
Abstract
Negative symptoms in schizophrenia respond poorly to antipsychotics, but may improve when these are augmented with selective serotonin reuptake inhibitors (SSRIs). The molecular mechanisms underlying the augmentation are unclear. Nevertheless, significant progress has been made, pointing to some candidate systems which may be involved in SSRI–antipsychotic synergism. Thus, the enhanced dopamine release by SSRI–antipsychotic treatment is modulated by specific serotonergic receptors and by tyrosine hydroxylase. There are modifications in gamma-aminobutyric acid system via glutamate decarboxylase 67, protein kinase C beta and the receptor for activated C-kinase 1 (Rack1). Some studies indicate the input of transcription and neurotrophic factors as phospho-cyclic adenosine monophosphate response element-binding protein, Fos and fibroblast growth factor-2. Alterations in calcium signaling (neurogranin, regulator of G-protein signaling and Rack1) and in cytokine receptors for interleukin-8 and chemokine have also been reported. While as yet limited in scope, the evidence suggests definable molecular targets which may be implicated in drug development based on SSRI–antipsychotic synergistic actions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Augmentation strategies in psychiatric disorders
Psychiatric disorders, such as schizophrenia, major depression, bipolar illness and obsessive-compulsive disorders (OCD), are characterized by a wide range of behavioral, emotional and cognitive abnormalities (Andreasen et al. 1994; Berman et al. 1997; Dell’Osso et al. 2007). This complex symptomatology often necessitates the use of more than one drug and many combinations of psychotropics have been tried in attempts to improve the clinical response of patients. Over the years, it has become evident that some combinations do induce a favorable clinical response, not achieved by each of the drugs given alone (Fava 2001; Silver 2001). Currently, the choice of drug combinations is based on a trial and error paradigm guided by clinical response. Understanding the biological principles, by which the combined treatments act, would enable a more “rational” selection of drugs and provide insights into the pathological mechanisms of the disorders.
This review stems from our interest in augmentation strategies, in particular the combination of selective serotonin reuptake inhibitor (SSRI) with antipsychotic drug. Our focus is on the use of this combination for treatment of negative symptoms of schizophrenia, although it has also been found effective in other psychiatric conditions (Table 1). While there is considerable research into the mechanisms of action of individual antipsychotic and antidepressant drugs, few studies examined the mechanism of augmentation strategies, hence there is a lack of a theoretical framework to guide research in this field. Our aim is to review the evidence for the synergistic effects of SSRI–antipsychotic treatment at clinical, biochemical and molecular levels and identify molecular targets which may mediate them and underlie the clinical effects.
Selective serotonin reuptake inhibitor–antipsychotic therapy for negative symptoms in schizophrenia
Schizophrenia is a chronic, disabling brain disorder comprising symptoms which can be classified into two groups: ‘positive symptoms’ (hallucinations and delusions) and ‘negative symptoms’ (apathy, anhedonia, flat affect, avolition and social withdrawal). Negative symptoms in particular are a challenge to available treatments. Thus, typical neuroleptics are effective against positive but not negative symptoms and the success of the newer generation, “atypical” drugs, is limited (Burton 2006). Augmentation of antipsychotic drugs with SSRI antidepressants may be a useful strategy for targeting negative symptoms as shown in controlled studies on addition of fluvoxamine to typical antipsychotic drug (Rummel et al. 2006; Silver et al. 2000; Silver and Nassar 1992; Silver and Shmugliakov 1998) and in open studies and case reports on augmentation of atypical drugs (clozapine and olanzapine) (Chaichan 2004; Hiemke et al. 2002; Lammers et al. 1999; Silver et al. 1995; Szegedi et al. 1999).
The mechanism of SSRI augmentation is unknown. Several SSRI antidepressants inhibit cytochrome P450 (CYP) enzymes, mainly CYP1A2 and CYP2D6 isozymes, which are metabolizers of antipsychotic drugs (Brosen 1998; Spina and de Leon 2007; Sproule et al. 1997). Therefore, combination of SSRI antidepressant with antipsychotic drug results frequently in higher plasma levels of the latter (Weigmann et al. 2001; Wetzel et al. 1998), which could potentially underlie the superior efficiency of the augmentation treatments. However, the improvement in negative symptomatology in schizophrenic patients under SSRI–haloperidol therapy did not correlate with the increase of antipsychotic levels in plasma (Avenoso et al. 1997; Hiemke et al. 2002; Yasui-Furukori et al. 2004). In addition, raising antipsychotic drug levels does not increase the efficacy of the drugs or the range of the responsive symptoms and may have adverse effects (Baldessarini et al. 1988; de Oliveira et al. 1995; Volavka et al. 2000). Moreover, in most of the combined antidepressant–antipsychotic treatments, the doses employed are lower than their therapeutic levels in monotherapy (Table 2). Finally, several effective augmentation treatments for schizophrenia used SSRI compounds (Poyurovsky et al. 2003; Weigmann et al. 2001; Wetzel et al. 1998) or other drugs (Goff et al. 2001; Goforth and Carroll 2007; Stoll and Haura 2000; Tsai et al. 2006) that have no pharmacokinetic interaction with antipsychotics. Thus, pharmacokinetic interactions cannot explain the clinical therapeutic efficacy of SSRI augmentation strategies for negative symptoms in schizophrenia.
