
Abstract
Huntington’s disease (HD) is a genetic neurodegenerative disorder. Oxidative stress, mitochondrial dysfunction, and protein metabolism impairment have been implicated in its pathogenesis. However, the contribution of these phenomena to HD onset or progression is not well known, and they have been less studied in peripheral blood. We analyzed plasma lipid peroxide (LPO) and lactate (LAC) concentrations as indicators of oxidative stress and mitochondrial dysfunction in symptomatic HD patients (sHD) and asymptomatic HD gene carriers (aHD). We also measured the plasma activity of aminopeptidases (APs), an important group of proteolytic enzymes. LPO and LAC concentrations were significantly elevated in sHD patients but not in aHD carriers. Aspartate and glutamate AP activities were significantly reduced in sHD patients and aHD carriers. These findings demonstrate that sHD patients are under oxidative stress, which may favor progression of the disease. Plasma AP activity was decreased before the appearance of HD symptoms and oxidative stress and may be related to protein metabolism impairment. These results indicate that therapy directed to improve oxidative stress and normalize AP activity may be useful in the treatment of HD. They also suggest that decreased plasma AP activity in aHD carriers may predict the future onset of HD symptoms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder (Petrasch-Parwez et al. 2007) with a low prevalence (5–10 cases/100,000). It is characterized by involuntary movements (chorea) due to a pronounced cell loss of striatal medium spiny neurons. HD is also accompanied by personality changes, dementia, and weight loss. The defective gene in HD is in chromosome 4 and contains a repeated expansion of CAG trinucleotide within its coding region that expresses a polyglutamine repeat in the huntingtin protein (htt). The number of CAG repeats is correlated with the age at onset and is indirectly related to the severity of disease. HD patients usually show around 40–50 CAG repeats, with >70 repeats observed in early onset or juvenile forms. Mutant htt tends to aggregate, producing insoluble polymers known as inclusion bodies (IBs).
Oxidative stress plays a major role in the etiology of HD. Mitochondrial dysfunction and electron transport chain impairment are the main mechanisms responsible for the production of reactive oxygen species (Trushina and McMurray 2007; Sayre et al. 2008). Mitochondria are thought to be especially vulnerable to oxidative injury, because most intracellular free radicals are generated in the mitochondrial transport chain. Mitochondria DNA is extremely susceptible to oxidative stress due to its localization in the mitochondrial matrix, lack of protective histones, and limited repair mechanisms. These oxidative stress conditions and the occurrence of iron-mediated Fenton reaction also result in oxidative damage to polyunsaturated lipids from lipoproteins and membranes. Consequently, the unsaturated lipid peroxides (LPO) generated have been proposed as in vivo biomarkers of oxidative stress in neurodegenerative diseases (Sayre et al. 2008).
Protein metabolism is also impaired in HD. The brain contains an important group of enzymes related to protein and peptide metabolism known as aminopeptidases (APs). APs are the most abundant proteolytic enzymes in the brain and play a key role in regulating the function of small bioactive peptides (Mitsui et al. 2004), but their importance in the striatum and in the physiopathology of neurodegenerative diseases has not been elucidated. Some neuropeptides, such as cholecystokinins, angiotensins, and enkephalins, coexist together with dopamine in the striatum. Cholecystokinin and other peptides are also present in other afferent fibers, which come from prefrontal cortical areas and modulate autonomic and endocrine functions. Once they are released by synaptic terminals and activate their respective receptors, these neuropeptides are mainly hydrolyzed by membrane-binding peptidases. Hence, it is highly relevant to study the enzymatic activities of APs in HD and to analyze their contribution to the onset and progression of the disease. Given the progressive formation and neuronal death of insoluble protein aggregates in HD, Alzheimer disease (AD), and Parkinson’s disease (PD), changes in AP activities may occur during progression of the disease. Therefore, data on their activities may also provide useful information on the disease stage.
The aims of this study were to compare oxidative stress among symptomatic HD patients (sHD), asymptomatic HD (aHD) gene carriers, and healthy controls and to explore the enzymatic activity of plasma APs in sHD and aHD groups.
