Sulfatides are P-selectin ligands and as such mediate platelet-leukocyte interactions via P-selectin and CD11b/CD18 (Mac-1), an integrin receptor localized at the surface of monocytes, neutrophils, and T-cells [28]. Sulfatides increase Mac-1 surface expression in neutrophils, which may contribute to the development of intimal hyperplasia after endothelial injury [95]. Further studies demonstrate that sulfatides contribute to the progress of neointimal thickening after vascular injury, which can eventually trigger atherosclerosis [45]. In the same context, erythrocyte membrane sulfatides significantly increase in sickle erythrocytes and play a relevant role in sickle cell adhesion to endothelial cells [125].
3.4.2 Cancer Diseases
Increased levels of sulfatides have been observed in renal cell carcinoma [90], well-differentiated endometrial adenocarcinoma [97], some types of lung tumors [29], brain tumors [61], and colon [74], hepatocellular [36], and ovarian cancers [62, 63]. Sulfatides have been proposed as early predictors of ovarian cancer [63]. Recently, using a combination of mass spectrometry metabolite analysis and gene expression profiles, it has been established that sulfatide levels are elevated in ovarian cancer compared to normal ovarian tissue [62]. Consistent with this observation, higher levels of mRNA that codifies for the enzymes CGT and CST, required for sulfatide synthesis, are also detected in epithelial ovarian carcinoma cells, whereas the levels of ASA, saposin, and galactosylceramidase remain unchanged [62]. Taken together, measurements of sulfatide levels using mass spectrometry analysis of tumor tissues represent an excellent and sensitive tool to be used as serum biomarkers for early tumors.
3.5 Conclusions and Future Perspectives
As summarized in this review, the role of membrane sulfatides in the nervous system, innate and adaptive immunity, platelet adhesion and aggregation, and bacterial and viral infection is clearly emerging. However, there are several questions about how membrane sulfatides signal that need to be addressed. For example, a precise measurement of membrane sulfatide levels elicited by external cues is required to understand sulfatide-mediated signaling. Also, the levels of the enzymes that participate in the synthesis and degradation of sulfatides should play a key role in the modulation of the membrane levels of the sphingolipid.
The number of identified sulfatide-binding proteins has substantially increased over the past 15 years. The general sulfatide binding mechanism consists of the formation of hydrogen bonds between the acyl chains of the sphingolipid with residues located in the hydrophobic cavity and accompanied by a few hydrogen bonds and electrostatic interactions between the side chain of basic residues of the protein and the negatively charged sulfate group of the galactose moiety. Perhaps, the key role of sulfatides center on the features of their acyl chains as they interact with protein hydrophobic cavities leaving, in some cases, the head group exposed at the surface of the protein. Thus, development of high-resolution methods for the discrimination of sulfatides with different fatty acid compositions is warranted. This is important as sulfatides with specific acyl chains lengths, unsaturation, or even hydroxylation modifications are tissue-dependent. Furthermore, predicting a sulfatide-binding site from the amino acid sequence of a protein is not an easy task. Whereas sulfatide-binding sites typically exhibit basic clusters of residues that follow the sequence BXBXBX or XBBXBX, some sulfatide-binding proteins exhibit unique sulfatide-binding basic motifs and some others do not employ basic residues at all.
With recent high-resolution structures of sulfatide-binding proteins we may also soon understand the role of sulfatides in protein membrane targeting as well as intra- and extracellular sulfatide-dependent protein dynamics. However, we still lack the information about sulfatide dynamics at membranes, its intracellular distribution of the glycosphingolipid, or its presence and relative concentration in lipid rafts. Moreover, the engagement of sulfatides in cardiovascular and cancer diseases makes this area of research clinically relevant. The identification of additional sulfatide-binding proteins and the appropriate measurement of sulfatide levels in serum and tumor tissues will certainly contribute to early prognosis.
