
Abstract
Huntington’s Disease (HD) is caused by trinucleotide CAG repeat expansion >36 in huntingtin (htt), a protein with several documented functions. The elongated polyglutamine (polyQ) stretch in the N-terminal region of htt leads to dysfunctional and degenerative events in neurons and peripheral tissues. In this study, by extending the analysis to several caspase activities (i.e. caspase 2, 3, 6, 8 and 9), we describe genotype- and time- dependent caspase activity abnormalities, decreased cell viability and a large set of alterations in mitochondria morphology, in cultured blood cells from HD patients. Patients homozygous for CAG repeat mutations and heterozygous with high size mutations causing juvenile onset (JHD) presented significantly increased caspase 2, 3, 6, 8 and 9 activities, decreased cell viability and pronounced morphological abnormalities, compared with cells carrying low mutation size and controls. After cyanide treatment, all caspases increased their activities in homozygous and highly expanded heterozygous cells, caspase 8 and 9 increased also in those cells carrying low-size mutations, remarking their key role as ‘caspase initiators’ in HD. The remarkable ageing-dependent abnormalities in peripheral cells carrying particularly toxic mutations (i.e. homozygotes’ and JHD’s blood cells) points out the potential dependence of clinical HD development and progression on either mutated htt dosage or missing wild type htt. Peripheral tissues (i.e. blood cells) may theoretically represent an important tool for studying HD mechanisms and searching for new biomarkers, according to the patients’ genotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Huntingtin (htt), the protein whose elongated poly(Q) in Huntington disease (HD) has gain of toxic effects, has also beneficial anti-apoptotic effects by its wild type form (Borrell-Pages et al. 2006; Cattaneo et al. 2001; Li and Li 2004). Missing wild-type htt or increasing levels of mutated htt represent both toxic events for cells, as described in vitro (Graham et al. 2006; Maglione et al. 2006; Rigamonti et al. 2000) and in vivo in both animal models (Leavitt et al. 2001) and patients (Squitieri et al. 2003). Patients missing wild-type htt and possessing a double dose (homozygous patients) of mutated protein (i.e. rare subjects homozygous for CAG repeat expansion mutation), manifest a more severe clinical and neuropathological course and morphological mitochondria abnormalities than heterozygotes with similar mutation length (Mormone et al. 2006; Squitieri et al. 2003, 2006). Similarly, patients with very long mutations in htt, thus manifesting juvenile HD (JHD), show a severe phenotype, with atypical and severe clinical manifestations and progression rate (Quarrell et al. 2009). Mitochondria from both homozygous (Squitieri et al. 2006) and JHD (Sawa et al. 1999) patients also show increased depolarization and possess severe morphological alterations. Caspase activity 3 and 9 are increased in lymphoblasts from homozygous (Maglione et al. 2006) and JHD patients (Sawa et al. 1999), caspase 8 also increased in homozygous subjects’ lymphobalsts (Maglione et al. 2006).
In this study, we simulated in vitro HD ageing- and toxic-input- related changes of caspase activities and analyzed their overtime changes and cell line viability in peripheral cultured cells from patients with different genotypes, including homozygotes for CAG mutation, JHD with long toxic mutations, adult HD with usually occurring CAG mutations, and controls.
Materials and methods
Cell cultures
Cell lines were obtained from patients and stored in bank, as we previously described (Maglione et al. 2006) and after requiring informed consent, in accordance to the Declaration of Helsinki (Br Med J 1991; 302; 1194) and to ethical approval of the local committee. Clinical characteristics and data are currently stored in a data bank. Cell lines included two unexpanded normal controls (ctrl), four heterozygotes, two with usually expected length (lowHD, range of expanded CAG repeats: 41–48) and two with highly expanded alleles (highHD, range of expanded CAG repeats: 70–120), and two homozygotes for the expanded allele (homoHD, genotypes: 42/44 and 40/46 CAG expanded repeats) (Table 1). These cells were used to compare controls. Heterozygous and homozygous patients. In order to validate the consistency of data obtained in a small (N = 2) cohort of lowHD and highHD cells we also report as electronic supporting files analogous data obtained from additional heterozygous patients. For this purpose, we report data from 4 additional cell lines from each cohort of heterozygous HD (lowHD and highHD) along with 4 additional control cell lines (Supplementary Figs. 1, 2). Cells were maintained in suspension in 25 cm2 flasks in RPMI 1640 (Gibco/Invitrogen, San Diego, CA, USA), supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin (Gibco/Invitrogen, San Diego, CA, USA), and l-glutamine (Gibco/Invitrogen, San Diego, CA, USA). For the longitudinal studies cells were serially passaged and kept in culture for 6 months as previously described (Cannella et al. 2009; Maglione et al. 2006; Squitieri et al. 2006). At each passage (twice per week), 3 × 106 cells were transferred to 8 ml fresh medium after thorough re-suspension. After 8–10 similar passages (15–20 doublings) indicated as “time”, a portion of the cells was used for the analysis. In total, these cells underwent about 60 passages during a 6-month culture.