Materials and methods
Subjects and plasma collection
Three study groups were formed: 24 symptomatic HD patients (sHD group), 7 asymptomatic HD gene carriers (aHD group), and a control group of 60 randomly selected healthy individuals (C group) with a similar age and sex distribution to that of the patients. HD was diagnosed by at least two neurologists, based on specific clinical features (see below) in individuals with a positive family history and a positive genetic analysis. All sHD patients were under treatment in the Alteration Movement Unit of our hospital. Asymptomatic HD gene carriers were defined as subjects with a proven increased number of CAG repeats (≥38) in the huntingtin gene but with no clinical symptoms of HD, although some of them showed pathological signs in neurological exploration, and they were recruited from among HD family members. HD patients were evaluated by clinical interview and neurological exploration. HD symptoms were assessed using the Unified Huntington’s Disease Rating Scale (UHDRS) (Huntington Study group 1996). Main clinical parameters are shown in Sect. “Results”. Blood samples were drawn from the cubital vein into EDTA tubes. DNA samples were analyzed in the Clinical Biochemistry Department of our hospital as previously described (Blanco et al. 2008). Plasma was isolated by centrifugation for 15 min at 3,000 rpm and stored at −80°C until analysis.
Determination of plasma lipid peroxides and lactate levels
Lipid peroxide (LPO) and lactate (LAC) levels were measured as indicators of oxidative stress and mitochondrial dysfunction by using PeroxiDetect (Sigma) and l-lactic acid Enzymatic Bioanalysis (Roche) kits, respectively, according to the manufacturer’s protocols. Samples were analyzed in duplicate, and results were expressed as nanomol of LPO per milliliter (nmol/ml) and gram of LAC per liter (g/l).
Enzyme activity assay
Most artificial substrates of aminopeptidases are amino acid amides containing a fluorophore, and their hydrolysis can be fluorometrically analyzed because hydrolysis products differ in their spectral properties (Stöckel-Maschek et al. 2003). Plasma activities of alanyl- (AlaAP), cystinyl- (CysAP), aspartyl- (AspAP), glutamyl- (GluAP), and leucyl-aminopeptidases (LeuAP) were measured fluorometrically in duplicate as described elsewhere (Banegas et al. 2004). Specific aminopeptidase activities were expressed as pmol of AlaNNap, CysNNap, AspNNap, GluNNap, and LeuNNap hydrolyzed per min per mg of protein. Fluorogenic assays were linear with respect to hydrolysis time and protein content.
Non-denaturing electrophoresis and Western blot analysis
Aminopeptidases enzymatic activities were determined by separating plasma samples using non-denaturing PAGE on a 10% separating gel. After electrophoresis, the gel was incubated with the specific substrate of the enzyme (aminoacyl-β-naphylamide) in 0.1 M phosphate buffer (6.1 pH) containing 0.15% Fast Black K (FBK) salt at 37°C for 30 min. After breaking down the substrate, the enzyme liberates β-naphylamide, which reacts with the salt and produces a purple dye. The gel was then washed for 30 min using 25% acetic acid, such that the dyed lanes correspond with the localization of the enzyme. The color intensity is proportional to the enzymatic activity.
Western blot analysis was also performed to determine specific aminopeptidase expression in plasma. Plasma samples were first subjected to depletion of albumin and immunoglobulin G proteins by immunoaffinity columns according to the kit instructions (Sigma). Then, plasma samples were separated by SDS/PAGE on a 12% separating gel and transferred to poly-vinylidene difluoride (PVDF) membranes (Bio Rad). Membranes were blocked with phosphate buffer solution (PBS) containing 0.1% tween 20 and 5% skim milk overnight at 4°C and were then incubated in PBS containing 0.1% tween 20, 0.5% skim milk, and rabbit anti-CD13/AlaAP antibody (Santa Cruz Technologies) diluted to 1/1,000 for 1 h at room temperature. Membranes were washed 3 times with PBS/tween and incubated for 1 h with HRP-conjugated anti-rabbit IgG antibody (Amersham) diluted to 1/5,000 in PBS/tween containing 0.5% skim milk. After washing the membranes 3 times with PBS/tween, blots were detected by an enhanced chemiluminescence method using an ECL plus Western blotting detection kit obtained from Amersham. Results were visualized by means of the Curix 60 Developing System using Agfa medical X-ray films.
