
Abstract
The major breakthrough discovery of enkephalins as endogenous opiates led our attempts to determine their inactivation mechanisms. Because the NH2-terminal tyrosine is absolutely necessary for the neuropeptides to exert analgesic effects, and aminopeptidase activities are extraordinarily high in the brain, a specific “amino-enkephalinase” should exist. Several aminopeptidases were identified in the central nervous system during the search. In fact, our laboratory found two novel neuron-specific aminopeptidases: NAP and NAP-2. NAP is the only functionally active brain-specific enzyme known. Its synaptic location coupled with its limited substrate specificity could constitute a “functional” specificity and contribute to enkephalin-specific functions. In addition, NAP was found to be essential for neuron growth, differentiation, and death. Thus, aminopeptidases are likely important for mental health and neurological diseases. Recently, puromycin-sensitive aminopeptidase (PSA) was identified as a modifier of tau-induced neurodegeneration. Because the enzymatic similarity between PSA and NAP, we believe that the depletion of NAP in Alzheimer’s disease (AD) brains plays a causal role in the development of AD pathology. Therefore, use of the puromycin-sensitive neuron-aminopeptidase NAP could provide neuroprotective mechanisms in AD and similar neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Proteolytic enzymes are fundamental for protein turnover and for metabolizing bioactive peptides. They are some of the most potent tools that catalyze an irreversible reaction—the breakdown of peptide bonds. Peptidases’ activity is excessively high in the brain, with a capability 100–1,000 times higher than that required for cerebral protein turnover [1].
Aminopeptidases
Aminopeptidases (EC 3.4.11.-) are a heterogeneous group of exopeptidases that catalyze the N-terminal cleavage of amino acid residues from proteins or peptides [2–4]. They are classified according to the number of amino acids cleaved from the NH2-terminus, their relative efficiency with which residues are removed, their location, their susceptibility to inhibitors, their metal ion content, their residues that coordinated the metal to the enzyme, and the pH for their maximal activity. Aminopeptidases are involved in numerous functions, particularly in the activation, modulation, and degradation of bioactive peptides [5–7]. The study of neurotransmitter/neuropeptide enzymes has substantially contributed to our understanding of synaptic biochemistry; some of their inhibitors are used as valuable therapeutics [7].
Opioid peptides
Enkephalins, endorphins, dynorphins, and orphanin belong to the family of pain-killing opioid peptides. They are neurotransmitters and neurohormones in the nervous and the endocrine system [8–15]. These peptides have a close structural relationship and share with opiate receptor-mediated actions in analgesia [16, 17], catalexy/epilepsy [16], memory [18], hypothermia [19], appetite [20], sexual behavior [21], hormones [22], mental diseases, and behavior [23–25]. It is remarkable that both the opioid peptides and aminopeptidases are involved in analgesia and memory [7].
Enkephalin inactivation
Immediately after discovering enkephalins, it was noted that they had extremely brief action. The neuropeptides were metabolized rapidly following systemic administration. The intact enkephalins were blocked from passing through the blood-brain barrier. Several research groups, including us, vigorously attempted to understand their inactivation mechanism [1]. It is believed that enkephalins must first bind to the opiate receptors in nerve cell membranes in order to trigger intracellular functions. Enkephalins are hydrolyzed in the brain and peripheral tissues with various mechanisms [1, 26]. The unique distribution of the CNS enkephalinergic neurons indicates that a particular enzyme specifically mediates enkephalin metabolism within and by neurons.
Very few peptidases have a high degree of substrate specificity [1]. An enkephalin-specific peptidase, the so-called “enkephalinase” has yet to be found. After binding to the opiate receptors on the outer membrane and triggering the subsequent reactions, enkephalin is apparently terminated by hydrolysis. Studies have found no uptake, internalization mechanism, N-acetylation, O-sulfation, phosphorylation, or glycosylation [26, 27]. In the CNS, several peptidases can cleave Met- and Leu-enkephalin (Tyr–Gly–Gly–Phe–Met, and Tyr–Gly–Gly–Phe–Leu) at different sites: Puromycin-sensitive aminopeptidase (PSA) and aminopeptidase N (APN) at the Tyr–Gly amide bond, dipeptidyl aminopeptidase (DAP) at the Gly–Gly bond, carboxylpeptidase at the Phe-Met bond, and angiotensin-converting enzyme (ACE) and endopeptidase 24.11 (NEP), at the Gly–Phe bond.
Carboxylpeptidase, ACE, DAP, and NEP
Because of their high catalytic K m (in mM level) for enkephalin hydrolysis, neither carboxylpeptidase nor ACE is likely a critical enzyme for enkephalin inactivation [28]. Although it has high activity in neurons and a low K m for enkephalins [29], DAP has a low activity in membranes and is uniformly distributed in the brain [30]. Inhibition of NEP by thiorphan induces analgesia [27, 31]. It is controversial whether NEP is a neuronal enzyme [32, 33]. But, molecular cloning of NEP 24.11 revealed its structural homology with the common acute lymphoblastic leukemia antigen (CD10), implying its potential primary role in immunology [34].
NH2-terminal hydrolysis
It has been suggested that enkephalin binding to the opiate receptor is coupled with subsequent aminopeptidase degradation [35]. Opioid peptides have a common NH2-terminal peptide sequence (Tyr–Gly–Gly–Phe–) [36]. The NH2-terminal tyrosine is an absolute requirement for these peptides to exert opiate effects [37]. With cell-free and intact-cell preparations, the liberation of Tyr is the major mode of enkephalin degradation for its inactivation [38, 39]. In addition, aminopeptidase inhibitor, bestatin or puromycin produces a dose-related, naloxone-reversible, analgesic effect [40–42].