To test the effect of ageing on cell viability, we designed the experiment in presence or absence of glucose without any medium refreshment for 4–6 days in non-6-month-passaged cell cultures. We measured live cells, at indicated time, by trypan blue exclusion test. Only cells that excluded the blue dye and had a well-defined cellular outline were scored as live. Data were plotted as percentage of viable cells relative to the total number of cells. Each experiment was performed in triplicate.
Measurement of caspase activities
Caspase activities were analyzed by commercial kits as described (Maglione et al. 2006). Briefly, cells were plated at a density of 1 × 106 cells per ml and then exposed to 1 mM cyanide (CN) (Fluka) for different times: 10 h for Caspase-8 activity, 12 h for Caspase-2, -6 and -9 activities and 20 h for Caspase-3 activity. We tested the longitudinal changes of caspase-2, -3, -6, and -8 activity at time 0 (after 2 weeks culture), 3 (after 3 months of culture) and 6 (after 6 months of culture). Caspase-3 activity in the wild type and R6/2 mouse brains, were analyzed at 4 and 12 weeks of age.
The brains were quickly removed on an ice-cold plate and they were homogenized in RIPA buffer (Maglione et al. 2006).
Huntingtin fragmentation
To analyze the level of the htt fragmentation, the cells were homogenized in lysis buffer containing 20 mM Tris–HCl pH 7.2, 150 mM NaCl, 1% NP40 and protease inhibitors (Sigma, St Louis, MO, USA). Identical protein concentration of each sample was assessed by Bradford method. Protein extracts were separated on SDS-PAGE gel and electrophoretically transferred on nitrocellulose membrane. Two independent antibodies against N-terminal Htt (MAB 2,166 for amino acids 414–503; Chemicon) and C-terminal domain (MAB 2,168 residues 2,146–2,541; Chemicon) were used for western blotting and provided closely similar results. Protein bands were visualized by enhanced chemiluminescence (ECL kit, Amersham Pharmacia) and exposed to Hyperfilm (Amersham Pharmacia).
Transmission electron microscopy (TEM)
For TEM examination, cells were fixed in 2% paraformaldehyde/3% glutaraldehyde in 0.1 M PBS (pH 7.4) and processed as previously described (Squitieri et al. 2006, 2010). Cells were selected by a double-blind method by an operator other than the one who carried out the morphometric analysis. Cell images were analyzed for area of mitochondria. For each cell line, we randomly chose two blocks (1,000 cells per block) that we further cut to obtain matched sections each containing about 100 detectable cells. Altered mitochondria, calculated over the total number of mitochondria in the average of 2,000 cells.
Statistical analyses
For statistical analysis, we used a nonparametric test (Mann–Whitney U) to study differences among the groups of cell lines according to different genotypes in cell viability, caspase activities and in the number of mitochondria and area. Significance at p < 0.05.
Results
Nutrient exhaustion and glucose deprivation induces reduction in cell viability in homo- and highHD cell lines
We plated cells in regular medium for 6 days and in glucose deprived medium for 4 days. During this time course, we never refreshed or changed the cell culture medium in order to induce a possible stress condition. At fixed time points, we counted viable cells by trypan blue exclusion method. We found a significant decrease in cell viability either in homo- and in highHD cell lines after 6 days culture in regular medium and 4 days culture in glucose deprived medium (Fig. 1a, b), indicating a genotype-related altered response to nutrient exhaustion, including glucose deprivation condition.