The ImageJ program (http://rsb.info.nih.gov/ij/index.html) was used for quantitative analysis of the bands.
Statistical analysis
Differences among groups were studied by using one-way ANOVA for each study variable. When significance was found, post hoc pair-wise comparisons were performed with Bonferroni’s penalization. Homogeneity of variances was attained by Neperian logarithm transformation. The Student’s t test was used to compare means between pairs of groups, and associations were studied by using a scatter plot to test linear association and by Pearson correlation coefficient analysis. Differences were considered significant if P values were 0.05 or less. STATA 10.1 software was used for the data analyses. All results were expressed as mean ± SEM.
Results
Characterization of subjects
Study groups did not differ in age or sex (see Table 1). Table 2 shows the values of the main clinical variables in the aHD and sHD groups. The number of CAG repeats was similar between the groups, but the motor UHDRS score was significantly higher in the sHD versus aHD group (P < 0.001), and the total functional capacity (TFC) score was significantly lower in the sHD group (P < 0.001).
Plasma lipid peroxides and lactate levels
Plasma LPO concentration may be affected by lipids, such as cholesterol and triglycerides. Analysis of the sHD, aHD, and C groups showed that they were all normolipemic, with no significant differences in concentrations.
The sHD group showed significantly higher plasma LPO (F exp = 12.74 [1, 73] df, P = 0.0019) and LAC (F exp = 8.07 [1, 65] df, P = 0.0180) concentrations in comparison to the C group. However, no differences were found between aHD and C groups. Plasma LPO and LAC concentrations were higher in the sHD group than in the aHD group, but the differences did not reach significance, likely due to the small number of aHD subjects (see Fig. 1). No significant correlations were found either in sHD or in aHD groups between LPO or LAC levels and time with disease, number of CAG repeats, motor UHDRS score, TFC score, or disease stage.

LPO and LAC concentrations of C (white bars), aHD (gray bars), and sHD (hatched bars) groups. LPO concentration is expressed in nmol/mL (left axis) and LAC concentration in g/L (right axis). Bars represent mean ± SEM; *P < 0.05 and **P < 0.01, sHD group versus C group
Plasma aminopeptidase activities
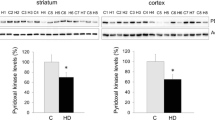
AspAP (aHD: F exp = 19.62 [1, 79] df, P = 0.0001; sHD: F exp = 8.22 [1, 79] df, P = 0.0159) and GluAP (aHD: F exp = 14.01 [1, 87] df, P = 0.0010; sHD: F exp = 13.31 [1, 87] df, P = 0.0013) activities were significantly lower in aHD and sHD groups than in C group. Moreover, AlaAP (F exp = 7.48 [1, 88] df, P = 0.0226) activity was significantly lower in sHD group than in C group. No differences in AP activities were found between sHD and aHD groups (see Fig. 2).

Plasma AlaAP, CysAP, AspAP, GluAP, and LeuAP activities expressed in picomoles of the corresponding aminoacyl-β-naphylamide hydrolyzed per minute per milligram of protein. Bars represent mean ± SEM of C group (white bars), aHD group (gray bars), and sHD group (hatched bars). ¤¤¤ P < 0.001, aHD versus C group; * P < 0.5 and ** P < 0.01, sHD versus C group
Non-denaturing electrophoresis and Western blot analysis
Figure 3a shows, as an example, the non-denaturing electrophoresis blot of AlaAP from plasma of C and sHD subjects, using non-denaturing gel electrophoresis in four different assays (I–IV). Band intensity is proportional to the enzymatic activity. From left to right, the first four strips correspond to the AlaAP activity displayed by 400 and 200 μg (both in duplicate) of plasma proteins from a C subject. The next four bands show AlaAP activity of the same amounts of sample (in duplicate) from a sHD subject. Figure 3b depicts the plot of the signal intensity data in Fig. 3a. AlaAP activity was lower in the sHD subject than in the C subject.