Membrane APN is involved in the metabolism of regulatory peptides in small intestinal and renal tubular epithelial cells, macrophages, and granulocytes [43]. The aminopeptidase is highly homologous to CD13, a 150-kD cell surface glycoprotein [43, 44]. Furthermore, its exclusive localization in the brain micro-vessels excludes APN as “enkephalinase” candidate [45].
PSA in enkephalin degradation
PSA is a neutral aminopeptidase that splits a single amino acid from the NH2-terminal of the peptides. It is most concentrated in the brain [46]. While the majority (80%) of the PSA resides in the cytosol, the rest is associated with the membranes. The soluble and membrane-associated forms of the enzyme are interchangeable [47]. The gene of PSA is 27–40% homologous to Zn++-binding aminopeptidases, including APN [48]. The PSA gene (GenePSA) is mapped to chromosome 17 at the interval 17q12–21 [49]. Through immunohistochemistry, the peptidase has been localized to the cytoplasm and the nucleus. It associates with microtubules of the spindle apparatus during mitosis [48]. PSA, a ubiquitous enzyme, participates in proteolytic events for cell growth and viability [48, 50]. Moreover, the enzyme is bound to intracellular membranes with the active sites inside the cytosol. It is believed that an “enkephalinase” is an ecto-enzyme that hydrolyzes extra-cellular enkephalins. Even though an anatomical distribution (specific location) associated with limited substrate specificity could constitute a ‘functional specificity’, an intracellular localization would preclude PSA from functioning as “enkephalinase” [39, 51]. The notion is further supported by the imparity that dwarfism is found in the GenePSA-deficient mice [52, 53] but not in the Geneenkephalin-deficient mice [54].
PSA structure and function relationship
Intense focus on PSA has yielded important knowledge of its protein structure, its substrates and cleavage activity, and the regulation of its activity [7]. In human PSA, glutamate 309 acts as a general acid/base catalyst. Its substitution with other amino acids resulted in a 5,000- to 500,000-fold reduction in catalytic activity [55]. Since aminopeptidases are complex proteins with pockets for substrate binding and catalysis, metal ion binding, and possibly protein–protein interactions [2, 56, 57], these peptide sequences could be targets for specific domain-selective inhibitors [58]. Recently, Tang et al. elegantly demonstrated that a peptidase could also be activated by structural modification [59]. Conformational modification of insulin-degrading enzyme made the enzyme 40-fold more active in accepting substrates and releasing products faster.
Natural substrates of aminopeptisases
PSA has a preference for a basic or hydrophobic residue in the P1 and P1′ sites and has subsite–subsite interactions that primarily affect binding [60]. Its active site extends beyond the S2′ position. It is highly possible that aminopeptidases, including PSA, have different activity towards physiologically important neuropeptides and endogenous inhibitors. The substrate specificity study of aminopeptidases will pinpoint their specific endogenous substrates and elucidate their biological function in neurotransmission. In addition to enkephalins, aminopeptidases sensitive to puromycin are involved in the inactivation of dynorphins, cholecystokinin, somatostatin, oxytocin, angiotensin, proctolin, and vasopressin [61]. Arg0-enkephalin, an immediate precursor of enkephalin [7, 30, 37, 62], can be converted to enkephalin by PSA [7].
PSA diverse functions
The broad substrate specificity and wide distribution of PSA in different tissues signify that it has many diversified functions. It participates in proteolytic events essential for cell mitosis, growth, and viability [63, 64]. Mice with mutations in the PSA (goku) gene developed as dwarfs, showed behavioral defects––anxiety and pain––and were sterile [52]. The fertility defects in goku mutant mice indicate that PSA participates in testosterone-mediated reproductive signaling pathways in testes and brain and plays a crucial role in maternal pregnancy recognition [53]. In Drosophila, PSA commands protein turnover jointly with other aminopeptidases. In addition, its expression is tightly regulated for normal development [65].
PSA in brain diseases
Recently, PSA was found to be crucial in mental and neurological diseases [7]. The PSA protein content in schizophrenic prefrontal cingulate and frontal cortices, thalamus, hippocampus, hypothalamus, and outer globus pallidus is less than its corresponding controls [66]. Moreover, PSA and amyloid precursor protein (APP) have been co-immunoprecipitated [67] and co-localized around senile plaques in the cerebral cortex and hippocampus of the Alzheimer’s disease (AD) brain [68].
Microtubule-associated protein tau in neurodegeneration
Neurofibrillary tangles (NFT) holding tau are a hallmark of neurodegenerative diseases––including AD. NFT burden correlates with cognitive decline and neurodegeneration in AD. The mechanisms of tau-induced neurodegeneration are not fully understood. Pathological aggregation of tau is a typical characteristic of many neurodegenerative diseases collectively called tauopathies [69, 70]. Either the gain of toxicity or loss of function hypotheses could explain tau’s pathological roles.
An essential role of tau in tauopathies became evident with the discovery that tau mutations cause inherited forms of frontotemporal dementia (FTD) with Parkinsonism linked to chromosome 17 (FTDP-17) [69–71]. The process of tau aggregation, its paired helical filament assembly, and its accumulation is not completely understood. While tau hyperphosphorylation evidently accelerates neurodegeneration [72–74)], the role of other posttranslational modifications, including proteolysis [73, 75], ubiquitination [76–78], and nitration and glycosylation [77–80], as well as the function of the tau amino terminus in this process, remain unclear [75, 81, 82].