Cell viability. Two cell lines for each genotype were used and cultured in nutrient exhaustion for 2 and 6 days (a) and in absence of glucose for 2 and 4 days (b). The percentage of viable cells was measured by trypan blue exclusion test. Cell viability was reduced in highHD, homoHD and lowHD cell lines compare to controls, * p < 0.05 (2 days). Cell viability was reduced in both highHD and homoHD cell lines compare to lowHDs and controls, * p < 0.05 (6 and 4 days)
Cyanide administration increases caspase activities in HD cells
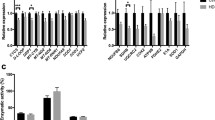
After treatment with 1 mM cyanide, caspase-3 activity was significantly increased in both homoHD and highHD cell lines compared to lowHDs and controls (Fig. 2a). We also found caspase-2 and caspase-6 activities significantly higher in homoHD and highHD cells than lowHD and control cell lines (Fig. 2b, c), whilst caspase-8 and caspase-9 activities were increased in all HD cell lines (Fig. 2d, e). Although in a less extend than in homoHD and highHD cell lines, lowHD cells showed increased cyanide-sensitive caspase-8 activation versus controls, while caspase-9 increased similarly according to a genotype-independent fashion in all HD cell lines (Fig. 2e). Conversely, lowHD cells never showed any significantly increased activation in caspase-3, -2 and -6 (Fig. 2a, c).

In vitro caspase activity following cyanide administration. Caspase activation after 1 mM cyanide (CN) administration. Two cell lines for each genotype were used, caspase -2, -3, -6, -8 and -9 activities were measured by colorimetric kit. (a) Caspase-3 activity was increased, in both homoHD and highHD cell lines compared with lowHDs and controls. (b, c) Caspase-2 and -6 activities were increased in homoHD and highHD cell lines compared with lowHDs and controls. (d, e) Caspase-8 and -9 activities were increased in all HD cell lines as compared with controls. * p < 0.05, ** p < 0.005, *** p < 0.0005
Data obtained in lowHD and highHD cell lines compared with controls were replicated in larger cohorts of patients (Supplementary Fig. 1).
Caspase activities increase in serially passaged HD cells and R6/2 HD mouse brain
We tested the caspase-2, -3, -6 and -8 activities in serially passaged lymphoblasts after 2 weeks (time 0), after 3 months (time 3), and after 6 months (time 6) in culture. For each caspase, we found a significant increase in highHD and homoHD compared to lowHD and controls at time 6 (Fig. 3a, d) and in particular, caspase 3 and 8 showed the most prominent increase (Fig. 3b, d). Interestingly the caspase-2 and -6 activities were already increased in high-and homoHD cell lines at time 0. We confirmed the effect of ageing on increased caspase-3 activity in the brain of old transgenic R6/2 mice at advanced HD stage (Fig. 4).

In vitro caspase activity over time. Caspase activation in serially passaged cell lines. Two cell lines for each genotype were used, caspase-2, -3, -6, and -8 activities were measured after 0, 3 and 6 months in culture by colorimetric kit. (a) Caspase-2 activity was increased in highHD and homoHD cell lines compared to lowHDs and controls. (b) Caspase-3 activity was increased in highHD and homoHD cell lines compared to lowHDs and controls. (c) Caspase-6 was increased in highHD and homoHD cell lines compared to lowHDs and controls. (d) Caspase-8 activity was increased in highHD and homoHD cell lines compared to lowHDs and controls. * p < 0.05

Brain caspase activity. Caspase-3 activation in WT and R6/2 mouse brain extracts. Caspase-3 activities were measured in brain extracts from 4- and 12-week-old animals by colorimetric kit. Caspase-3 activity was increased in 12-week-old R6/2 mice as compared with age-matched controls and in 12-week-old R6/2 mice as compared to 4-week-old R6/2 mice. * p < 0.05
For what concern data obtained in lowHD and highHD cell lines compared with controls these were validated in larger cohorts of patients (Supplementary Fig. 2).
Increased htt fragmentation in highHD cell lines as a consequence of increased caspase activity
We analyzed the pattern of htt fragmentation in non-passaged cell lines at 2 weeks (time 0) and in passaged cell lines after 6 months (time 6) culture. Cell lines with lowHD genotype did not show evidence of increased fragmentation even after many passages and long-term culture, whilst highHD cells showed a typical pattern of multiple bands, some with increased mutation size (Fig. 5).