References
Adams JC, Lawler J (2011) The thrombospondins. CSH Perspect Biol 3(10):a009712
Ahn VE, Faull KF, Whitelegge JP, Fluharty AL, Prive GG (2003) Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc Natl Acad Sci USA 100:38–43
Alajlouni R, Drahos KE, Finkielstein CV, Capelluto DG (2011) Lipid-mediated membrane binding properties of Disabled-2. Biochim Biophys Acta 1808:2734–2744
Alderson NL, Rembiesa BM, Walla MD, Bielawska A, Bielawski J, Hama H (2004) The human FA2H gene encodes a fatty acid 2-hydroxylase. J Biol Chem 279:48562–48568
Andrews RK, Booth WJ, Bendall LJ, Berndt MC (1995) The amino Acid sequence glutamine-628 to valine-646 within the A1 repeat domain mediates binding of von Willebrand factor to bovine brain sulfatides and equine tendon collagen. Platelets 6:245–251
Barral DC, Brenner MB (2007) CD1 antigen presentation: how it works. Nat Rev Immunol 7:929–941
Bendelac A, Savage PB, Teyton L (2007) The biology of NKT cells. Annu Rev Immunol 25:297–336
Blomqvist M, Rhost S, Teneberg S, Lofbom L, Osterbye T, Brigl M, Mansson JE, Cardell SL (2009) Multiple tissue-specific isoforms of sulfatide activate CD1d-restricted type II NKT cells. Eur J Immunol 39:1726–1735
Borthakur G, Cruz MA, Dong JF, McIntire L, Li F, Lopez JA, Thiagarajan P (2003) Sulfatides inhibit platelet adhesion to von Willebrand factor in flowing blood. J Thromb Haemost 1:1288–1295
Bruun JM, Roeske-Nielsen A, Richelsen B, Fredman P, Buschard K (2007) Sulfatide increases adiponectin and decreases TNF-alpha, IL-6, and IL-8 in human adipose tissue in vitro. Mol Cell Endocrinol 263:142–148
Calderon RO, Attema B, DeVries GH (1995) Lipid composition of neuronal cell bodies and neurites from cultured dorsal root ganglia. J Neurochem 64:424–429
Chen M, Geng JG (2006) P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch Immunol Ther Exp (Warsz) 54:75–84
Chi S, Qi Z (2006) Regulatory effect of sulphatides on BKCa channels. Br J Pharmacol 149:1031–1038
Choi BK, Schifferli DM (1999) Lysine residue 117 of the FasG adhesin of enterotoxigenic Escherichia coli is essential for binding of 987P fimbriae to sulfatide. Infect Immun 67:5755–5761
Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Popko B (1996) Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell 86:209–219
D’Hooge R, Van Dam D, Franck F, Gieselmann V, De Deyn PP (2001) Hyperactivity, neuromotor defects, and impaired learning and memory in a mouse model for metachromatic leukodystrophy. Brain Res 907:35–43
De Libero G, Mori L (2012) Novel insights into lipid antigen presentation. Trends Immunol 33:103–111
Drahos KE, Welsh JD, Finkielstein CV, Capelluto DG (2009) Sulfatides partition disabled-2 in response to platelet activation. PLoS One 4:e8007
Dupree JL, Coetzee T, Suzuki K, Popko B (1998) Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J Neurocytol 27:649–659
Eckhardt M (2008) The role and metabolism of sulfatide in the nervous system. Mol Neurobiol 37:93–103
Eckhardt M, Hedayati KK, Pitsch J, Lullmann-Rauch R, Beck H, Fewou SN, Gieselmann V (2007) Sulfatide storage in neurons causes hyperexcitability and axonal degeneration in a mouse model of metachromatic leukodystrophy. J Neurosci 27:9009–9021
Fantini J, Hammache D, Delezay O, Pieroni G, Tamalet C, Yahi N (1998) Sulfatide inhibits HIV-1 entry into CD4-/CXCR4+ cells. Virology 246:211–220