a Non-denaturing gel electrophoresis blots for the detection of AlaAP performed in four different assays using 400 and 200 μg in duplicate of plasma proteins of C (lines1–4) and sHD (lines5–8) subjects. b Quantification of the AlaAP activity reported in a. Bars represent the mean (±SEM) of the signal intensity expressed as percentage
Figure 4a shows the Western blot of AlaAP from plasma of C and sHD subjects in three different assays. Assay I—from left to right, the first two strips correspond to 150 and 100 ng, respectively, of commercial AlaAP with a molecular weight of 140 kDa. The next two strips are AlaAP contained in 90 and 75 μg, respectively, of plasma proteins from a C subject. The last two strips correspond to the AlaAP in the same amounts of plasma proteins from a sHD subject. Assays II and III—from left to right, the first strip corresponds to 150 ng of commercial AlaAP with a molecular weight of 140 kDa. The next two strips are AlaAP contained in 60 and 30 μg, respectively, of plasma proteins from a C subject. The last two strips correspond to the AlaAP in the same amounts of plasma proteins from a sHD subject. Figure 4b shows a plot of the relative intensity data in Fig. 4a. With the exception of assay II, the AlaAP concentration was lower in the sHD subject than in the control.

a Western blots for the detection of AlaAP performed in three different assays: I-AlaAP detected in 90 and 75 μg of plasma proteins from C (lines3, 4) and sHD (5, 6) subjects. II and III-AlaAP detected in 60 and 30 μg of plasma proteins from C (lines2, 3) and sHD (4, 5). b Quantification of the AlaAP concentrations reported in a. Bars represent the relative intensity of each sample, expressed as percentage, using the standard as the common point of comparison. Relative intensity <1 indicates that sample has less AlaAP protein than the standard
Discussion
The pathogeneses of HD, AD, and PD are all reported to include mitochondrial dysfunction and oxidative stress (Andersen 2004; Trushina and McMurray 2007), protein aggregation (Spires and Hannan 2007), inflammatory phenomena (Li and Li 2004), and apoptosis (Gil and Rego 2008). Hence, although HD, AD, and PD have different etiologies and clinical features, they share some of the pathological mechanisms that lead to selective neuronal loss and neurodegeneration.
The brain is particularly susceptible to oxidation-mediated insults due to its high energy requirements and lipid concentrations. Free radicals, oxidative-induced injury, and impaired cellular metabolism are known to be implicated in HD (Browne et al. 1999; Saft et al. 2005), but there is scant direct evidence of these phenomena in the peripheral blood of HD patients. We studied changes in plasma LPO concentrations and found significantly elevated plasma LPO in sHD but not aHD patients. These results suggest that although oxidative stress is not evident in early stage HD patients, it increases with progression of the disease. Chen et al. (2007) also reported increased plasma oxidative stress in HD patients, with higher values in patients with more severe HD symptoms. Moreover, Klepac et al. (2007) found significantly higher plasma LPO and significantly lower plasma reduced glutathione (GSH) levels in HD patients than in healthy controls. However, unlike the present results, they also found higher LPO and lower GSH levels in asymptomatic HD gene carriers versus healthy controls. A study with a larger number of aHD carriers may be required to evaluate oxidative stress before the onset of clinical symptoms.