Huntingtin (htt) fragmentation. Huntingtin (htt) fragmentation in Juvenile HD blood cells. NH2-terminal htt fragments were visualized by immunoblot using MAB 2166 monoclonal antibody. C-terminal domain were visualized using MAB 2168 (residues 2,146–2,541) (only MAB 2166 shown). The htt fragmentation was increased in highHD at time 6 as indicated by the rows
Mitochondrial size is altered in highHD and homoHD cell lines
We examined the number and size of mitochondria in non-passaged and in serially passaged lymphoblasts at time 0 and at time 6. Coherently with previous results (Mormone et al. 2006; Squitieri et al. 2006, 2010) we did not find any alteration in mitochondrial size and/or morphology in control and lowHD cell lines either at basal level or after 6 months in culture. HighHD showed a significant increase in summed total area of mitochondria per cell after 6 months in culture as compared to controls and lowHD cells (Figs. 6, 7a), while the homoHD cell line showed increased mitochondrial area since time 0. Such significant increase persisted at time 6 (Fig. 6a). Conversely, we failed to find any significant difference in the number of organelles among the different groups of cell lines (Fig. 7b). TEM showed a visible abnormal morphology of mitochondria and material accumulation in the cytoplasm of homoHD cells. Such abnormalities greatly increased in passaged cells after a 6-month culture (Fig. 7) with evidence of vacuoles and organelle cannibalism. HighHD cells also showed abnormalities in mitochondrial size and morphology (swelling mitochondria) with particularly altered organelle distribution (mitochondrial coalescence), compared to lowHD and controls (data not shown).

Mitochondrial area. Mitochondrial area is increased in serially passaged HD cell lines. Two cell lines for each genotype were used, total and maximum mitochondrial area per cell were measured after 0 (time 0) and 6 (time 6) months in culture. (a) Total area was increased in homoHD cell lines at time 0, and in highHD at time 6 compared to lowHDs and controls (ctrl) cell lines. Results are expressed as the mean ± SEM of mitochondrial area of a total of 2,000 cells per group (see “Materials and methods”) (b) Maximum mitochondrial area was increased in homoHD at time 0, and in highHD at time 6 compared to lowHDs and controls (ctrl). * p < 0.05, ** p < 0.01

Morphological alterations. Representative micrographs of morphological alteration in serially passaged HD cell lines. Mitochondrial clustering was visible in highHD cell lines after 6 months in cultures (time 6) compared with the beginning of cell culture (time 0). Each micrograph report an insert at higher magnification showing the cluster of mitochondria
Discussion
In this study, we describe aging-related biochemical and ultrastructural changes in cultured peripheral cells from HD patients with different genotype and CAG mutation size. Our strategy aimed to mirror the pathological process occurring in the nervous system of HD patients in the attempt to search for peripheral, easy-to-get, markers reflecting the apoptotic pathway occurring in HD. We therefore analyzed peripheral patients’ and control subjects’ cells (i.e. blood cells) whose culture can easily survive for long times. Biochemical, biophysical and morphological changes of HD lymphoblasts and their organelles have been widely described (Almeida et al. 2008; Maglione et al. 2006; Mormone et al. 2006; Nagata et al. 2004; Panov et al. 2002; Sawa et al. 1999; Squitieri et al. 2006; Toneff et al. 2002) and, recently, also confirmed in primary patients’ cell cultured with a genotype dependent pattern (Squitieri et al. 2010). On the other hand, the proteolytic cleavage of htt and the caspase-dependent proapoptotic pathway represent key mechanisms in the pathogenesis of HD (Goldberg et al. 1996; Wellington et al. 1998) and other neurodegenerative diseases (Gervais et al. 1999). Such biological processes occur early in HD (Wellington et al. 2002) and could therefore potentially offer clues to early preventive therapies to slow down the HD progression (Graham et al. 2006; Ona et al. 1999; Wellington et al. 2000).
In our study, patients cell cultures showed different viability according to their genotype, cells with high toxic CAG repeat expansions and homozygotes for mutations showing increased rate of cell death compared to controls and to cell lines with usually occurring mutation sizes (Fig. 1). This is in accordance with the clinical observations on more severe disease course in these patients (Cannella et al. 2004; Squitieri et al. 2003) and with a protective role of wild-type htt in HD (Cattaneo et al. 2001). To test caspase activities changes in these cells, we treated them by cyanide, a toxic drug acting against mitochondrial function (Sawa et al. 1999) and analyzing the different caspase pattern activation according to their genotype. Interestingly, we noticed an increased caspase 2, 3, 6, 8 and 9 activity after treatment in homoHD and highHD cells compared to control. Caspase 8 and 9 were also up-regulated in low mutated cell lines compared to controls, coherently with the hypothesis that they represent ‘initiator caspases’ in the HD pathological process (Kiechle et al. 2002; Sanchez et al. 1999; Vis et al. 2005). Accordingly, in our model, even cells carrying mutations with low CAG size show increased activities of caspase initiators, after a toxic input. Coherently we may postulate that our model reflects at least partially in periphery, although after artificial toxic stimulus, what documented in the central nervous system (Sanchez Mejia and Friedlander 2001). The demonstration of htt fragment accumulation is suggestive of a consequential effect of the increased caspase activation on protein cleavage also in peripheral cells as already described (Maglione et al. 2006; Toneff et al. 2002) and in a different fashion according to the patients’ genotype. The selective increase of caspase 2, 3 and 6 observed in high and homoHD lymphoblasts also showing the most remarkable protein fragmentation (Fig. 5) may offer clues to further study the different behaviour of such proteases in HD.