Numerous in vitro studies have identified potential proteases that are active against tau, including calpain, caspases, and thrombin [75, 83, 84]. However, their connection to neurodegeneration in tauopathy is vague. Thus, identifying the causes that influence tau aggregation or degradation in vivo and modulate tau-induced neurodegeneration has important implications for understanding tau-induced neurodegeneration and designing potential therapeutic interventions [70, 73, 85–87].
Aminopeptidases in Alzheimer’s disease
NH2-terminal caspase-mediated cleavage of tau in vitro and in AD may be involved in FTD formation [75, 81]. Moreover, FTDP-17 mutations could render tau protein less susceptible to proteolysis, increasing the propensity of tau to form aggregates. Analogous to the role of proteolytic processing of other potentially toxic moieties, such as proteolytic processing of amyloid by presenilins or secretases [88, 89], cerebral aminopeptidases can also control tau toxicity in vivo by regulating tau-induced neurodegeneration.
Regulation of APP and its proteolytic fragments plays a critical role in AD pathogenesis [89]. Similarly, it is clear from the human FTD-causing mutations that even relatively subtle changes in tau isoform levels can cause neurodegeneration [90]. Conversely, turning off an inducible mutant tauP301L transgene after the onset of severe tau pathology in the mouse, thereby reducing mutant tau levels, can reverse the neurodegenerative process [87]. We speculate that factors, such as brain aminopeptidases, that act to modulate tau levels or splicing, are candidates for playing a causal or contributory role in disease, and may represent potential targets for therapeutics. Factors that lead to brain aminopeptidases down-regulation in humans would be expected to increase tau, which in turn could contribute to disease susceptibility. Neurodegeneration in FTD is known to primarily affect the superficial cortical laminae. Therefore, even within brain regions, different neurons are likely to show distinct patterns of vulnerability to neurodegeneration. These observations highlight a potential role for brain aminopeptidases that warrant analysis in human patient samples.
Identification of PSA as an inhibitor of tau-induced neurodegeneration
Most recently, Karsten et al. used DNA microarrays to find genes that were more activated in certain regions than others in the brains of transgenic tauP301L mice [91]. The mice were engineered to have a mutant form of human tau that causes neurodegeneration. The gene for PSA was among those identified as more being activated in the resistant cerebellum.
In fruit fly Drosophila, the group later found that suppression of PSA gene worsened neurodegeneration. With in vitro study, they found that PSA degraded tau peptides. Karsten et al. then compared levels of PSA gene expression in samples of brain tissue from the cortex and cerebellum of both normal humans and those with fibrillary tangle disorders. In both, a 5-fold elevation of PSA was found in the cerebellum as compared to the cortex. Although PSA was known to be highly brain enriched, its role vis-a-vis tau degradation or modification of tau-induced neurodegeneration had first been characterized. It was concluded that PSA might play a pivotal role in preventing tau-induced neurodegeneration, most likely by direct cleavage of tau. The work not only reveals a significant protective factor in fibrillary tangle disorders, it provides new direction for further study of other such factors. Disabling the mutant, pathological form of tau in mice after the mice showed neuronal pathology could reverse neurodegeneration. Geschwind and co-workers [91] thus speculated that factors that act to modulate tau levels or splicing, such as PSA, are candidates for playing a causal or contributory role in disease.
PSA (also known as Npepps), a highly conserved protein, protects against tau-induced neurodegeneration in vivo, and its loss of function exacerbates neurodegeneration [91]. Although PSA does not directly alter APP levels in vitro, brain aminopeptidases could provide a link between amyloid deposition and tau pathology observed in AD. While this and other questions relevant to the causal role of PSA in human diseases involving tau are important to answer, discovering that it is a protease potentially mediating tau degradation is an important step in understanding the pathogenesis of tauopathies and developing new therapeutic interventions in these fatal neurodegenerative diseases.
Sengupta et al. extended the observations using human recombinant PSA purified from Escherichia coli [92]. The enzymatic activity and characteristics of the purified PSA were verified using chromogenic substrates, metal ions, and several specific and nonspecific protease inhibitors, including puromycin. PSA was shown to digest recombinant human full-length tau in vitro, and this activity was hindered by puromycin. The mechanism of amino terminal degradation of tau was confirmed using a novel N-terminal cleavage-specific tau antibody (Tau-C6g, specific for cleavage between residues 13–14) and a C-terminal cleavage-specific tau antibody (Tau-C3). Additionally, PSA was able to digest soluble tau purified from normal human brain to a greater extent than either soluble or PHF tau purified from AD brain, indicating that post-translational modifications and/or polymerization of tau may affect its digestion by PSA. The results were consistent with observations that PSA modulates tau levels in vivo and may be involved in tau degradation in human brain.
Puromycin inhibition targets
Cerebral administration of puromycin induces memory loss and amnesia, characteristic symptoms of AD [93–95]. The effects are neither due to the inhibition of protein synthesis nor the impairment of cholinergic transmission at central synapses [94, 95]. The puro-peptide complexes persisted in synaptosomes and not other organelles [96]. Recently, we found that puromycin represses neuron growth and differentiation in vitro by inhibiting neuronal aminopeptidases [97]. The study raises a number of interesting questions: How many puromycin-sensitive aminopeptidases are in the brain? Is there a brain-specific PSA? What is puromycin’s primary inhibitor target in the brain?