Defects in energy metabolism are well documented in HD (Grünewald and Beal 1999). Glucose metabolism is markedly reduced in the basal ganglia and cerebral cortex of symptomatic HD patients (Browne et al. 1999), and biochemical studies in postmortem HD tissues found selective dysfunction in mitochondrial components of the oxidative phosphorylation pathway in targeted brain regions (Gu et al. 1996). These impairments probably resulted from free radical-induced oxidative damage to mitochondria and other cellular components. Aerobic metabolism alterations promote the exit of pyruvate from mitochondria with a subsequent reduction to LAC, leading to a loss of energy production (Martin et al. 2005). Therefore, plasma LAC may be a useful marker of mitochondrial dysfunction in neurodegenerative diseases (Finsterer 2005). We found significantly higher plasma LAC concentrations in sHD patients than in controls, whereas concentrations were similar between aHD patients and controls. Similar data were obtained for LPO, indicating that mitochondrial dysfunction and oxidative stress take place simultaneously in HD, with each contributing to the development of the other. Increased plasma LAC levels were proposed as a marker of energy metabolism deficit in HD. Using localized proton magnetic resonance imaging (MRI), a threefold elevation of LAC was found in occipital cortex of symptomatic HD patients, and brain LAC concentrations were correlated with time with the disease (Jenkins et al. 1993). Harms et al. (1997) applied the same technique and reported LAC peaks in frontal cortex in 8/17 symptomatic HD patients and 4/4 asymptomatic gene carriers but not in controls. A recent MRI study by Martin et al. (2007) reported higher LAC/creatine and LAC/N-acetyl aspartate ratios in parieto-occipital and cerebellar regions of HD patients. Therefore, the elevation of plasma LAC levels in HD appears to be well established, although no clear relationship has been found between LAC elevation and the clinical course of the disease.
A further characteristic of HD patients is a severe impairment of protein metabolism, and we studied the activity of a group of enzymes related to protein and peptide metabolism, APs, which are abundant in the brain. There have been very few studies of AP activity in neurodegenerative diseases. Overall AP activity was lower in the aHD and sHD groups, although significant differences with controls were only found for AspAP and GluAP activities in the aHD group and for AlaAP, AspAP, and GluAP activities in the sHD group. AlaAP activity was similar between the aHD group and controls but significantly lower in the sHD group than in controls. Although the AlaAP mean activity was also lower in the sHD than in the aHD group, this difference did not reach significance, probably due to the small number of asymptomatic carriers. There have been few studies on APs and Huntington’s disease, and the role of these enzymes in the pathology of HD has yet to be established. Butterworth (1986) also reported the lower activity of three peptidases (angiotensin converting enzyme, thermolysin-like metalloendopeptidase, and AlaAP) in the caudate nucleus of HD patients. These enzymes are also involved in enkephalin/substance P activation and metabolism (Hui 2007), and their reduction is related to the decreases in enkephalin, dynorphin, and substance P observed in HD patients (Dawbarn et al. 1986; Iadarola and Mouradian 1989). AlaAP hydrolyzes the neuropeptides methionine- and leucine-enkephalin in healthy brains, thereby regulating the ligand concentration for opioid receptors. Enkephalins bind with high affinity to opioid receptors expressed on the surface of neural cells. Activation of opioid receptors leads to inhibition of adenylyl cyclase activity, with a subsequent decrease in cAMP (Potolicchio et al. 2005). APs are also involved in activation of the central renin-angiotensin system (RAS), the main role of which is to regulate blood pressure and body fluid homeostasis in the brain (Banegas et al. 2006). AlaAP and GluAP are responsible for the production of angiotensin (Ang) III and angiotensin IV, respectively, from the inactive decapeptide angiotensin I. Several studies in the striatum of rats reported that Ang II and Ang IV promote dopamine release via a pre-synaptic mechanism (Mendelshon et al. 1993; Stragier et al. 2004). In this context, Mota et al. (2008) found high levels of GluAP in human basal ganglia and suggested that it might play a role in the conversion of brain AngII–AngIII as well as in the control of dopamine release. Although further research is required on this issue, it is possible that these AP-regulated mechanisms are impaired in HD.