One interesting finding shows a progressive increase of all tested caspase activities in cells with particularly toxic genetic conditions (high and homoHD cells) reflecting the increased rate of cell death observed in these cells after glucose deprivation, the increased protein fragmentation during time in the same cells and, conclusively, the remarkable clinical severity expressed by these particular patients (Gutekunst et al. 1999; Hackam et al. 1998; Scherzinger et al. 1999; Squitieri et al. 2003; Zeron et al. 2001). The progressive increased caspase-3 activity we document in a mouse model typically showing features of JHD fits perfectly with our findings and with the hypothesis that the proteolytic HD process is a biological dynamic event progressively affecting life span (Chen et al. 2000; Ona et al. 1999; Kiechle et al. 2002) (Fig. 4).
TEM confirmed a more evident basal toxic condition in homoHD and highHD cell lines showing increased mitochondrial area (swelling mitochondria) together with impressive morphological changes. Specifically high HD cell lines showed an aberrant nuclear mitochondria clustering likely derived from altered mitochondrial metabolism and potentially associated with the increased protease activities. This observation is in line with it has been described in other peripheral tissues, i.e. muscular system (Chaturvedi et al. 2009) and in primary cell culture from patients (Squitieri et al. 2010).
Our study highlights, to our knowledge for first time in a HD peripheral tissue, a possible relationship between the increasing time dependent caspase activities (Fig. 3) and the increasing time dependent sub-cellular morphological abnormalities (Fig. 7). Interestingly, such changes depended on genotype, thus giving further strength to the intriguing hypothesis that additional/different mechanisms may contribute to HD, according to specific rare genotypes (i.e. homozygotes for CAG mutation and highly expanded mutation carriers). This adds on previous evidence we obtained showing analogous results for dose-dependency of HD alteration in mitochondrial size (Squitieri et al. 2006).
Our study show obvious limitations. First, this is an artificial in vitro model and cannot perfectly replicate what is occurring in vivo in a longer life span time. Second, we are here testing just some of the cleavage protease events specifically related to caspase activities, which only in part contribute to htt fragmentation. Further experiments testing the contribution of proteases, other than caspases, to the htt fragmentation, would be required to elucidate this point. However, notwithstanding these obvious limitations, we are here describing longitudinal progressive time dependent pathological phenomena in a model showing the toxic effects of CAG mutation in its physiological context.
References
Almeida S, Sarmento-Ribeiro AB, Januário C, Rego AC, Oliveira CR (2008) Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington’s disease patients. Biochem Biophys Res Commun 374(4):599–603
Borrell-Pages M, Zala D, Humbert S, Saudou F (2006) Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci 63(22):2642–2660
Cannella M, Gellera C, Maglione V, Giallonardo P, Cislaghi G, Muglia M, Quattrone A, Pierelli F, Di Donato S, Squitieri F (2004) The gender effect in juvenile Huntington disease patients of Italian origin. Am J Med Genet B Neuropsychiatr Genet 125B(1):92–98
Cannella M, Maglione V, Martino T, Ragona G, Frati L, Li GM, Squitieri F (2009) DNA instability in replicating Huntington’s disease lymphoblasts. BMC Med Genet 10:11
Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001) Loss of normal huntingtin function: new developments in Huntington’s disease research. Trends Neurosci 24:182–188
Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, Starkov A, Kiaei M, Cannella M, Sassone J, Ciammola A, Squitieri F, Beal MF (2009) Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum Mol Genet 18(16):3048–3065
Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6:797–801
Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW (1999) Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 97:395–406
Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR (1996) Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet 13(4):442–449
Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125(6):1179–1191
Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ (1999) Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci 19(7):2522–2534
Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR (1998) The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol 141(5):1097–1105
Kiechle T, Dedeoglu A, Kubilus J, Kowall NW, Beal MF, Friedlander R, Hersch SM, Ferrante RJ (2002) Cytochrome C and caspase-9 expression in Huntington’s disease. Neuromolecular Med 1(3):183–195
