
Abstract
Intraneuronal accumulation of abnormally hyperphosphorylated tau in the brain is a histopathological hallmark of Alzheimer’s disease and a family of related neurodegenerative disorders collectively called tauopathies. At present there is no effective treatment available for these progressive neurodegenerative diseases which are clinically characterized by dementia in mid to old-age. Here we report the treatment of 14–17-months-old 3xTg-AD mice with tau antibodies 43D (tau 6–18) and 77E9 (tau 184–195) to the N-terminal projection domain of tau or mouse IgG as a control by intraperitoneal injection once a week for 4 weeks, and the effects of the passive immunization on reduction of hyperphosphorylated tau, Aβ accumulation and cognitive performance in these animals. We found that treatment with tau antibodies 43D and 77E9 reduced total tau level, decreased tau hyperphosphorylated at Ser199, Ser202/Thr205 (AT8), Thr205, Ser262/356 (12E8), and Ser396/404 (PHF-1) sites, and a trend to reduce Aβ pathology. Most importantly, targeting N-terminal tau especially by 43D (tau 6–18) improved reference memory in the Morris water maze task in 3xTg-AD mice. We did not observe any abnormality in general physical characteristics of the treated animals with either of the two antibodies during the course of this study. Taken together, our studies demonstrate for the first time (1) that passive immunization targeting normal tau can effectively clear the hyperphosphorylated protein and possibly reduce Aβ pathology from the brain and (2) that targeting N-terminal projection domain of tau containing amino acid 6–18 is especially beneficial. Thus, targeting selective epitopes of N-terminal domain of tau may present a novel effective therapeutic opportunity for Alzheimer disease and other tauopathies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common type of dementia and is characterized by progressive loss of memory and other cognitive functions (Thies et al. 2013). The two major histopathological hallmarks in brains of AD patients are extracellular senile plaques consisting of amyloid-β (Aβ) peptides (Glenner and Wong 1984) and intracellular neurofibrillary tangles (NFTs), composed of abnormally hyperphosphorylated tau protein (Grundke-Iqbal et al. 1986). The tau pathology made up of the hyperphosphorylated tau is also a hallmark of several neurodegenerative disorders which include frontolobar dementias, the corticobasal degeneration, Progressive supranuclear palsy, Pick disease, Guam Parkinsonism dementia complex, and dementia pugilistica. The density of tau lesions directly correlates with dementia. At present, there is no effective treatment available for AD and related tauopathies. Most therapeutic approaches for AD mainly focused on reducing Aβ levels in the brain which included blocking the formation of Aβ by modulating β-secretase and/or γ-secretase, promoting the clearance of Aβ, preventing aggregation of Aβ and destabilizing Aβ oligomers; however, to date, none of these approaches have yielded a successful outcome (Giacobini and Gold 2013; Reitz 2012). The first tau immunotherapy targeting active immunization against truncated tau, tau 151–391, is now in Phase I human clinical trial (Axon Neuroscience, Bratislava, Slovakia).
Harnessing the immune system to prevent or remove the Aβ and tau aggregates is an emerging and promising disease-modifying approach for AD (Winblad et al. 2014). In the last decade, Aβ immunotherapy progressed from preclinical studies in transgenic mouse models of AD to clinical trials in humans (Lambracht-Washington and Rosenberg 2013). While immunization with Aβ increased the clearance of Aβ, it failed to reduce neurofibrillary pathology and prevent progressive neurodegeneration (Holmes et al. 2008). Furthermore, Aβ immunotherapies showed little cognitive benefit in mild-to-moderate AD patients (Doody et al. 2014; Salloway et al. 2014). Importantly, multivariate analyses indicate that neurofibrillary tangles, neuron number loss, and synapse loss, but not amyloid load, strongly correlate with cognitive impairment in AD patients (Alafuzoff et al. 1987; Arriagada et al. 1992; Giannakopoulos et al. 2003; Terry et al. 1991; Tomlinson et al. 1970). These findings have led to the belief that targeting tau pathology might be more effective than Aβ-directed therapy for AD.
Intracellular aggregates of tau locate inside of neuron, which complicates its targeting for clearance. However, active immunization with recombinant α-synuclein in a transgenic mouse model was found to decrease aggregates of α-synuclein, an intracellular synaptic protein that accumulates in the brains of patients with Parkinson’s Disease and AD (Masliah et al. 2005). This finding supported that intracellular proteins could also be potential targets for immunotherapy. Indeed, immunotherapy targeting pathological tau has been tested in several AD transgenic mouse models with different phospho-tau peptides. Accumulating evidence from these preclinical studies has shown that active immunization in transgenic tauopathy mouse models using tau phospho-peptides reduce tau pathology (Asuni et al. 2007; Bi et al. 2011; Boimel et al. 2010; Boutajangout et al. 2011; Gu et al. 2013; Troquier et al. 2012) and rescue or slow the cognitive decline (Asuni et al. 2007; Boutajangout et al. 2011; Troquier et al. 2012). Passive immunotherapy using antibodies against pathology of tau has also been shown to slow disease progression (Boutajangout et al. 2011; Chai et al. 2011; Yanamandra et al. 2013).