Fredman P, Mansson JE, Rynmark BM, Josefsen K, Ekblond A, Halldner L, Osterbye T, Horn T, Buschard K (2000) The glycosphingolipid sulfatide in the islets of Langerhans in rat pancreas is processed through recycling: possible involvement in insulin trafficking. Glycobiology 10:39–50
Frenette PS, Johnson RC, Hynes RO, Wagner DD (1995) Platelets roll on stimulated endothelium in vivo: an interaction mediated by endothelial P-selectin. Proc Natl Acad Sci U S A 92:7450–7454
Frenette PS, Moyna C, Hartwell DW, Lowe JB, Hynes RO, Wagner DD (1998) Platelet-endothelial interactions in inflamed mesenteric venules. Blood 91:1318–1324
Frenette PS, Wagner DD (1996) Adhesion molecules–Part 1. N Engl J Med 334(23):1526–1529
Frenette PS, Wagner DD (1996) Adhesion molecules–Part II: blood vessels and blood cells. N Engl J Med 335:43–45
Gahmberg CG (1997) Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr Opin Cell Biol 9:643–650
Gnewuch C, Jaques G, Havemann K, Wiegandt H (1994) Re-assessment of acidic glycosphingolipids in small-cell-lung-cancer tissues and cell lines. Int J Cancer 8:125–126
Goncalves C, Berthiaume F, Mourez M, Dubreuil JD (2008) Escherichia coli STb toxin binding to sulfatide and its inhibition by carragenan. FEMS Microbiol Lett 281:30–35
Guchhait P, Shrimpton CN, Honke K, Rumbaut RE, Lopez JA, Thiagarajan P (2008) Effect of an anti-sulfatide single-chain antibody probe on platelet function. Thromb Haemost 99:552–557
Guo NH, Krutzsch HC, Negre E, Vogel T, Blake DA, Roberts DD (1992) Heparin- and sulfatide-binding peptides from the type I repeats of human thrombospondin promote melanoma cell adhesion. Proc Natl Acad Sci U S A 89:3040–3044
Halder RC, Jahng A, Maricic I, Kumar V (2007) Mini review: immune response to myelin-derived sulfatide and CNS-demyelination. Neurochem Res 32:257–262
Harrison D, Hussain SA, Combs AC, Ervasti JM, Yurchenco PD, Hohenester E (2007) Crystal structure and cell surface anchorage sites of laminin alpha1LG4-5. J Biol Chem 282:11573–11581
Hirahara Y, Tsuda M, Wada Y, Honke K (2000) cDNA cloning, genomic cloning, and tissue-specific regulation of mouse cerebroside sulfotransferase. Eur J Biochem 267:1909–1917
Hiraiwa N, Fukuda Y, Imura H, Tadano-Aritomi K, Nagai K, Ishizuka I, Kannagi R (1990) Accumulation of highly acidic sulfated glycosphingolipids in human hepatocellular carcinoma defined by a series of monoclonal antibodies. Cancer Res 50:2917–2928
Hohenester E, Tisi D, Talts JF, Timpl R (1999) The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Mol Cell 4:783–792
Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, Taniguchi N (2002) Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci USA 99(7):4227–4232. doi:10.1073/pnas.032068299
Honke K, Zhang Y, Cheng X, Kotani N, Taniguchi N (2004) Biological roles of sulfoglycolipids and pathophysiology of their deficiency. Glycoconj J 21:59–62
Hu R, Li G, Kamijo Y, Aoyama T, Nakajima T, Inoue T, Node K, Kannagi R, Kyogashima M, Hara A (2007) Serum sulfatides as a novel biomarker for cardiovascular disease in patients with end-stage renal failure. Glycoconj J 24:565–571
Huang CL, Cheng JC, Liao CH, Stern A, Hsieh JT, Wang CH, Hsu HL, Tseng CP (2004) Disabled-2 is a negative regulator of integrin alpha(IIb)beta(3)-mediated fibrinogen adhesion and cell signaling. J Biol Chem 279:42279–42289