Leavitt BR, Guttman JA, Hodgson JG, Kimel GH, Singaraja R, Vogl AW, Hayden MR (2001) Wild-Type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet 68(2):313–324
Li SH, Li XJ (2004) Huntingtin and its role in neuronal degeneration. Neuroscientist 10(5):467–475
Maglione V, Cannella M, Gradini R, Cislaghi G, Squitieri F (2006) Huntingtin fragmentation and increased caspase 3, 8 and 9 activities in lymphoblasts with heterozygous and homozygous Huntington’s disease mutation. Mech Ageing Dev 127(2):213–216
Mormone E, Matarrese P, Tinari A, Cannella M, Maglione V, Farrace MG, Piacentini M, Frati L, Malorni W, Squitieri F (2006) Genotype-dependent priming to self- and xeno-cannibalism in heterozygous and homozygous lymphoblasts from patients with Huntington’s disease. J Neurochem 98(4):1090–1099
Nagata E, Sawa A, Ross CA, Snyder SH (2004) Autophagosomelike vacuole formation in Huntington’s disease lymphoblasts. Neuroreport 15(8):1325–1328
Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM (1999) Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature 399(6733):263–267
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT (2002) Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 5(8):731–736
Quarrell OWJ, Brewer HM, Squitieri F, Barker RA, Nance MA, Landwehrmeyer GB (2009) Juvenile Huntington’s Disease and other trinucleotide repeat disorders. Oxford University press
Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Covoni S, Vincenz C, Cattaneo E (2000) Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci 20(10):3705–3713
Sanchez Mejia RO, Friedlander RM (2001) Caspases in Huntington’s disease. Neuroscientist 7(6):480–489
Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J (1999) Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron 22(3):623–633
Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF Jr, Greenamyre JT, Snyder SH, Ross CA (1999) Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med 5(10):1194–1198
Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci USA 96(8):4604–4609
Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, Almqvist EW, Turner D, Bachoud-Levi AC, Simpson SA, Delatycki M, Maglione V, Hayden MR, Donato SD (2003) Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain 126:946–955 Pt4
Squitieri F, Cannella M, Sgarbi G, Maglione V, Falleni A, Lenzi P, Baracca A, Cislaghi G, Saft C, Ragona G, Russo MA, Thompson LM, Solaini G, Fornai F (2006) Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech Ageing Dev 127(2):217–220
Squitieri F, Falleni A, Cannella M, Orobello S, Fulceri F, Lenzi P, Fornai F (2010) Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J Neural Transm 117(1):77–83
Toneff T, Mende-Mueller L, Wu Y, Hwang SR, Bundey R, Thompson LM, Chesselet MF, Hook V (2002) Comparison of huntingtin proteolytic fragments in human lymphoblast cell lines and human brain. J Neurochem 82(1):84–92
Vis JC, Schipper E, de Boer-van Huizen RT, Verbeek MM, de Waal RM, Wesseling P, ten Donkelaar HJ, Kremer B (2005) Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol 109(3):321–328
Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, Hayden MR (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem 273(15):9158–9167
Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR (2000) Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem 275(26):19831–19838
Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, Loubser O, van Raamsdonk J, Singaraja R, Yang YZ, Gafni J, Bredesen D, Hersch SM, Leavitt BR, Roy S, Nicholson DW, Hayden MR (2002) Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J Neurosci 22(18):7862–7872
Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR, Raymond LA (2001) Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci 17(1):41–53
Acknowledgments
We thank the European Huntington’s Disease Network, all patients and their families (contribute of 5 × 1000 to Associazione Italiana Corea di Huntington-Neuromed to FS), IRCCS Neuromed (contribute of 5 × 1000 to SO), the Italian Society of Neurologists (SNO—Lascito Gobessi), for their kind cooperation and support to FS. FS is a member of the Euro HD-Network and of Huntington Study Group.
Author information
Authors and Affiliations
Corresponding author
Additional information
F. Squitieri and V. Maglione contributed equally to this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Squitieri, F., Maglione, V., Orobello, S. et al. Genotype-, aging-dependent abnormal caspase activity in Huntington disease blood cells. J Neural Transm 118, 1599–1607 (2011). https://doi.org/10.1007/s00702-011-0646-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-011-0646-1