Tau pathology is believed to spread transcellularly (Avila et al. 2014). The abnormally hyperphosphorylated/oligomeric tau released in the extracellular space from the affected neurons is suspected to serve as the seeds for the spread of tau pathology by the ingesting cells (Clavaguera et al. 2009; Flight 2013; Frost et al. 2009). Therefore, tau immunotherapy may clear extracellular tau that is involved in the spreading of tau pathology (Zilka et al. 2008). Yanamandra et al. (2013) screened tau antibodies with the ability to block seeding activity present in P301S brain; infusion of tau antibodies specific for blocking P301S tau seeds into the lateral ventricle of P301S mice for 3 months reduced hyperphosphorylated, aggregated and insoluble tau, blocked development of tau seeding activity, and improved cognitive deficits. This finding indicated that tau immunotherapy can target its transcellular propagation.
Tau protein consists of an N-terminal projection region, a proline-rich domain, a microtubule-binding domain, and a C-terminal region (Mandelkow et al. 1996). Although the role of tau in regulating microtubule dynamics is extensively established, much less is known about the functional role of the N-terminal domain of tau on neuron survival. A 17-kD N-terminal tau fragment generated by calpain cleavage, comprising residues amino acid 45–230, was proposed to mediate Aβ-induced toxicity (Park and Ferreira 2005), and mediate tau neurotoxicity in Drosophila tauopathy model (Reinecke et al. 2011). However, the toxicity and in vivo relevance of this 17 kD fragment are debated. Garg et al. (2011) reported that this 17 kD fragment cleaved by calpain is tau 125–230, which is much shorter than previously reported tau 45–230. Furthermore, both tau 125–230 and 45–230 fragments showed no toxicity in Chinese hamster ovary (CHO) cells, neuroblastoma cells (N2a), and in primary hippocampal neurons. Other N-terminal tau fragments including tau 1–44, 26–44, 26–230, and 1–156 were reported to cause an NMDAR-mediated powerful toxicity in cerebellar granule neurons, but tau 45–230 exerted a toxicity with unknown mechanism (Amadoro et al. 2004, 2006; Corsetti et al. 2008). Tau 1–230 was also reported to protect neuron from apoptosis (Amadoro et al. 2004), which indicated that N-terminal domain of tau can be either neuroprotective or neurotoxic according to its length.
Previously we discovered that the Alzheimer abnormally hyperphosphorylated tau, instead of interacting with tubulin and promoting its assembly into microtubules, sequesters normal tau, forming oligomers and consequently filaments which can be sedimented at 100,000–200,000×g (Alonso et al. 1994, 1996; Iqbal et al. 1986; Kopke et al. 1993). This finding which formed the basis of tau spread studies (Clavaguera et al. 2009, 2013; Sanders et al. 2014) led us to test the removal of pathological tau by passive immunization using antibodies to normal tau.
In the present study, we evaluated the effects of an antibody specific to the amino terminal region of human tau (amino acid residues 6–18) and an antibody targeting a tau more distal amino terminal projection tau region in aged 3xTg-AD mice at moderate to severe stage of pathology. We found that while passive immunization with both antibodies reduced total tau and tau hyperphosphorylated at several sites, without effecting Aβ pathology, the immunization with 43D targeting tau 6–18, but not 77E9 targeting tau 184–195 improved cognition in aged 3xTg-AD mice.
Materials and methods
Antibodies and reagents
Primary antibodies used in this study are listed in Table 1. Tau antibodies 43D against tau 6–18 epitope and 77E9 against tau 184–195 epitope were generated at New York State Institute for Basic Research, Staten Island. Peroxidase-conjugated anti-mouse and anti-rabbit IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The enhanced chemiluminescence kit was from Pierce (Rockford, IL, USA). Human Aβ1–40 enzyme-linked immunosorbent assay (ELISA) kits were from Invitrogen (Carlsbad, CA, USA). DPBS buffer was from ThermoScientific, MA, USA. Other chemicals were from Sigma (St. Louis, MO, USA).
Animals and antibody injections
Animal studies were approved by our Institutional Animal Care and Use Committee (IACUC) and were according to US PHS NIH guidelines.
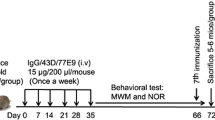
The aged female 3xTg-AD mice harboring PS1M146V, APPSWE, and tauP301L transgenes, created in the laboratory of Dr. Frank LaFerla, represent one of the most biologically relevant mouse models for AD (Oddo et al. 2003b). These mice develop amyloid plaques starting at 6 months of age and NFTs starting 12 months age, respectively, where the pathologies are predominantly restricted to the hippocampus, amygdala, and the cerebral cortex (Oddo et al. 2003a). The female 3xTg-AD mice (9–10 mice/group) were injected at 14–17 months of age intraperitoneally (i.p.) with 100 µg of 43D or 77E9 antibodies in 100 µL saline once a week for 4 weeks. Mice treated identically but with vehicle (saline) only or mouse IgG in saline were treated as controls. One day after the fourth injection, the animals were tested for spatial learning and memory by Morris water maze task. At the end of the reference memory test mice were killed, one half of the brain was fixed in 4 % paraformaldehyde for histological and immunohistological studies, and the other half was dissected into hippocampus and cerebral cortex and saved at −75 °C for biochemical analysis (Fig. 1a).