Brain-specific neuron aminopeptidases
PSA (GenePSA-dependent puromycin-sensitive aminopeptidase) accounts for only 59% of the total brain aminopeptidase activity, followed by the GenePSA-independent puromycin-sensitive aminopeptidase for 29%, and puromycin-insensitive aminopeptidase for 12% [52, 98]. Robust aminopeptidase activity was located in the cell bodies of the CNS neurons [99]. PSA transcripts were detected in rat CNS neurons by in situ hybridization [48]. This preferential expression in neurons indicates that specific neuron aminopeptidase(s) have dedicated functions. Aminopeptidases with over-lapping substrate specificities are present in the CNS. In order to find specific aminopeptidases in the brain, we developed a post-column continuous-flow aminopeptidase detector by exploiting the highly sensitive substrate β-nathylamides (βNA). Its conjugation with an FPLC provides a fast, sensitive, specific, and reliable method for aminopeptidase screening and quantitation. The method separates an aminopeptidase of interest from the interfering peptidases, activators, and endogenous inhibitors [100].
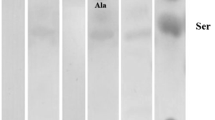
We first compared the different aminopeptidases in the brain and the peripheral tissues of rat. We found two novel neuron-specific aminopeptidases: NAP and NAP-2. NAP and NAP-2 were present only in the CNS but not in peripheral tissues, serum, or sciatic nerve [7, 101]. NAP was found in rat, mouse, bovine, and human brain. NAP-2 was found only in rat and mouse (Fig. 1). Neither NAP nor NAP-2 was detected in chicken. In the rat cerebrocortical neuron cultures, the activity of NAP was 33% of the total aminopeptidase activity, 12% in the cerebellar granule cells, and absent in the astrocytes. NAP was also absent in the glioma C6 and the neuroblastoma SK-N-SH cells [101]. The distribution of NAP and NAP-2 was different in the rat brain; the highest NAP activity was found in the hippocampus, whereas the highest NAP-2 activity was found in the colliculus. NAP activity was always double that of NAP-2 in all the tissues and cell cultures. Hypothalmus had the highest ratio of NAP/NAP-2. Both aminopeptidases were concentrated in synaptosomes with the NAP activity being greater than that of NAP-2 (Hui and Hui, unpublished observations).

Aminopeptidase Zymograms of of Rat (upper) and Human (lower) by FPLC-aminopeptidase Analyzer. Enzyme activity was determined by measuring the fluorescence of the released β-napthylamine
NAP expression parallels neuron growth
NAP was found in the rat hippocampus of all ages, ranging from prenatal to 3-month old [97]. Its concentration was lower in immature rat; the 19th embryonic-day fetus had the least. NAP increased steeply during the prenatal through the neonatal period, 9-fold by the first month. The growth speed diminished subsequently, increasing 20% in the second month and 13% in the third. The age-dependent increase in NAP activity paralleled its protein expression. The specific hydrolytic activity/NAP antigenicity in newborn, 15-day-old, and 30-day-old were 1.00, 0.88, and 1.00, respectively. Its growth profile was distinct from that of the leveled PSA. A similar pattern between NAP and PSA was also found in the developing primary cerebellar granule cells. NAP increased linearly, whereas PSA remained unchanged, from 1–7 days after the seeding of the cells. By the 9th day, NAP activity increased by 4-fold. Adding puromycin or bestatin, a general aminopeptidase inhibitor, to the medium on the 6th day inhibited neurite growth and caused cell death. The data indicate that NAP is essential for the neuron growth, differentiation, and cell death.
NAP biochemistry
NAP was purified from rat brain about 5,000-fold to homogeneity with 9% recovery [101]. The neutral aminopeptidase hydrolyzes βNAs of amino acids with aliphatic, polar uncharged, positively charged, or aromatic side chains. It has a K m of 95 μM and a k cat of 7.8 s−1 on Met-enkephalin, releasing only the N-terminal tyrosine. The thiol-dependent metallo-enzyme is most sensitive to amastatin inhibition with a K i of 0.04 μM, and is the aminopeptidase most sensitive to puromycin. The purified single-chain enzyme was estimated to be 110 kDa in molecular mass. It has a pI of 5.25 and a pH optimum of 7.0. Only Mg (II) restores the activity of the apoenzyme. The purified enzyme is distinct from all known aminopeptidases, including PSA. The NAP enrichment in the synaptosomes suggests that this neuron-specific peptidase plays a role in neuron growth, neurotransmission, and synaptic differentiation. Because the substrate and inhibitor specificity of NAP and PSA are rather similar [101], their hydrolysis of tau is expected to be comparable.
We cloned a candidate 1,404-bp cDNA (63.2% identity to mouse PSA) encoding the N-terminal section of rat NAP [7]. The nucleotide segment position at 875–1,404 is homologous to position 1,087–1,613 of PSA (96.4% identity), containing a sequence encoding a divalent metal-binding motif, HEXXH(X)18E, of aminopeptidases [102]. The sequence at the 5′- end 1–874, with an identity 44.9% to PSA, is novel. It is void of the sequence encoding a universal N-terminal PENKRPFERLPTEVSPINY of PSA [47, 48, 63]. The blocked N-terminal residue, though, has not been identified. The cloning data imply that NAP is a novel aminopeptidase and a possible member of the PSA superfamily located in chromosome 17.