Another pathological feature of HD is htt aggregates (Walker 2007). Mutant htt causes a severe mitochondrial defect, which may produce oxidative stress (Goswami et al. 2006; Trushina and McMurray 2007). This oxidative stress would disturb proteasome function by increasing the number of damaged proteins or by reducing ATP production. However, the role of htt aggregates in cell death is controversial. Numerous in vitro studies have indicated that htt toxicity may be due to small fragments produced by an incomplete N-terminal degradation in the proteasome rather than to the formation of intracellular inclusions (Arrasate et al. 2004). Bhutani et al. (2007) recently reported that cytosolic AlaAP is the main enzyme able to metabolize proteasome-released polyQ sequences. Therefore, the ineffective clearance of extended polyQ fragments by AlaAP may promote the formation of cytosolic inclusions. Based on the above results, it is possible that the reduced AP activity found in HD is related to the impairment of protein metabolism. All of these findings indicate that the etiology of HD is not only related to htt but also to other alterations in protein metabolism, especially changes in enzymatic activity, including the AP alterations reported in the present study. Our results suggest that these changes may be related to degenerative processes in HD. Hence, plasma AP activity may have diagnostic potential in this disease.
In conclusion, this study demonstrates the presence of oxidative stress markers and altered AP activity in the plasma of symptomatic HD patients. Plasma LPO and LAC are elevated in symptomatic HD patients but not in asymptomatic HD subjects, while AP activities are reduced both in symptomatic and in asymptomatic HD subjects. These changes may be related to the progression of the disease. A therapy directed to improve oxidative stress and normalize AP activity may be useful in the treatment of HD.
References
Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Rev Neurosci 5(S10):S18–S25
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Banegas I, Prieto I, Vives F, Alba F, Duran R, Segarra AB, de Gasparo M, Ramírez M (2004) Plasma aminopeptidase activities in rats after left and right intrastriatal administration of 6-hydroxydopamine. Neuroendocrinology 80:219–224
Banegas I, Prieto I, Vives F, Alba F, de Gasparo M, Segarra AB, Hermoso F, Duran R, Ramírez M (2006) Brain aminopeptidases and hypertension. J Renin Angiotensin Aldosterone Syst 7(3):129–134
Bhutani N, Venkatraman P, Goldberg AL (2007) Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J 26:1385–1396
Blanco S, Suarez A, Gandia-Pla S, Gómez-Llorente C, Antúnez A, Gómez-Capilla JA, Fárez-Vidal E (2008) Use of capillary electrophoresis for accurate determination of CAG repeats causing Huntington disease. An oligonucleotide design avoiding shadow bands. Scand J Clin Lab Invest 68:577–584
Browne SE, Ferrante RJ, Beal MF (1999) Oxidative stress in Huntington’s disease. Brain Pathol 9:147–163
Butterworth J (1986) Changes in nine enzyme markers for neurons, glia, and endothelial cells in agonal state and Huntington’s disease caudate nucleus. J Neurochem 47:583–587
Chen CM, Wu YR, Cheng ML, Liu JL, Lee YM, Le PW, Soong BW, Chiu DTY (2007) Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem Biophys Res Commun 359:335–340
Dawbarn D, Zamir N, Waters CM, Hunt SP, Emson PC, Brownstein MJ (1986) Peptides derived from prodynorphin are decreased in basal ganglia of Huntington’s disease brains. Brain Res 372:155–158
Finsterer J (2005) Lactate stress testing in sporadic amyotrophic lateral sclerosis. Int J Neurosci 115:583–591
Gil JM, Rego AC (2008) Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci 27:2803–2820
Goswami A, Dikshit P, Mishra A, Mulherkar S, Nukina N, Jana NR (2006) Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction. Biochem Biophys Res Commun 342:184–190
Grünewald T, Beal MF (1999) Bioenergetics in Huntington’s disease. Ann NY Acad Sci 893:203–213
Gu M, Gash MT, Mann VM, Javay-Agid F, Cooper JM, Schapira AHV (1996) Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 39:385–389