Design of study (a) and b–d immunohistochemical data which show that passive immunization targeting the N-terminal projection domain of tau with 43D and 77E9 antibodies decreases tau hyperphosphorylation at Ser202/Thr205. a Antibodies and, as control, mouse IgG or saline were administered intraperitoneally. Saline injected control group was used for behavioral studies, and IgG injected control animals were used for immunohistochemical and biochemical analysis. b Immunostaining of subiculum (SC) and CA1 from aged 3xTg-AD mice immunized with 43D, 77E9 or control IgG using AT8 antibody. c, d Scatter plots show the quantification of AT8 immunostaining load in total subiculum and CA1 region from 3 to 4 serial sections from four mice per group. Scale bar 100 μm; data are shown as percentage of mouse IgG-treated animals (100 %), mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired two-tailed t test
General examination
A general examination of all the mice was conducted in the home cages throughout the whole study. Any gross abnormalities in overall health, home cage nesting, sleeping, feeding, grooming, and condition of the fur of animals were noted; body weight was measured once a week.
Morris water maze
Morris water maze (MWM) task was used to evaluate spatial learning and memory of the mice (D’Hooge and De Deyn 2001; Morris et al. 1982). The test was performed in a circular white pool (with a diameter of 180 cm and a height of 60 cm) filled with white dye tinted water and maintained at room temperature (20 ± 1 °C). The maze was designated of two virtual principal axes with each line bisecting the maze perpendicular to the other one to divide the maze into four equal quadrants. The end of each line demarcates four cardinal points: north (N), south (S), east (E) and, west (W). A platform was positioned in the middle of one of the quadrants submerged 1 cm below water surface. Each mouse performed four trials per day for four consecutive days from semi-random start positions (Vorhees and Williams 2006) to find the hidden platform. Each trial was terminated as soon as the mouse climbed onto the hidden platform. If a mouse failed to find the platform within 90 s, it was gently guided to it. At the end of each trial, the mouse was left on the platform for 20 s, then removed, dried and returned it to its home cage. A 60 s probe test without platform was performed 24 h after the last trial. The swim path, swim distance (cm), escape latency (s), swim speed (cm/s), time spent in each quadrant (s), and distance traveled in each quadrant (cm) were recorded through an automated tracking system (Smart video tracking system, version 2.0.14, Panlab; Harvard Apparatus).
Tissue processing
After completion of the Morris water maze task, all mice were sacrificed by cervical dislocation. Forebrain cortex and hippocampus were detached immediately from the left hemisphere and frozen in dry ice for biochemical analysis. The right hemisphere was fixed in 4 % paraformaldehyde in 100 mM phosphate buffered saline (PBS) for at least 24 h at room temperature. Tissues were then post-fixed in a 30 % sucrose solution at 4 °C for overnight. Forty µm sagittal sections of the entire half hemisphere were cut using a freezing microtome. The sections were stored in glycol anti-freeze solution (ethylene glycol, glycerol and 100 mM PBS in 3:3:4 ratio) at −20 °C until further processing.
Human Aβ40 measurements by ELISA
The tissue from forebrain cortex was homogenized in 10 volumes of ice-cold guanidine hydrochloride buffer (50 mM Tris–HCl, pH 8.0, 5.0 M guanidine.HCl). The homogenate was mixed for 4 h at room temperature, and then stored at −20 °C. For ELISA, each brain homogenate was diluted 1:25 with ice-cold reaction buffer (5 % BSA, 0.03 % Tween-20, 2.1 mM AEBSF, 20 µg/mL aprotinin, 20 μg/mL leupeptin, 2.0 mM EDTA, pH 7.4] in DPBS (ThermoScientific, prod #28344) and centrifuged at 16,000×g for 20 min at 4 °C. The final concentration of AEBSF was 1 mM to prevent proteolysis of the Aβ peptides, and the final concentration of guanidine hydrochloride was 0.1 M. The supernatant was further diluted 1:1(v/v) with standard diluent buffer and assessed using ELISA kit specific for Human Aβ40 and calibrated with synthetic Aβ peptides from Invitrogen (Cat #KHB3482) according to the manufacturer’s instructions. The Aβ40 peptide standards were prepared with the same composition of the buffer used for the dilution of the samples.
Western blot analysis
Mouse brain tissue was homogenized in pre-chilled buffer containing 50 mM Tris–HCl, pH 7.4, 0.25 M sucrose, 2 mM EDTA, 10 mM β-mercaptoethanol, 0.5 mM AEBSF, 10 µg/mL aprotinin, 10 µg/mL leupeptin, 4 µg/mL pepstatin, 5 mM benzamidine, 20 mM β-glycerophosphate, 50 mM sodium fluoride, and 1 mM sodium vanadate. Each homogenate was boiled in Laemmli’s buffer for 5 min and protein concentration was measured by modified Lowry method (Bensadoun and Weinstein 1976). The samples were resolved in 10 % or 12.5 % SDS-PAGE and electro-transferred onto Immobilon-P membrane (Millipore, Bedford, MA, USA). The blots were then probed with primary antibody (Table 1) and developed with the corresponding horseradish peroxidase–conjugated secondary antibody and ECL kit (Pierce, Rockford, IL). Densitometrical quantification of protein bands in Western blots were analyzed using the Multi Gauge V3.0 software (Fuji Photo Film Co., Ltd).