NAP-2 biochemistry
Lately, rat NAP-2 was purified to apparent homogeneity by ammonium sulfate fractionation followed by column chromatography successively on Phenyl-Sepharose, Sephadex G-200, and twice on Mono Q with FPLC (Hui and Hui, unpublished observations). The purified aminopeptidase is a single polypeptide with an apparent molecular mass of 110 kD and a pI of 5.6. It splits βNAs of amino acids with aliphatic, polar uncharged, positively charged, and aromatic side chains. The enzyme also hydrolyses Met-enkephalin with a K m of 106 μM and k cat of 2.6 s−1, and Leu-enkephalin with a K m 82 μM and and k cat of 1.08 s−1. It is highly susceptible to model aminopeptidase inhibitors and most sensitive to amastatin with an IC50 of 0.05 μM. The glyco-, cysteine-, and metallo-aminopeptidase is a phosphorylated enzyme that dephosphorylates spontaneously. Its properties, natural occurrence, and developmental profile are distinct from PSA and NAP. With the completion of the Human Genome Project, it is becoming clear that there is a high degree of genomic similarity between species. Yet only rodents have NAP-2. The additional neuronal-specific aminopeptidase indicated that NAP is different between species; human NAP is different from the rat NAP.
NAP activity depletes in AD brains
Brain-specific proteins are powerful biomarkers and helpful tools in studies of neural mechanisms and neuropathology. Amongst the more than 100 brain-specific proteins, only NAP and the neuron-specific enolase are enzymes [7, 101, 103]. To our knowledge, NAP is the only functional brain-specific enzyme (Table 1). The independent identification of PSA as a potential protector against tauopathies reinforces our belief that the brain-specific and puromycin-sensitive neuron-aminopeptidase NAP has an imperative role in AD.
We studied the post-microsomal extracts of human brain tissues with an automatic FPLC-aminopeptidase analyzer [100]. After the injection, the column was washed with Bicine buffer then eluted with a NaCl gradient in the same buffer increasing linearly from 0 to 0.21 M at 12 min, 0.23 M at 26 min, 0.24 M at 36 min, 0.29 M at 56 min, and 0.5 M at 61 min. The enzyme elution from the FPLC was mixed with Leu βNA and incubated on-line in a delaying coil for 3 min. Finally, the sample was measured fluorometrically (Ex. 250/ Em. 389 nm). hNAP (detected at 41.5 min) and the PSA (36 min) peak were separated from each other (Fig. 1). The NAP activity was 14% of the total aminopeptidase activity [101]. To our surprise, in a preliminary study, its activity in the frontal cortex from the postmortem AD brains was drastically reduced to less than 5% of the total brain aminopeptidase activity.
Conclusions
Aminopeptidases are highly active enzymes for enkephalin and tau degradation. We found two novel neuron-specific aminopeptidases: One of them, NAP, has been the first functionally active brain-specific enzyme identified. In an independent study, we uncovered what appears to be a natural protective mechanism of aminopeptidases against a central cause of neuronal death in AD and similar neurodegenerative diseases. We theorize that it is possible to use drugs to enhance that mechanism to alleviate AD pathology. Typical AD symptoms, memory loss and amnesia, can be induced by puromycin inhibition. NAP is the possible primary target of puromycin in the brain. In AD brains, we found that NAP was depleted more than PSA. We believe that NAP is a leading/controlling factor in tau degradation. Thus, we are studying whether NAP is a possible biomarker for neurodegeneration, whether there are environmental (microbial) and endogenous inhibitors for the aminopeptidase, and how these inhibitors are linked to neurodegeneration.
References
Hui K-S, Lajtha A (1983) Neuropeptides. In: Lajtha A (ed) Handbook of neurochemistry, 2nd edn., vol. 4. Plenum, New York, pp 1–19
Taylor A (1993) Aminopeptidases: structure and function. FASEB J 7:290–298
Taylor A (1993) Aminopeptidases: towards a mechanism of action. Trends Biochem Sci 18:167–171
Yao T, Cohen RE (1999) Giant proteases: beyond the proteasome. Curr Biol 9: R551–R553
Lendeckel U, Arndt M, Frank K, Spiess A, Reinhold D, Ansorge S (2000) Modulation of WNT-5A expression by actinonin: linkage of APN to the WNT-pathway? Adv Exp Med Biol 477:35–41
Stoltze L, Schirle M, Schwarz G, Schroter C, Thompson MW, Hersh LB, Kalbacher H, Stevanovic S, Rammensee HG, Schild H (2000) Two new proteases in the MHC class I processing pathway. Nat Immunol 1:413–418
Hui K-S (2007) Neuropeptidases. In: Lajtha A, Banik NL (eds), Handbook of neurochemistry and molecular neurobiology: neural protein metabolism and function, 3rd edn., vol. 7. Springer-Verlag, Berlin, Heidelberg
Nakanish S, Inoue A, Kita T, Nakamura M, Chang ACY, Cohen SN, Numa S (1979) Nucleotide sequence of cloned cDNA for bovine corticotropin-β-lipotropin precursor. Nature 278:423–424
Gubler U, Seeburg P, Hoffman BJ, Gage LP, Udenfriend S (1982) Molecular cloning establishes proenkephalin as precursor of enkephalin-containing peptides. Nature 295:206–208