The contribution of SSRI drugs as augmentation agents for negative symptoms appears to be distinct from “nonspecific” antidepressant action. First, patients having prominent depressive symptoms were excluded from controlled clinical studies and the score of depression parameters for those individuals included in the study was low and did not change by the combined treatment. In addition, tricyclic antidepressants, which are equally effective antidepressants, do not improve negative symptoms in schizophrenic patients, though they can be useful when depression is associated with the disease (Siris 1993). Notably, SSRIs acting through serotonergic receptors, but not maprotiline which acts via noradrenergic receptors, are effective augmentative agents for negative symptoms (Silver and Shmugliakov 1998), suggesting the mechanism requires manipulation of the serotonergic system (Siris 1993). Paradoxically, clozapine and fluvoxamine–haloperidol combination produce a similar clinical effect despite their different pharmacology: clozapine, a “gold standard” in the treatment of negative symptoms in schizophrenia, is a serotonin (5HT) antagonist while the antidepressant fluvoxamine elevates 5HT receptor action. Furthermore, adding SSRI to clozapine may improve effectiveness (Silver et al. 1995, 1996). Thus, the mechanisms mediating the clinical effects are likely to be located downstream from the initial pharmacological effects of the drugs at the receptor/transporter levels.
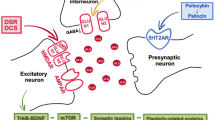
In this review, we will discuss evidence examining some of the potential targets relevant to the mechanism of augmentative treatments (Fig. 1).

Molecular targets potentially involved in the therapeutic synergistic effect of SSRI–antipsychotic treatment. The synergistic therapeutic effect of the combination of SSRI and antipsychotic drug is hypothesized to be reflected at the molecular level. The review discuses some targets that may be involved in the mechanism of action of this augmentation strategy
Candidate targets and mechanism of action of SSRI–antipsychotic combined treatment
Dopamine system
Cortical dopaminergic transmission
One of the most consistent animal findings on combined antipsychotic–SSRI treatments is the synergistic and selective increase in frontal cortex extracellular concentrations of dopamine (DA) compared to each individual drug (Ago et al. 2005; Denys et al. 2004b; Dremencov et al. 2007; Gobert et al. 1997; Huang et al. 2006a; Koch et al. 2004; Yoshino et al. 2004; Zhang et al. 2000).
Several lines of evidence support the therapeutic relevance of enhanced DA release. Reduced monoaminergic activity in the frontal cortex is associated with negative symptoms of schizophrenia (Heinz et al. 1998; Weinberger and Berman 1996). Atypical antipsychotics such as clozapine, olanzapine and amperozide, which may improve cognitive and negative symptoms, enhance DA efflux in the frontal cortex while typical drugs, such as haloperidol, ineffective against negative symptoms do not induce DA release (Advokat 2005; Devoto et al. 2003; Ichikawa et al. 2002; Kuroki et al. 1999; Li et al. 1998; Moghaddam and Bunney 1990; Nomikos et al. 1994; Westerink et al. 2001; Yamamoto and Cooperman 1994; Youngren et al. 1999; Zhang et al. 2000). Furthermore, adding the monoamine oxidase inhibitor B (MAOI-B), selegiline, to antipsychotic may improve negative symptoms in schizophrenia patients (Bodkin et al. 2005).
The mechanism underlying the increase in cortical DA following SSRI–antipsychotic treatment is not clear but it is generally accepted that the 5HT system plays an important modulatory role. The effect of SSRI augmentation on DA neurotransmission appears to involve selective changes in activation of 5HT receptors throughout the brain rather than in cortical 5HT output (Ago et al. 2005; Huang et al. 2006a). Activation of 5HT-1A receptors (Ago et al. 2005; Gobert et al. 1997; Millan et al. 2000; Rollema et al. 2000; Yoshino et al. 2004) and antagonism at 5HT-2 heteroreceptors, mainly 5HT-2A and 5HT-2C, have been implicated in enhanced cortical DA/NE release (Bonaccorso et al. 2002; Cremers et al. 2007; Dremencov et al. 2007; Huang et al. 2006a; Liegeois et al. 2002; Marek et al. 2005; Pehek et al. 2006; Szabo and Blier 2002; Westerink et al. 2001). The findings of down-regulation of 5HT-2A following combined antipsychotic–fluvoxamine treatment in the peripheral mononuclear cells (PMC) of schizophrenia patients, parallel to the improvement in negative symptoms (Chertkow et al. 2007b) and the reduced expression in rat brain after administration of the atypical antipsychotic olanzapine (Huang et al. 2006b), support involvement of this receptor subtype in the mechanism of action of drugs enhancing cortical DA. Notably, 5HT acting on the widespread 5HT-2C receptors exerts a tonic inhibitory influence on DAergic neurotransmission, whereas it stimulates DAergic release, under facilitated conditions, through 5HT-2A receptors, located mainly in the cortex (Landen and Thase 2006).