Immunohistochemical staining
Immunohistochemical quantification of abnormally hyperphosphorylated tau was performed on 3–4 sections from minimum 4 mice per group. Free-floating sagittal sections were washed in 10 mM PBS (15 min × 3) and then incubated in 0.5 % Triton X-100 for 20 min. The sections were again washed in 10 mM PBS (15 min x 3) and blocked in a solution containing 5 % normal goat serum and 0.1 % Triton X-100 for 30 min. Sections were then incubated at 4 °C overnight with the mouse monoclonal antibody AT8 which recognizes tau phosphorylation at Ser202/Thr 205 (1:500, ThermoScientific, Rockford, IL, USA). The next day, after washing three times for 15 min with 10 mM PBS, Alexa 488-conjugated goat anti-mouse IgG antibody (1:500, Molecular Probes, Carlsbad, CA, USA) in 10 mM PBS with 0.05 % Tween-20 was used as secondary antibody for 2 h at room temperature. Sections were subsequently washed, mounted, and cover slipped using Fluorogel mounting medium (Electron Microscopy Sciences, Hatfield, PA, USA). Only brain regions showing overt positive specific staining, namely the CA1 of the hippocampus and the subiculum, were evaluated. Maximum projection images were generated based on confocal z-stacks using Nikon 90i fluorescent microscope equipped with Nikon C1 three-laser confocal system and a Nikon DS U1 digital camera. The AT8 immunoreactive load was quantified using NIH Image J (v.1.46r) as described previously (Kazim et al. 2014; Rosario et al. 2006).
For thioflavin-S positive (TS+) plaque load quantification, 5–7 serial sections were selected from minimum 4 mice per group. Thioflavin-S staining was performed as described before (Kazim et al. 2014; Vallet et al. 1992). Briefly, free floating brain sections were rinsed in water for 6 min and then incubated in 0.25 % KMnO4 for 4 min. Sections were washed in water for another 6 min, and incubated in 1 % K2S2O5 and 1 % oxalic acid until the brown color completely faded. Sections were stained with 0.05 % thioflavin-S in water in dark for 8 min. Finally, sections were wash in 80 % ethanol for 2 min and in water for 3 min, and mounted and cover slipped using Fluorogel mounting medium (Electron Microscopy Sciences, Hatfield, PA, USA). The maximum projection images were taken and TS +plaque load was quantified in hippocampus CA1 and subiculum using NIH Image J (v.1.46r).
Statistical analysis
Data were analyzed using GraphPad Prism version 5.0 (GraphPad Software Inc, La Jolla, CA, USA) and one-way or two-way ANOVA (as appropriate) followed by a Bonferroni’s post hoc test. Further intergroup comparisons were also performed using unpaired two-tailed t test. All data are presented as mean ± SEM, and p < 0.05 was considered statistically significant.
Results
Passive immunization with antibodies targeting the N-terminal projection domain of tau reduces both total and hyperphosphorylated tau in aged 3xTg-AD mice
Triple transgenic AD mice are known to develop tau pathology starting around 12 months of age which is first apparent in the hippocampus and then progresses to the cerebral cortex (Oddo et al. 2003b). To assess whether passive immunization with tau antibodies 43D against tau 6–18 and 77E9 against tau 184–195 reduces tau pathology in aged 3xTg-AD mice, level of tau phosphorylated at Ser202/Thr205 sites was first determined with AT8 antibody by immunohistochemistry (Fig. 1).We found that passive immunization with 43D and 77E9 antibodies dramatically decreased tau hyperphosphorylation at Ser202/Thr205 in subiculum and CA1 area (Fig. 1b–d).
Furthermore, we investigated the levels of total and hyperphosphorylated taus by Western blots and found that passive immunization with 77E9 antibody significantly reduced levels of both total tau (R134d) and its hyperphosphorylation at Ser199, Ser202/Thr205 (AT8), Thr205, Ser262/356 (12E8), and Ser396/404 (PHF-1) sites in hippocampus compared with mouse IgG-treated control animals. Immunization with 43D against tau 6–18 also showed a decrease in total tau level, though this reduction did not reach statistical significance and significantly decreased tau phosphorylation at Ser199, Ser202/Thr205 (AT8), Ser262/356 (12E8), and Ser396/404 (PHF-1) sites with the exception at Thr205 site (Fig. 2). Similar results were found in forebrain cortex (Fig. 3). Especially, passive immunization with 43D antibody also decreased tau phosphorylation at Thr205 site in forebrain cortex. Interestingly, passive immunization targeting tau 184–195 by 77E9 decreased more total and hyperphosphorylated taus than that by 43D antibody (Figs. 2, 3). All together, these data clearly indicated that immunization targeting tau 6–18 and 184–195 can dramatically reduce tau pathology.