Boileu G, Barbeau C, Jeannotte L, Chretien M, Drouin J (1983) Complete structure of the porcine proopiomelanocortin mRNA derived from the nucleotide sequence of cloned cDNA. Nucleic Acids Res 11:8063–8071
Yoshikawa K, Williams C, Sabol SL (1984) Rat brain preproenkephalin mRNA, cDNA cloning, primary structure, and distribution in the central nervous system. J Biol Chem 259:14301–14308
Civelli O, Douglass J, Goldstein A, Herbert E (1985) Sequence and expression of the rat prodynorphin gene. Proc Natl Acad Sci USA 82:4291–4295
Nothacker HP, Reinschied RK, Mansour A, Henningsen RA, Ardati A, Monsma FJ, Watson SJ, Civelli O (1996) Primary structure and tissue distribution of the orphanin FQ precursor. Proc Natl Acad Sci USA 93:8677–8682
Molleraeu C, Simons MJ, Soulariue P, Liners F, Vassart G, Meunier JC, Parmentier M (1996) Structure tissue distribution and chromosomal localization of the prepronociceptin gene. Proc Natl Acad Sci USA 93:8666–8670
Hook VYH, Reisine TD (2001) Endorphin. In: Creighton TE (ed) Encyclopedia of molecular medicine, pp 1161–1164
Frenk H, McCarty BC, Lieberkind JC (1978) Different brain areas mediate the analgesic and epileptic properties of enkephalin. Science 200:335–337
Lee JJ, Hahm ET, Min BI, Cho YW (2004) Activation of protein kinase C antagonizes the opioid inhibition of calcium current in rat spinal dorsal horn neurons. Brain Res 1017:108–119
De Wied D, Bohus B, van Ree JM, Urban I (1978) Behavioral and electrophysiological effects of peptides related to lipotropin (β-LPH) J. Pharmacol Exp Ther 204:570–580
Holaday JH, Loh HH, Li CH (1978) Unique behavioral effects of β-endorphin and their relationship to thermoregulation and hypothalamic function. Life Sci 22:1525–1536
Brands B, Thornhill JB, Hirst M, Gowdey CW (1979) Suppression of food intake and body weight gain by naloxone in rats. Life Sci 24:1773–1778
Meyerson BJ, Terenius L (1977) β-endorphin and male sexual behavior. Eur J Pharmacol 42:191–192
Morley JE. 1981. The endocrinology of the opiates and opioid peptides. Metabolism 30:195–209
Bloom F, Segal D, Ling N, Guillemin R (1976) Endorphins: profound behavioral effects in rats suggest new etiological factors in mental illness. Science 194:630–632
Jacquet YF, Marks N (1976) The C-fragment of β-lipotropin: an endogenous neuroleptic or antipsychotogen? Science 194:632–635
Matsuzaki S, Ikeda H, Akiyama G, Sato M, Moribe S, Suzuki T, Nagase H, Cools AR, Koshikawa N (2004) Role of mu- and delta-opioid receptors in the nucleus accumbens in turning behaviour of rats. Neuropharmacology 46:1089–1096
Goodman RR, Fricker LD, Snyder SH (1983) Enkephalins. In: Krieger DT, Brownstein MJ, Martin JB (eds) Brain peptides. John Wiley & Sons, New York, pp 827–849
Patey G, De La Baume S, Schwartz J-C, Gros C, Roques B-P, Fournie-Zaluski M-C, Soroca-Lucas E (1981) Selective protection of methionine enkephalin release from brain slices by enkephalinase inhibition. Science 212:1153–1155
Schwartz J-C (1983) Metabolism of enkephalins and the inactivating neuropeptidase concept. Trends Neurosci 6:15–18
Lee C-M, Snyder SH (1982) Dipeptidyl-aminopeptidase III of rat brain. J Biol Chem 257:12043–12050
Horsthemke B, Hamprecht B., Bauer K (1983) Heterogeneous distribution of enkephalin-degrading peptidases between neuronal and glial cells. Biochem Biophys Res Comm 115:423–429
Llorens C, Cacel G, Swerts J-P, Perdrisot R, Fournie-Zaluski K-C, Schwartz J-C, Roques B-P (1980) Rational design of enkephalinase inhibitor: substrate specificity of enkephalinase studied from inhibitory potency of various dipeptides. Biochem Biophys Res Commun 96:1710–1716
Barnes K, Turner AJ, Kenny AJ (1988) Electronmicroscopic immunocytochemistry of pig brain shows that endopeptidase 24.11 is localized in neuronal membranes. Neurosci Lett 94:64–69
Skidgel RA, Erdos EG (2004) Angiotensin-converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides 25:521–525
Howell S, Murray H, Scott CS, Turner AJ, Kenny AJ (1991) A highly sensitive EL.IS.A. for endopeptidase-24.11, the common acute-lymphoblastic-leukaemia antigen (CALLA, CD-10), applicable to material of porcine and human origin. Biochem J 278:417–421
Knight M, Klee WA (1978) The relationship between enkephalin degradation and opiate receptor occupancy. J Biol Chem 253:3843–3847
Cox BM (1982) Endogenous opioid peptides: a guide to structures and terminology. Life Sci 31:1645–1658
Amar C, Vilkas E, Laurent S, Gautray B, Schmitt H (1983) A new enkephalin analogue: trans-4-hydroxycinnamoyl-glycyl-glycyl-phenylalanyl-leucine synthesis and biological properties. Int J Peptide Protein Res 22:434–436
Lentzen H., Palenker J (1983) Localization of the thiorphan-sensitive endopeptidase, termed enkephalinase A, on glial cells. FEBS Lett 153:93–97
Schwartz J-C, Costentin J, Lecomte J-M (1985) Pharmacology of enkephalinase inhibitors. Trends Pharmacol Sci 6:472–476
Zhang A-Z, Yang H-Y, Costa E (1982) Nociception enkephalin content and dipeptidyl carboxypeptidase activity in brain of mice treated with exopeptidase inhibitors. Neuropharmacol 21:625–630