Huang CL, Cheng JC, Stern A, Hsieh JT, Liao CH, Tseng CP (2006) Disabled-2 is a novel alphaIIb-integrin-binding protein that negatively regulates platelet-fibrinogen interactions and platelet aggregation. J Cell Sci 119:4420–4430
Huesca M, Borgia S, Hoffman P, Lingwood CA (1996) Acidic pH changes receptor binding specificity of Helicobacter pylori: a binary adhesion model in which surface heat shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect Immun 64:2643–2648
Huesca M, Goodwin A, Bhagwansingh A, Hoffman P, Lingwood CA (1998) Characterization of an acidic-pH-inducible stress protein (hsp70), a putative sulfatide binding adhesin, from Helicobacter pylori. Infect Immun 66(9):4061–4067
Inoue T, Taguchi I, Abe S, Li G, Hu R, Nakajima T, Hara A, Aoyama T, Kannagi R, Kyogashima M, Node K (2010) Sulfatides are associated with neointimal thickening after vascular injury. Atherosclerosis 211(1):291–296
Isaac G, Pernber Z, Gieselmann V, Hansson E, Bergquist J, Mansson JE (2006) Sulfatide with short fatty acid dominates in astrocytes and neurons. FEBS J 273:1782–1790
Ishizuka I (1997) Chemistry and functional distribution of sulfoglycolipids. Prog Lipid Res 36(4):245–319
Iwamura C, Shinoda K, Endo Y, Watanabe Y, Tumes DJ, Motohashi S, Kawahara K, Kinjo Y, Nakayama T (2012) Regulation of memory CD4 T-cell pool size and function by natural killer T cells in vivo. Proc Natl Acad Sci U S A 109:16992–16997
Jackson SP (2007) The growing complexity of platelet aggregation. Blood 109:5087–5095
Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V (2004) Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med 199:947–957
Jansson L, Tobias J, Jarefjall C, Lebens M, Svennerholm AM, Teneberg S (2009) Sulfatide recognition by colonization factor antigen CS6 from enterotoxigenic Escherichia coli. PLoS One 4:e4487
Jeon SB, Yoon HJ, Park SH, Kim IH, Park EJ (2008) Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J Immunol 181:8077–8087
Kalb E, Engel J (1991) Binding and calcium-induced aggregation of laminin onto lipid bilayers. J Biol Chem 266:19047–19052
Kamlekar RK, Gao Y, Kenoth R, Molotkovsky JG, Prendergast FG, Malinina L, Patel DJ, Wessels WS, Venyaminov SY, Brown RE (2010) Human GLTP: three distinct functions for the three tryptophans in a novel peripheral amphitropic fold. Biophys J 99:2626–2635
Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, Steinman L, Robinson WH (2006) Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med 12:138–143
Kolter T, Sandhoff K (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol 21:81–103
Kushi Y, Arita M, Ishizuka I, Kasama T, Fredman P, Handa S (1996) Sulfatide is expressed in both erythrocytes and platelets of bovine origin. Biochim Biophys Acta 1304(3):254–262
Kwiatkowska K (2010) One lipid, multiple functions: how various pools of PI(4,5)P(2) are created in the plasma membrane. Cell Mol Life Sci 67:3927–3946
Kyogashima M (2004) The role of sulfatide in thrombogenesis and haemostasis. Arch Biochem Biophys 426:157–162
Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, Wagner DD, Furie B (1989) PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 59:305–312
Li J, Pearl DK, Pfeiffer SE, Yates AJ (1994) Patterns of reactivity with anti-glycolipid antibodies in human primary brain tumors. J Neurosci Res 39:148–158