Passive immunization targeting the N-terminal projection domain of tau with 43D and 77E9 antibodies reduces levels of total and hyperphosphorylated taus in hippocampus of aged 3xTg-AD mice. a Representative Western blots of hippocampus developed with R134d against total tau and several phosphorylation-dependent and site-specific tau antibodies. b–g Densitometrical quantification of the blots after normalized with the GAPDH levels. Data are percentage of mouse IgG (100 %)-treated animals reported as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired two-tailed t test

Passive immunization targeting the N-terminal projection domain of tau with 43D and 77E9 antibodies decreases levels of total and hyperphosphorylated taus in forebrain cortex of aged 3xTg-AD mice. a Representative Western blots of forebrain cortex developed with R134d against total tau and several phosphorylation-dependent and site-specific tau antibodies. b–g Densitometrical quantification of the blots after normalized with the GAPDH levels. Data are reported as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired two-tailed t test
Passive immunization with tau antibodies does not markedly affect Aβ accumulation in moderate to severe stage of plaque pathology
Triple transgenic AD mice develop amyloid plaques starting around 6 months of age which is first apparent in the cortex and progresses to the hippocampus with age (Oddo et al. 2003b). We investigated whether passive immunization with tau antibodies 43D and 77E9 could alter the level of Aβ. We found that immunization with tau antibodies targeting tau 6–18 and 184–195 epitopes showed a trend but statistical insignificant reduction in amyloid plaque load in subiculum and CA1 region (Fig. 4a–c). In line with the immunohistochemistry results, the level of Aβ1-40 determined by ELISA showed only a small reduction in 43D and 77E9 treated mice (Fig. 4d). These data suggested that immunotherapy targeting tau 6–18 or 184–195 does not have a significant effect on the amyloid plaque load in aged 3xTg-AD mice.

Passive immunization targeting the N-terminal projection domain of tau with 43D and 77E9 antibodies does not affect significantly Thioflavin-S positive (TS+) amyloid plaques and level of Aβ40. a Immunostaining of subiculum (SC) and CA1 from aged 3xTg-AD mice immunized with 43D, 77E9, and control IgG using Thioflavin-S. Representative pictures of TS+ staining from subiculum (SC) and CA1 area. Scale bar 100 µm. Scatter plots of the quantification of plaque load in total subiculum (b), and CA1 (c), region from 5 to 7 serial sections from four mice per group. d The level of Aβ40 was quantified by ELISA. Data in b–d are shown as mean ± SEM. Data were analyzed by unpaired two-tailed t test
Passive immunization with tau antibodies targeting the N-terminal projection domain of tau does not affect the general behavioral phenotypes in aged 3xTg-AD mice
During the 5-week period of the study, we carefully monitored the general condition of mice daily and weighed the mice once a week. We did not note any abnormality in general physical characteristics, including grooming, posture, and clasping reflex. Immunization with 43D and 77E9 did not cause significant change in body weight (Fig. 5a). These data indicated that passive immunization targeting N-terminal tau 6–18 by 43D and tau 184–195 by 77E9 does not cause any neurological deficits in aged female 3xTg-AD mice.

Passive immunization with tau antibody 43D improves cognitive performance without any side effects in aged 3xTg-AD mice. Effect of the passive immunization on body weight (a) and reference memory in Morris water maze task (b–e). a The body weights of the mice were measured once a week. b The escape latency (s) to reach the hidden platform during acquisition phase for 4 days. c Percent time in the quadrant during the probe trial. d The average swim speed during the water maze training. e Distance covered during the probe trial. Data are reported as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control mice by two-way ANOVA followed by a Bonferroni’s posthoc test
Passive immunization with tau antibodies targeting the N-terminal projection domain of tau improves cognitive performance in aged 3xTg-AD mice
Triple transgenic AD mice are known to show strong deficits in learning and memory in the Morris water maze task starting at 6 months of age (Clinton et al. 2007). In the present study, the animals were subjected to Morris water maze task to determine the effect of passive immunization with tau antibodies on the spatial reference memory. We found that 3xTg-AD mice treated with 43D antibody took significant less time than control mice to find the hidden platform (Fig. 5b) in acquisition phase. More importantly, in the probe trial, 3xTg-AD mice treated with 43D antibody also spent a significant longer time than control mice in the target quadrant that formerly contained the platform (Fig. 5c). Unlike the mice treated with 43D antibody, the performance of 3xTg-AD mice treated with 77E9 was similar to that of control mice (Fig. 5b, c). Additionally, no significant difference was observed in swim speed (Fig. 5d) and distance covered (Fig. 5e) during probe trial among 43D, 77E9, and saline-treated 3xTg-AD mice. These data demonstrated that passive immunization with tau antibodies 43D against N-terminal projection domain tau 6–18, but not 77E9 targeting tau 184–195 could improve cognitive performance in aged 3xTg-AD mice.