De La Baume S. Yi CC, Schwartz J-C, Marcais-Collado H, Costentin J (1983) Participation of both “enkephalinase” and aminopeptidase activities in the metabolism of endogenous enkephalins. Neurosci 8:143–151
Herman ZS,Strachura Z, Laskawiec G, Kowalski J, Obuchowicz E (1985) Antinociceptive effects of puromycin and bacitracin. Pol J Pharmacol 37:133–140
Look AT, Ashmun RA, Shapiro LH, Peiper SC (1989) Human myeloid plasma membrane glycoprotein CD13 (gp150) is identical to aminopeptidase. N J Clin Invest 83:1299–1307
Razak K, Newland AC (1992) Induction of CD13 expression on fresh myeloid leukaemia: correlation of CD13 expression with aminopeptidase-N activity. Leuk Res 16:625–630
Hersh LB, Aboukhair N, Watson S (1987) Immunohistochemical localization of aminopeptidase M in rat brain and periphery: relationship of enzyme localization and enkephalin metabolism. Peptides 8:523–532
McLellan S, Dyer SH, Rodriguez G, Hersh LB (1988) Studies on the tissue distribution of the puromycin-sensitive enkephalin-degrading aminopeptidases. J Neurochem 51:1552–1559
Dyer SH, Slaughter CA, Orth K, Moomaw CR, Hersh LB (1990) Comparison of the soluble and membrane-bound forms of the puromycin-sensitive enkephalin-degrading aminopeptidases from rat. J Neurochem 54:547–554
Tobler AR, Constam DB, Schmitt-Graff A, Malipiero U, Schlapbach R, Fontana A (1997) Cloning of the human puromycin-sensitive aminopeptidase and evidence for expression in neuron. J Neurochem 68:889–897
Thompson MW, Tobler A, Fontana A, Hersh LB (1999) Cloning and analysis of the gene for the human puromycin-sensitive aminopeptidase. Biochem Biophys Res Commun 258:234–240
Brooks DR, Hooper NM, Isaac RE (2003) The Caenorhabditis elegans orthologue of mammalian puromycin-sensitive aminopeptidase has roles in embryogenesis and reproduction. J Biol Chem 278:42795–42801
Roques BP (1985) Enkephalinase inhibitors and molecular exploration of the differences between active sites of enkephalinase and the angiotensin conversion enzyme. J Pharmacol (Paris) 16:5–31
Osada T, Ikegami S, Takiguchi-Hayashi K, Yamazaki Y, Katoh-Fukui Y, Higashinakagawa T, Sakaki Y, Takeuchi T (1999) Increased anxiety and impaired pain response in puromycin-sensitive aminopeptidase gene-deficient mice obtained by a mouse gene-trap method. J Neurosci 19:6068–6078
Osada T, Watanabe G, Kondo S, Toyoda M, Sakaki Y, Takeuchi T (2001) Male reproductive defects caused by puromycin-sensitive aminopeptidase deficiency in mice. Mol Endocrinol 15:960–971
Konig M, Zimmer AM, Steiner H, Holmes PV, Crawley J, Brownstein MJ, Simmer A (1996) Pain responses anxiety and aggression in mice deficient in pre-enkephalin. Nature 383:535–538
Thompson MW Govindaswami M, Hersh LB (2003) Mutation of active site residues of the puromycin-sensitive aminopeptidase: conversion of the enzyme into a catalytically inactive binding protein. Arch Biochem Biophys 413:236–242
Wilce MCJ, Bond CS, Dixon NE, Freeman HC, Guss JM, Lilley PE, Wilce JA (1998) Structure and mechanism of a proline-specific aminopeptidase from Escherichia coli. Proc Nat Acad Sci USA 95:3472–3477
Thunnissen MMGM, Nordlund P, Haeggström JZ (2001) Crystal structure of human leukotrine A4 hydrolyse, a bifunctional enzyme in inflammation. Nat Struct Biol 8:131–135
Wei L, Alhenc-Gelas F, Corvol P, Clauser E (1991) The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J Biol Chem 266:9002–9008
Shen Y, Joachimiak A, Rosner MR, Tang W-J (2006) Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 443:870–874
Johnson GD, Hersh LB (1990) Studies on the subsite specificity of the rat brain puromycin-sensitive aminopeptidase. Arch Biochem Biophys 276:305–309
Hui K-S, Hui M, Lajtha A (1983) Properties of a brain membrane aminoenkephalinase: inhibition studies. In: Sun G, Bazan N, Wu J-Y, Porcellati G, Sun AY (eds) Neural membranes. Humana Press, NJ, pp 375–393
Turner AJ, Murphy LJ, Medeios MS, Barnes K (1996) Endopeptidase-24.11(neprilysin) and relatives: twenty years on. Adv Exptl Med Biol 389:141–148
Constam DB, Tobler AR, Rensing-Ehl A, Kemler I, Hersh LB, Fontana A (1995) Puromycin-sensitive aminopeptidase: sequence analysis, expression, and functional characterization. J Biol Chem 270:26931–26939
Sekine K, Fujii H, Abe F (1999) Induction of apoptosis by bestatin (ubenimex) in human leukemic cell lines. Leukemia 13:729–734
Schulz C, Perezgasga L, Fuller MT (2001) Genetic analysis of dPSA, the Drosophila orthologue of puromycin-sensitive aminopeptidase, suggests redundancy of aminopeptidases. Dev Genes Evol 211:581–588
Hui M, Palkovits M, Lajtha A, Budai D, Hui K-S (1995) Changes in puromycin-sensitive aminopeptidase in postmortem schizophrenic brain regions. Neurochem Int 27:433–441
Schonlein C, Loffler J, Huber G (1994) Purification and characterization of a novel metalloprotease from human brain with the ability to cleave substrates derived from the N-terminus of beta-amyloid protein. Biochem Biophys Res Commun 201:45–53