Liu Y, Chen Y, Momin A, Shaner R, Wang E, Bowen NJ, Matyunina LV, Walker LD, McDonald JF, Sullards MC, Merrill AH Jr (2010) Elevation of sulfatides in ovarian cancer: an integrated transcriptomic and lipidomic analysis including tissue-imaging mass spectrometry. Mol Cancer 9:186
Makhlouf AM, Fathalla MM, Zakhary MA, Makarem MH (2004) Sulfatides in ovarian tumors: clinicopathological correlates. Int J Gynecol Cancer 14:89–93
Malinina L, Malakhova ML, Kanack AT, Lu M, Abagyan R, Brown RE, Patel DJ (2006) The liganding of glycolipid transfer protein is controlled by glycolipid acyl structure. PLoS Biol 4:e362
Malinina L, Malakhova ML, Teplov A, Brown RE, Patel DJ (2004) Structural basis for glycosphingolipid transfer specificity. Nature 430:1048–1053
Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL (2006) Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia 53:372–381
Matsushita T, Meyer D, Sadler JE (2000) Localization of von willebrand factor-binding sites for platelet glycoprotein Ib and botrocetin by charged-to-alanine scanning mutagenesis. J Biol Chem 275:11044–11049
Mattjus P (2009) Glycolipid transfer proteins and membrane interaction. Biochim Biophys Acta 1788:267–272
Matyas GR, Beck Z, Karasavvas N, Alving CR (2009) Lipid binding properties of 4E10, 2F5, and WR304 monoclonal antibodies that neutralize HIV-1. Biochim Biophys Acta 1788:660–665
Merten M, Beythien C, Gutensohn K, Kuhnl P, Meinertz T, Thiagarajan P (2005) Sulfatides activate platelets through P-selectin and enhance platelet and platelet-leukocyte aggregation. Arterioscler Thromb Vasc Biol 25:258–263
Merten M, Thiagarajan P (2000) P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation 102:1931–1936
Merten M, Thiagarajan P (2001) Role for sulfatides in platelet aggregation. Circulation 104:2955–2960
Moody DB, Zajonc DM, Wilson IA (2005) Anatomy of CD1-lipid antigen complexes. Nat Rev Immunol 5:387–399
Morichika H, Hamanaka Y, Tai T, Ishizuka I (1996) Sulfatides as a predictive factor of lymph node metastasis in patients with colorectal adenocarcinoma. Cancer 78(1):43–47
Nakajima T, Kamijo Y, Yuzhe H, Kimura T, Tanaka N, Sugiyama E, Nakamura K, Kyogashima M, Hara A, Aoyama T (2013) Peroxisome proliferator-activated receptor alpha mediates enhancement of gene expression of cerebroside sulfotransferase in several murine organs. Glycoconj J in press
Nakayama T, Matsushita T, Yamamoto K, Mutsuga N, Kojima T, Katsumi A, Nakao N, Sadler JE, Naoe T, Saito H (2008) Identification of amino acid residues responsible for von Willebrand factor binding to sulfatide by charged-to-alanine-scanning mutagenesis. Int J Hematol 87:363–370
Norton WT, Autilio LA (1965) The chemical composition of bovine CNS myelin. Ann N Y Acad Sci 122:77–85
Patel O, Pellicci DG, Gras S, Sandoval-Romero ML, Uldrich AP, Mallevaey T, Clarke AJ, Le Nours J, Theodossis A, Cardell SL, Gapin L, Godfrey DI, Rossjohn J (2012) Recognition of CD1d-sulfatide mediated by a type II natural killer T cell antigen receptor. Nat Immunol 13:857–863
Peng L, Suzuki K (1987) Ultrastructural study of neurons in metachromatic leukodystrophy. Clin Neuropathol 6:224–230
Perino J, Foo CH, Spehner D, Cohen GH, Eisenberg RJ, Crance JM, Favier AL (2011) Role of sulfatide in vaccinia virus infection. Biol Cell 103:319–331