Discussion
Alzheimer’s disease is the sixth leading cause of death, and estimated 11 % of people aged 65 and older have AD in the US (Thies et al. 2013). To date, none of the treatments available for AD slow or stop the death and malfunction of neurons in the brain that causes Alzheimer’s symptoms and make the disease fatal. Immunotherapies targeting the two pathophysiological hallmarks of AD—amyloid-β (Aβ) peptides and hyperphosphorylated tau protein—have been speculated to provide effective treatment for AD patients. In the present study, we treated aged 3xTg-AD mice with antibodies 43D against tau 6–18 and 77E9 against tau 184–195 once a week for 4 weeks, and found that passive immunization targeting tau 6–18 and 184–195 both decreased total tau and tau hyperphosphorylated at Ser199, Ser202/Thr205 (AT8), Thr205, Ser262/356 (12E8), and Ser396/404 (PHF-1) sites in the forebrain. Most importantly, 3xTg-AD mice treated with 43D antibody showed better performance in Morris water maze task than the saline-treated control mice, which indicates that immunotherapy targeting normal N-terminal projection domain of tau, tau 6–18 improves cognition in aged 3xTg-AD mice. On the contrary, targeting tau 184–195, reduced levels of hyperphosphorylated tau, but failed to rescue cognitive deficits. Immunization with these two antibodies showed a trend but did not significantly reduce Aβ accumulation in the aged 3xTg-AD mice. Although this finding is consistent with the study which showed that tau pathology precedes Aβ pathology in aged and AD brains (Braak and Braak 1997; Braak et al. 2013), at present we do not know the mechanism by which tau immunotherapy may influence Aβ pathology. We speculate that the reduction in Aβ pathology observed in the present study is probably due to proteolysis of Aβ as a bystander effect of the activation of the complement system produced by tau immunization. However, a reduction of APP synthesis and or its amyloidogenic processing are other possibilities which, at present, we cannot rule out. To our knowledge the present study is the first to show a trend to decrease Aβ and plaque load by tau immunotherapy in a mouse model.
Indeed, accumulating evidence from preclinical studies indicates that pathological tau based immunotherapy decreases the tau pathology and rescues the functional impairment (Asuni et al. 2007; Bi et al. 2011; Boimel et al. 2010; Boutajangout et al. 2010, 2011; Chai et al. 2011; Gu et al. 2013; Troquier et al. 2012; Yanamandra et al. 2013). The present study shows that antibodies to N-terminal projection domain of tau 6–18 and 184–195 not only decrease total tau level, but also dramatically reduce the hyperphosphorylated protein in the advanced stage of the pathology.
A central objective of a promising preclinical treatment for AD is to stop or at least effectively modify the course of AD. Several preclinical studies indicated that tau based immunotherapies improved functional deficits in several tau transgenic models when the treatment was initiated prior to the onset of tau pathology (Asuni et al. 2007; Boutajangout et al. 2010, 2011; Chai et al. 2011; Troquier et al. 2012). Boutajangout et al. (2011) injected intraperitoneally PHF1 antibody in 9–12 weeks old JNPL3 P301L mice at a dose of 250 µg/125 µL once a week for 13 weeks and found that the treated mice performed better than control mice on the traverse beam task. These data were supported by another passive immunotherapy study employing PHF1 antibody at 15 mg/kg three times a week for 2 months and then at 10 mg/kg twice a week for another 2 months starting treatment in 2 month old JNPL3 mice which improved functional deficits tested by Rota-Rod test after completion of antibody injection (Chai et al. 2011). Passive immunization with a mixture of three tau antibodies: HJ9.4 (tau 7–13), HJ8.5 (tau 25–30), and HJ9.3 (tau 306–320) in 6 month old P301S tau transgenic mice by continuous intracerebroventricular infusion for 3 months using Alzet subcutaneous osmotic minipump rescued cognitive deficits assessed by the fear conditioning test (Yanamandra et al. 2013).
3xTg-AD mice develop tau pathology starting at 12 months of age and Aβ accumulation as early as at 6 months of age (Oddo et al. 2003a). In the present study, we started treatment with 43D and 77E9 antibodies at 100 µg/100 µL once a week for 4 weeks when the 3xTg-AD mice were at 14–17 months old, which correspond to moderate to late stages of AD. Our data clearly demonstrate that immunization targeting tau 6–18 by 43D antibody but not using anti-tau 184–195 improves cognition using Morris water maze task, which suggests that the tau epitope specificity is important for tau based immunotherapy to prevent or attenuate the cognitive decline. Thus, immunotherapy targeting tau 6–18 epitope may present a promising therapeutic opportunity for AD patients.