Minnasch P, Yamamoto Y, Ohkubo I, Nishi K (2003) Demonstration of puromycin-sensitive alanyl aminopeptidase in Alzheimer disease brain. Leg Med (Tokyo) 5 (supply 1): S285–S287
Ingram EM, Spillantini MG (2002) Tau gene mutations: dissecting the pathogenesis of FTDP-17. Trends Mol Med 8:555–562
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917
Kosik KS, Shimura H (2005) Phosphorylated tau and the neurodegenerative foldopathies. Biochim Biophys Acta 1739:298–310
Lee VM, Balin BJ, Otvos L Jr, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science 251:675–678
Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci USA 100:10032–10037
Bancher C, Grundke-Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM (1991) Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res 539:11–18
Gong CX, Liu F, Grundke-Iqbal I, Iqbal K (2005) Posttranslational modifications of tau protein in Alzheimer’s disease. J Neural Transm 112:813–838
Iqbal K, Grundke-Iqbal I (1991) Ubiquitination and abnormal phosphorylation of paired helical filaments in Alzheimer’s disease. Mol Neurobiol 5:399–410
Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Merkle RK, Gong CX (2002) Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett 512:101–106
Wang JZ, Grundke-Iqbal I, Iqbal K (1996) Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease. Nat Med 2:871–875
Amadoro G, Serafino AL, Barbato C, Ciotti MT, Sacco A, Calissano P, Canu N (2004) Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ 11:217–230
Chen F, David D, Ferrari A, Gotz J (2004) Posttranslational modifications of tau–role in human tauopathies and modeling in transgenic animals. Curr Drug Targets 5:503–515
Arai T, Guo JP, McGeer PL (2005) Proteolysis of nonphosphorylated and phosphorylated tau by thrombin. J Biol Chem 280:5145–5153
Mercken M, Grynspan F, Nixon RA (1995) Differential sensitivity to proteolysis by brain calpain of adult human tau, fetal human tau and PHF-tau. FEBS Lett 368:10–14
Kosik KS, Ahn J, Stein R, Yeh LA (2002) Discovery of compounds that will prevent tau pathology. J Mol Neurosci 19:261–266
Price DL, Tanzi RE, Borchelt DR, Sisodia SS (1998) Alzheimer’s disease: genetic studies and transgenic models. Annu Rev Genet 32:461–493
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Sisodia SS, St George-Hyslop PH (2002) Gamma-Secretase Notch, Abeta and Alzheimer’s disease: where do the presenilins fit in? Nat Rev Neurosci 3:281–290
D′Souza I, Schellenberg GD (2005) Regulation of tau isoform expression and dementia. Biochim Biophys Acta 1739:104–115
Karsten SL, Sang T-K, Gehman LT, Chatterjee S, Liu J, Lawless GM, Sengupta S, Berry RW, Pomakian J, Oh HS, Schulz C, Hui K-S, Wiedau-Pazos M, Vinters HV, Binder LI, Geschwind DH, Jackson GR (2006) A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-Induced neurodegeneration. Neuron 51:549–560
Sengupta S, Horowitz PM, Karsten SL, Jackson GR, Geschwind DH, Fu Y, Berry RW, Binder LI (2006) Degradation of Tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry 45:15111–15119
Springer AD, Agranoff BW (1976) Electroconvulsive shock––or puromycin––induced retention deficits in goldfish given two-avoidance sessions. Behav Biol 18:309–324
Flexner JB, Flexner LB, Stellar E (1963) Memory in mice as affected by intracerebral puromycin. Science 141:57–59
Eisenstein EM, Altman HJ, Barraco DA, Barraco RA, Lovell KL (1983) Brain protein synthesis and memory: the use of antibiotic probes. Fed Proc 42:3080–3085
Flexner LB, Gambetti P, Flexner JB, Roberts RB (1971) Studies on memory: distribution of peptidyl-puromycin in subcellular fractions of mouse brain. Proc Natl Acad Sci USA 68:26–28
Hui M, Hui KS (2003) Neuron-specific aminopeptidase and puromycin-sensitive aminopeptidase in rat brain development. Neurochem Res 28:855–860
Osada T, Watanabe G, Sakaki Y, Takeuchi T (2001) Puromycin-sensitive aminopeptidase is essential for the maternal recognition of pregnancy in mice. Mol Endocrinol 15:882–893
Shaw SG, Cook WF (1978) Localization and characterisation of aminopeptidase in the CNS and the hydrolysis of enkephalin. Nature 274:816–817
Hui K-S, Hui M (1996) An automatic continuous-flow aminopeptidase detector and its applications. Anal Biochem 242:271–273
Hui K-S, Saito M, Hui M (1998) A novel neuron-specific aminopeptidase in Rat brain synaptosomes: its identification, purification, and characterization. J Biol Chem 273:31053–31060
Shannon JD, Baramova EN, Bjarnason JB, Fox JW (1989) Amino acid sequence of a Crotalus atrox venom metalloproteinase which cleaves type IV collagen and gelatin. J Biol Chem 264:11575–11583
Hui M, Lajtha A, Hui K-S (1993) A new soluble brain-specific protein: identification and partial purification. Brain Res 606:36–43
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue in honor of Naren Banik.
Rights and permissions
About this article
Cite this article
Hui, KS. Brain-Specific Aminopeptidase: From Enkephalinase to Protector Against Neurodegeneration. Neurochem Res 32, 2062–2071 (2007). https://doi.org/10.1007/s11064-007-9356-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-007-9356-3