Pernber Z, Molander-Melin M, Berthold CH, Hansson E, Fredman P (2002) Expression of the myelin and oligodendrocyte progenitor marker sulfatide in neurons and astrocytes of adult rat brain. J Neurosci Res 69:86–93
Plow EF, Pierschbacher MD, Ruoslahti E, Marguerie GA, Ginsberg MH (1985) The effect of Arg-Gly-Asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets. Proc Natl Acad Sci U S A 82:8057–8061
Pouyani T, Seed B (1995) PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell 83:333–343
Ramakrishnan H, Hedayati KK, Lullmann-Rauch R, Wessig C, Fewou SN, Maier H, Goebel HH, Gieselmann V, Eckhardt M (2007) Increasing sulfatide synthesis in myelin-forming cells of arylsulfatase A-deficient mice causes demyelination and neurological symptoms reminiscent of human metachromatic leukodystrophy. J Neurosci 27:9482–9490
Roberts DD, Haverstick DM, Dixit VM, Frazier WA, Santoro SA, Ginsburg V (1985) The platelet glycoprotein thrombospondin binds specifically to sulfated glycolipids. J Biol Chem 260:9405–9411
Roberts DD, Rao CN, Liotta LA, Gralnick HR, Ginsburg V (1986) Comparison of the specificities of laminin, thrombospondin, and von Willebrand factor for binding to sulfated glycolipids. J Biol Chem 261:6872–6877
Roeske-Nielsen A, Fredman P, Mansson JE, Bendtzen K, Buschard K (2004) Beta-galactosylceramide increases and sulfatide decreases cytokine and chemokine production in whole blood cells. Immunol Lett 91:205–211
Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, McIntire LV, Berndt MC, Lopez JA (1999) The glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J Exp Med 190:803–814
Saadat L, Dupree JL, Kilkus J, Han X, Traka M, Proia RL, Dawson G, Popko B (2010) Absence of oligodendroglial glucosylceramide synthesis does not result in CNS myelin abnormalities or alter the dysmyelinating phenotype of CGT-deficient mice. Glia 58:391–398
Sakakibara N, Gasa S, Kamio K, Makita A, Nonomura K, Togashi M, Koyanagi T, Hatae Y, Takeda K (1991) Distinctive glycolipid patterns in Wilms’ tumor and renal cell carcinoma. Cancer Lett 57:187–192
Samygina VR, Popov AN, Cabo-Bilbao A, Ochoa-Lizarralde B, Goni-de-Cerio F, Zhai X, Molotkovsky JG, Patel DJ, Brown RE, Malinina L (2011) Enhanced selectivity for sulfatide by engineered human glycolipid transfer protein. Structure 19:1644–1654
Sandhoff R, Grieshaber H, Djafarzadeh R, Sijmonsma TP, Proudfoot AE, Handel TM, Wiegandt H, Nelson PJ, Grone HJ (2005) Chemokines bind to sulfatides as revealed by surface plasmon resonance. Biochim Biophys Acta 1687:52–63
Sarlieve LL, Zalc B, Neskovic NM, Zanetta JP, Rebel G (1984) Structure and immunological localization of spleen sulfolipid. Biochim Biophys Acta 795:166–168
Shamshiev A, Gober HJ, Donda A, Mazorra Z, Mori L, De Libero G (2002) Presentation of the same glycolipid by different CD1 molecules. J Exp Med 195:1013–1021
Shimazawa M, Kondo K, Hara H, Nakashima M, Umemura K (2005) Sulfatides, L- and P-selectin ligands, exacerbate the intimal hyperplasia occurring after endothelial injury. Eur J Pharmacol 520:118–126
Simonis D, Schlesinger M, Seelandt C, Borsig L, Bendas G (2010) Analysis of SM4 sulfatide as a P-selectin ligand using model membranes. Biophys Chem 150:98–104
Sugiyama T, Miyazawa M, Mikami M, Goto Y, Nishijima Y, Ikeda M, Hirasawa T, Muramatsu T, Takekoshi S, Iwamori M (2012) Enhanced expression of sulfatide, a sulfated glycolipid, in well-differentiated endometrial adenocarcinoma. Int J Gynecol Cancer 22:1192–1197