Tau is a highly soluble, natively unfolded microtubule-associated protein that normally promotes tubulin assembly, microtubule stability, and cytoskeletal integrity. Therefore, another major concern associated with tau based immunotherapy is potential toxicity because of cellular uptake of antibodies and binding to normal tau may result in destabilization of the microtubules and subsequent interference with axonal transport and cytoskeletal integrity (Sigurdsson 2008). Currently, there is limited information available on adverse effects from targeting pathological tau, particularly the phosphorylated tau (Asuni et al. 2007; Bi et al. 2011; Boimel et al. 2010; Boutajangout et al. 2010, 2011; Chai et al. 2011; Troquier et al. 2012), or tau oligomers (Castillo-Carranza et al. 2014) in various tau transgenic mice models. Only one study reported that immunization of female C57BL/6 mice with full length recombinant tau led to neurological deficits with an NFT-like morphology, axonal damage, gliosis, mononuclear infiltration, and neurological deficits such as limp tail and limb paralysis (Rosenmann et al. 2006), suggesting that targeting normal tau could damage neurons and induce encephalitis. However, tau-deficient mice develop normally indicating that other microtubule associated proteins can perform similar functions (Denk and Wade-Martins 2009), and reducing endogenous tau has been shown to ameliorate Aβ induced dysfunction in transgenic mice (Roberson et al. 2007). Therefore, the key question for tau immunotherapy is the tau specific epitope for targeting. Indeed, passive immunization with the DA31 and MC1 monoclonal antibodies in P301L mutant tau mice demonstrated that the antibody MC1, which targets PHF-tau, was more efficacious than the high affinity DA31 antibody against tau 150–190, which targets normal tau, which suggest that specificity is more important than affinity in therapeutic applications (d’Abramo et al. 2013). In our study, 43D and 77E9 target tau 6–18 and 184–195, respectively. The different specificity of targeting tau epitope by 43D and 77E9 may be one possible explanation for immunization with 77E9 antibody reduced more both total tau and the hyperphosphorylated protein than 43D antibody at most of the tested phosphorylation sites, but treatment with 43D antibody and not 77E9 improved cognition in aged 3xTg-AD mice. We cannot rule out that this discrepancy could be due to neutralization of the beneficial effect of immunization with 77E9 antibody by its toxicity, though we did not observe significant abnormality in general physical characteristics, including grooming, posture, and clasping reflex. N-terminal tau fragments including tau 1–44, 26–44, 26–230, 1–156, and 45–230 exert powerful toxicity (Amadoro et al. 2004, 2006; Corsetti et al. 2008). Contrarily, tau 1–230 protects neuron from apoptosis (Amadoro et al. 2004), which indicates that N-terminal projection domain of tau can be either neuroprotective or neurotoxic according to its length. The therapeutic beneficial effect of passive immunization with tau antibody 43D to tau 6–18 found in the present study is consistent with a toxic domain in this region of the protein. However, without testing experimentally it is not possible to predict whether immunotherapy with tau 19–230 will be neuroprotective or deleterious and mediate Aβ toxicity. In the present study, we did not observe any deleterious effect of immunization with antibody 77E9 to tau 184–195.
In summary, passive immunization targeting N-terminal projection domain of tau 6–18 and 184–195 with 43D and 77E9 antibodies once a week for 4 weeks can effectively decrease both total and hyperphosphorylated taus at an advanced stage of the disease in 3xTg-AD mice. Importantly, short-term treatment with 43D antibody targeting tau 6–18 rescued cognitive impairments possibly by reduction of the neurotoxic region of tau. The passive immunization targeting N-terminal projection domain of tau, however, had a trend but no statistically significant ameliorating effect on the advanced stage Aβ pathology in the transgenic mice. Overall, our studies suggest (1) that passive immunization targeting N-terminal projection domain of tau can present a promising treatment opportunity for AD; (2) that targeting a normal tau epitope can effectively clear the hyperphosphorylated tau; and (3) that more attention should be paid to select the effective tau epitopes for targeting.
References
Alafuzoff I, Iqbal K, Friden H, Adolfsson R, Winblad B (1987) Histopathological criteria for progressive dementia disorders: clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol (Berl) 74:209–225
Alonso AD, Zaidi T, Grundke-Iqbal I, Iqbal K (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA 91:5562–5566
Alonso AD, Grundke-Iqbal I, Iqbal K (1996) Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 2:783–787
Amadoro G, Serafino AL, Barbato C, Ciotti MT, Sacco A, Calissano P, Canu N (2004) Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cel Death Differ 11:217–230. doi:10.1038/sj.cdd.4401314
Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N (2006) NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci USA 103:2892–2897. doi:10.1073/pnas.0511065103
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42:631–639
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM (2007) Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 27:9115–9129
Avila J, Simon D, Diaz-Hernandez M, Pintor J, Hernandez F (2014) Sources of extracellular tau and its signaling. J Alzheimers Dis 40(Suppl 1):S7–S15. doi:10.3233/JAD-131832
Bensadoun A, Weinstein D (1976) Assay of proteins in the presence of interfering materials. Anal Biochem 70:241–250
Bi M, Ittner A, Ke YD, Gotz J, Ittner LM (2011) Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One 6:e26860. doi:10.1371/journal.pone.0026860
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H (2010) Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol 224:472–485. doi:10.1016/j.expneurol.2010.05.010
Boutajangout A, Quartermain D, Sigurdsson EM (2010) Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci 30:16559–16566. doi:10.1523/JNEUROSCI.4363-10.2010
Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM (2011) Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem 118:658–667. doi:10.1111/j.1471-4159.2011.07337.x
Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357
Braak H, Zetterberg H, Del Tredici K, Blennow K (2013) Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-beta changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol 126:631–641. doi:10.1007/s00401-013-1139-0