Harms L, Meierkord H, Timm G, Pfeiffer L, Ludolph AC (1997) Decreased N-acetyl-aspartate/choline ratio and increased lactate in the frontal lobe of patients with Huntington’s disease group: a proton magnetic resonance spectroscopy study. J Neurol Neurosurg Psychiatry 62:27–30
Hui K (2007) Brain-specific aminopeptidase: from enkephalinase to protector against neurodegeneration. Neurochem Res 32:2062–2071
Huntington Study Group (1996) Unified Huntington’s disease rating scale: reliability and consistency. Mov Disord 11:136–142
Iadarola MJ, Mouradian MM (1989) Decrease in a proenkephalin peptide in cerebrospinal fluid in Huntington’s disease and progressive supranuclear palsy. Brain Res 479:397–401
Jenkins BC, Koroshetz WJ, Beal MF, Rosen BR (1993) Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 43:2689–2695
Klepac N, Relja M, Klepac R, Hecimovic S, Babic T, Trkulja V (2007) Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects. J Neurol 254:1676–1683
Li SH, Li XJ (2004) Huntingtin–protein interactions and the pathogenesis of Huntington’s disease. Trends Genet 20:146–154
Martin E, Rosenthal RE, Fiskum G (2005) Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res 79:240–247
Martin WR, Wieler M, Hanstock CC (2007) Is brain lactate increased in Huntington’s disease? J Neurol Res 263:70–74
Mendelshon FA, Jenkins TA, Berkevic SF (1993) Effects of angiotensin II on dopamine and serotonine turnover in the striatum of conscious rats. Brain Res 613:221–229
Mitsui T, Nomura S, Itakura A, Mizutani S (2004) Role of aminopeptidases in the blood pressure regulation. Biol Pharm Bull 27:768–771
Mota N, Iturrioz X, Claperon C, Bodineau L, Fassot C, Roques BP, Palkovits M, Llorens-Cortes C (2008) Human brain aminopeptidase A: biochemical properties and distribution in brain nuclei. J Neurochem 106:416–428
Petrasch-Parwez E, Nguyen H, Löbbecke-Schumacher M, Habbes HW, Wieczorek S, Riess O, Andres KH, Dermietzel R, Von Hörsten S (2007) Cellular and subcellular localization of Huntington aggregates in the brain of a rat transgenic for Huntington disease. J Comp Neurol 501(5):716–730
Potolicchio I, Carven GJ, Xu S, Stipp C, Riese RJ, Stern LJ, Santambrogio L (2005) Proteomic analysis of microglia-derived exosomes. Metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J Immunol 175(4):2237–2243
Saft C, Zange J, Andrich J, Müller K, Lindenberg K, Landwehrmeyer B, Vorgerd M, Kraus PH, Przuntek H, Schöls L (2005) Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov Disord 20:674–679
Sayre LM, Perry G, Smith MA (2008) Oxidative stress and neurotoxicity. Chem Res Toxicol 21:172–188
Spires TL, Hannan AJ (2007) Molecular mechanisms mediating pathological plasticity in Huntington’s disease and Alzheimer’s disease. J Neurochem 100:874–882
Stöckel-Maschek A, Stiebitz B, Koelsch R, Neubert K (2003) A continuous fluorimetric assay for aminopeptidase P detailed analysis of product inhibition. Anal Biochem 322:60–67
Stragier B, Sarre S, Vanderheyden P, Vauquelin G, Fournie-Zaluski MC, Ebinger G, Michotte Y (2004) Metabolism of angiotensin II is required for its in vivo effect on dopamine release in the striatum of the rat. J Neurochem 90:1251–1257
Trushina E, McMurray CT (2007) Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145:1233–1248
Walker FO (2007) Huntington’s disease. Lancet 369:218–228
Acknowledgments
The authors thank the Junta de Andalucia for its support to PAI group CTS 438. R. Duran was supported by a grant of Formacion de Personal Docente e Investigador, Junta de Andalucia, Spain and postdoctoral research contract of the University of Granada. We are grateful to the Department of Clinical Biochemistry of San Cecilio University Hospital for the analysis of lipids and Huntington genetics. All subjects signed their informed consent to participate in the study, which was approved by the Ethical Committee for Human Research of our hospital.
Conflict of interest statement
Authors have no conflict of interest with any commercial company or sources of funding.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Duran, R., Barrero, F.J., Morales, B. et al. Oxidative stress and plasma aminopeptidase activity in Huntington’s disease. J Neural Transm 117, 325–332 (2010). https://doi.org/10.1007/s00702-009-0364-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-009-0364-0