Sundell IB, Halder R, Zhang M, Maricic I, Koka PS, Kumar V (2010) Sulfatide administration leads to inhibition of HIV-1 replication and enhanced hematopoeisis. J Stem Cells 5:33–42
Suzuki T, Sometani A, Yamazaki Y, Horiike G, Mizutani Y, Masuda H, Yamada M, Tahara H, Xu G, Miyamoto D, Oku N, Okada S, Kiso M, Hasegawa A, Ito T, Kawaoka Y, Suzuki Y (1996) Sulphatide binds to human and animal influenza A viruses, and inhibits the viral infection. Biochem J 318:389–393
Svennerholm L, Bostrom K, Fredman P, Jungbjer B, Mansson JE, Rynmark BM (1992) Membrane lipids of human peripheral nerve and spinal cord. Biochim Biophys Acta 1128:1–7
Takahashi T, Murakami K, Nagakura M, Kishita H, Watanabe S, Honke K, Ogura K, Tai T, Kawasaki K, Miyamoto D, Hidari KI, Guo CT, Suzuki Y, Suzuki T (2008) Sulfatide is required for efficient replication of influenza A virus. J Virol 82:5940–5950
Talts JF, Andac Z, Gohring W, Brancaccio A, Timpl R (1999) Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J 18:863–870
Taraboletti G, Rao CN, Krutzsch HC, Liotta LA, Roberts DD (1990) Sulfatide-binding domain of the laminin A chain. J Biol Chem 265:12253–12258
Theilmeier G, Lenaerts T, Remacle C, Collen D, Vermylen J, Hoylaerts MF (1999) Circulating activated platelets assist THP-1 monocytoid/endothelial cell interaction under shear stress. Blood 94:2725–2734
Thudichum JL (1884) A treatise on the chemical constitution of the brain. Balliere, Tindall, and Cox, London
Tjong SC, Wu PL, Wang CM, Huang WN, Ho NL, Wu WG (2007) Role of glycosphingolipid conformational change in membrane pore forming activity of cobra cardiotoxin. Biochemistry 46:12111–12123
van Zyl R, Gieselmann V, Eckhardt M (2010) Elevated sulfatide levels in neurons cause lethal audiogenic seizures in mice. J Neurochem 112:282–295
Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA (2010) Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci USA 107:11993–11998
Vos JP, Lopes-Cardozo M, Gadella BM (1994) Metabolic and functional aspects of sulfogalactolipids. Biochim Biophys Acta 1211:125–149
Wang CH, Liu JH, Lee SC, Hsiao CD, Wu WG (2006) Glycosphingolipid-facilitated membrane insertion and internalization of cobra cardiotoxin. The sulfatide.cardiotoxin complex structure in a membrane-like environment suggests a lipid-dependent cell-penetrating mechanism for membrane binding polypeptides. J Biol Chem 281:656–667
Wang L, Kamijo Y, Matsumoto A, Nakajima T, Higuchi M, Kannagi R, Kyogashima M, Aoyama T, Hara A (2011) Kidney transplantation recovers the reduction level of serum sulfatide in ESRD patients via processes correlated to oxidative stress and platelet count. Glycoconj J 28:125–135
Watarai S, Onuma M, Yamamoto S, Yasuda T (1990) Inhibitory effect of liposomes containing sulfatide or cholesterol sulfate on syncytium formation induced by bovine immunodeficiency virus-infected cells. J Biochem 108:507–509
Welsh JD, Charonko JJ, Salmanzadeh A, Drahos KE, Shafiee H, Stremler MA, Davalos RV, Capelluto DG, Vlachos PP, Finkielstein CV (2011) Disabled-2 modulates homotypic and heterotypic platelet interactions by binding to sulfatides. Br J Haematol 154:122–133