Castillo-Carranza DL et al (2014) Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34:4260–4272. doi:10.1523/JNEUROSCI.3192-13.2014
Chai X et al (2011) Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem 286:34457–34467. doi:10.1074/jbc.M111.229633
Clavaguera F et al (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11:909–913
Clavaguera F et al (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 110:9535–9540. doi:10.1073/pnas.1301175110
Clinton LK et al (2007) Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis 28:76–82
Corsetti V et al (2008) Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol Cell Neurosci 38:381–392. doi:10.1016/j.mcn.2008.03.011
d’Abramo C, Acker CM, Jimenez HT, Davies P (2013) Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One 8:e62402. doi:10.1371/journal.pone.0062402
Denk F, Wade-Martins R (2009) Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging 30:1–13. doi:10.1016/j.neurobiolaging.2007.05.010
D’Hooge R, De Deyn PP (2001) Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev 36:60–90
Doody RS et al (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370:311–321. doi:10.1056/NEJMoa1312889
Flight MH (2013) Neurodegenerative disease: tau immunotherapy targets transcellular propagation. Nat Rev Drug Discov 12:904. doi:10.1038/nrd4179
Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852. doi:10.1074/jbc.M808759200
Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y (2011) Cleavage of Tau by calpain in Alzheimer’s disease: the quest for the toxic 17 kD fragment. Neurobiol Aging 32:1–14. doi:10.1016/j.neurobiolaging.2010.09.008
Giacobini E, Gold G (2013) Alzheimer disease therapy-moving from amyloid-beta to tau. Nat Rev Neurol. doi:10.1038/nrneurol.2013.223
Giannakopoulos P et al (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60:1495–1500
Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917
Gu J, Congdon EE, Sigurdsson EM (2013) Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J Biol Chem 288:33081–33095. doi:10.1074/jbc.M113.494922
Holmes C et al (2008) Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372:216–223
Iqbal K et al (1986) Defective brain microtubule assembly in Alzheimer’s disease. Lancet 2:421–426
Kazim SF, Blanchard J, Dai CL, Tung YC, LaFerla FM, Iqbal IG, Iqbal K (2014) Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. doi:10.1016/j.nbd.2014.07.001
Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 268:24374–24384
Lambracht-Washington D, Rosenberg RN (2013) Anti-amyloid beta to tau-based immunization: developments in immunotherapy for Alzheimer disease. Immuno Targets Ther 2013:105–114. doi:10.2147/ITT.S31428
Mandelkow EM, Schweers O, Drewes G, Biernat J, Gustke N, Trinczek B, Mandelkow E (1996) Structure, microtubule interactions, and phosphorylation of tau protein. Ann N Y Acad Sci 777:96–106
Masliah E et al (2005) Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46:857–868. doi:10.1016/j.neuron.2005.05.010
Morris RG, Garrud P, Rawlins JN, O’Keefe J (1982) Place navigation impaired in rats with hippocampal lesions. Nature 297:681–683
Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM (2003a) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging 24:1063–1070
Oddo S et al (2003b) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421
Park SY, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 25:5365–5375. doi:10.1523/JNEUROSCI.1125-05.2005
Reinecke JB et al (2011) Implicating calpain in tau-mediated toxicity in vivo. PloS One 6:e23865. doi:10.1371/journal.pone.0023865
Reitz C (2012) Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis 2012:369808. doi:10.1155/2012/369808
Roberson ED et al (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316:750–754
Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ (2006) Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer’s disease. J Neurosci 26:13384–13389. doi:10.1523/JNEUROSCI.2514-06.2006
Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O (2006) Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol 63:1459–1467. doi:10.1001/archneur.63.10.1459
Salloway S et al (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370:322–333. doi:10.1056/NEJMoa1304839
Sanders DW et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. doi:10.1016/j.neuron.2014.04.047
Sigurdsson EM (2008) Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimers Dis 15:157–168
Terry RD et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Thies W, Bleiler L, Alzheimer’s A (2013) 2013 Alzheimer’s disease facts and figures. Alzheimers Dement 9:208–245. doi:10.1016/j.jalz.2013.02.003
Tomlinson BE, Blessed G, Roth M (1970) Observations on the brains of demented old people. J Neurol Sci 11:205–242
Troquier L et al (2012) Targeting phospho-Ser422 by active Tau Immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res 9:397–405
Vallet PG, Guntern R, Hof PR, Golaz J, Delacourte A, Robakis NK, Bouras C (1992) A comparative study of histological and immunohistochemical methods for neurofibrillary tangles and senile plaques in Alzheimer’s disease. Acta Neuropathol 83:170–178
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858. doi:10.1038/nprot.2006.116
Winblad B, Graf A, Riviere ME, Andreasen N, Ryan JM (2014) Active immunotherapy options for Alzheimer’s disease. Alzheimers Res Ther 6:7. doi:10.1186/alzrt237
Yanamandra K et al (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80:402–414. doi:10.1016/j.neuron.2013.07.046
Zilka N, Kontsekova E, Novak M (2008) Chaperone-like antibodies targeting misfolded tau protein: new vistas in the immunotherapy of neurodegenerative foldopathies. J Alzheimers Dis 15:169–179
Acknowledgments
We are thankful to Ms. Janet Murphy for secretarial assistance. Studies described in this publication were supported by the New York State Office of People with Developmental Disabilities (OPWDD). Tau antibodies 43D and 77E9 employed for this study were generated and characterized under the supervision of Dr. Inge Grundke-Iqbal who passed away on September 22, 2012.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Dai and X. Chen contributed equally to this work.
For special issue of Journal of Neural Transmission in memory of Professor Sigfried Hoyer.
Rights and permissions
About this article

Cite this article
Dai, Cl., Chen, X., Kazim, S.F. et al. Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm 122, 607–617 (2015). https://doi.org/10.1007/s00702-014-1315-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-014-1315-y