Winzeler AM, Mandemakers WJ, Sun MZ, Stafford M, Phillips CT, Barres BA (2011) The lipid sulfatide is a novel myelin-associated inhibitor of CNS axon outgrowth. J Neurosci 31:6481–6492
Wu PL, Chiu CR, Huang WN, Wu WG (2012) The role of sulfatide lipid domains in the membrane pore-forming activity of cobra cardiotoxin. Biochim Biophys Acta 1818:1378–1385
Xiao S, Charonko JJ, Fu X, Salmanzadeh A, Davalos RV, Vlachos PP, Finkielstein CV, Capelluto DG (2012) Structure, sulfatide binding properties, and inhibition of platelet aggregation by a Disabled-2 protein-derived peptide. J Biol Chem 287:37691–37702
Yaghootfam A, Sorkalla T, Haberlein H, Gieselmann V, Kappler J, Eckhardt M (2007) Cerebroside sulfotransferase forms homodimers in living cells. Biochemistry 46:9260–9269
Yang H, Lang S, Zhai Z, Li L, Kahr WH, Chen P, Brkic J, Spring CM, Flick MJ, Degen JL, Freedman J, Ni H (2009) Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression. Blood 114:425–436
Yuki D, Sugiura Y, Zaima N, Akatsu H, Hashizume Y, Yamamoto T, Fujiwara M, Sugiyama K, Setou M (2011) Hydroxylated and non-hydroxylated sulfatide are distinctly distributed in the human cerebral cortex. Neuroscience 193:44–53
Zajonc DM, Elsliger MA, Teyton L, Wilson IA (2003) Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 A. Nat Immunol 4:808–815
Zajonc DM, Maricic I, Wu D, Halder R, Roy K, Wong CH, Kumar V, Wilson IA (2005) Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J Exp Med 202:1517–1526
Zeng Z, Castano AR, Segelke BW, Stura EA, Peterson PA, Wilson IA (1997) Crystal structure of mouse CD1: an MHC-like fold with a large hydrophobic binding groove. Science 277:339–345
Zhang X, Nakajima T, Kamijo Y, Li G, Hu R, Kannagi R, Kyogashima M, Aoyama T, Hara A (2009) Acute kidney injury induced by protein-overload nephropathy down-regulates gene expression of hepatic cerebroside sulfotransferase in mice, resulting in reduction of liver and serum sulfatides. Biochem Biophys Res Commun 390:1382–1388
Zhang XL, Rafi MA, DeGala G, Wenger DA (1990) Insertion in the mRNA of a metachromatic leukodystrophy patient with sphingolipid activator protein-1 deficiency. Proc Natl Acad Sci USA 87:1426–1430
Zhou Z, Thiagarajan P, Udden M, Lopez JA, Guchhait P (2011) Erythrocyte membrane sulfatide plays a crucial role in the adhesion of sickle erythrocytes to endothelium. Thromb Haemost 105:1046–1052
Acknowledgements
We thank Janet Webster for critical reading and comments on the manuscript. Work in the Capelluto laboratory is supported by the American Heart Association, the Thomas F. and Kate Miller Jeffress Memorial Trust, the National Science Foundation (IOS), and the National Institutes of Health (NICHD). C. V. Finkielstein’s research is funded by the National Science Foundation CAREER Award and by the Avon Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Xiao, S., Finkielstein, C.V., Capelluto, D.G.S. (2013). The Enigmatic Role of Sulfatides: New Insights into Cellular Functions and Mechanisms of Protein Recognition. In: Capelluto, D. (eds) Lipid-mediated Protein Signaling. Advances in Experimental Medicine and Biology, vol 991. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6331-9_3
Download citation
DOI: https://doi.org/10.1007/978-94-007-6331-9_3
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6330-2
Online ISBN: 978-94-007-6331-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)