An indirect serotonergic effect on cortical neurotransmission can occur via 5HT-2 receptors located in gamma-aminobutyric acid (GABA) and glutamate pathways, originating at the frontal cortex and projecting to the locus coeruleus (LC) and ventral tegmental area (VTA). These neuronal tracks, in turn, modulate both 5HT cell bodies at the LC or at the VTA and mesocortical DA neurons (Alex and Pehek 2007; Millan et al. 2000; Pehek et al. 2006), thus forming feedback/compensatory long-loop circuitry. Indeed, it was proposed that combined antipsychotic–antidepressant treatments potentiate DA/NE release in the frontal cortex through non-cortical serotonergic action (Ago et al. 2005; Seager et al. 2004, 2005).
Dopamine turnover and tyrosine hydroxylase (TH) modulation
Dopamine turnover is extensively altered by antipsychotic treatment. Combined haloperidol and fluvoxamine treatment, but not the individual drugs, reduced DA turnover in rat cerebellum and increased DA metabolites and TH protein level in the striatum (Chertkow et al. 2007a). Similarly, olanzapine combined with fluoxetine induced TH in rat LC (Ordway and Szebeni 2004) compared to the individual compounds. TH catalyzes the rate-limiting step in DA synthesis and may thereby affect the function of dopaminergic circuits, including DA neural firing (Melia et al. 1992; Mereu et al. 1983). Typical and atypical antipsychotic drugs show different, region specific effects on TH activity. Thus, clozapine and risperidone increased TH immunoreactivity in both medial prefrontal cortex (mPFC) and LC, while haloperidol caused a smaller increase in TH protein expression in the LC, and did not alter its levels in the mPFC (Verma et al. 2007).
Gamma-aminobutyric acid–glutamate systems
Postmortem studies demonstrated loss of specific GABAergic inhibitory neurons (Reynolds et al. 2001), reduction in the GABA synthesizing enzyme, glutamate decarboxylase 67 (GAD67) (Blum and Mann 2002; Hashimoto et al. 2008; Kalkman and Loetscher 2003; Volk et al. 2000) and compensatory upregulation of GABA-A receptor (Benes et al. 1996; Deng and Huang 2006) in brains of schizophrenia patients. In addition, hypofunction of the ionotropic glutamate N-methyl-d-aspartate (NMDA) receptor (Javitt 2006; Shim et al. 2008) may play an important role in the pathophysiology and treatment of negative symptoms and lead to inhibition of GABAergic interneurons (Konradi and Heckers 2003).
We recently reported that SSRI augmentation of antipsychotic decreased components involved in the modulation of GABA-A receptor activity, including GAD67 protein and protein kinase C (PKC) beta in the rat frontal cortex (Chertkow et al. 2006). In a pilot clinical study, we found reduced expression of receptor for activated C-kinase 1 (Rack1), involved in GABA-A receptor phosphorylation, in PMC of schizophrenia patients undergoing SSRI augmentation treatment (Chertkow et al. 2007b).
Antipsychotic drugs can modify GABA and glutamate system elements including GABA-A receptor, extracellular GABA levels, GAD67 expression, glutamate transporters (Schmitt et al. 2003), neuregulin-1 (which regulates the expression of NMDA and GABA-A alpha receptors) (Zhang et al. 2007) as well as ionotropic and metabotropic glutamate receptors (Fumagalli et al. 2008; Tarazi et al. 2003). Addition of SSRI to antipsychotic modifies these effects (Chertkow et al. 2006, 2007b). Interestingly, atypical antipsychotics differ from typical ones in the effect on GABA system including hippocampal and cortical GABA-A receptor density (Zink et al. 2004b), GABA transporter expression (Zink et al. 2004a) and GAD67 levels (Zink et al. 2004b). Possible mechanisms mediating drug-induced modification of GABA system include inhibition of DA innervations on the GABA and glutamate neurons, activation of PKC anchored to Rack1 (Feng et al. 2001), and increase in brain levels of allopregnanolone (Allo), a potent positive allosteric modulator of GABA at GABA-A receptor (Pinna et al. 2006).
There is clinical evidence linking GABA/glutamate agents with negative and cognitive symptoms in schizophrenia. Silver et al (2005) have demonstrated correlation between dehydroepiandrosterone (DHEAS) level and cognitive function in schizophrenia patients (Silver et al. 2005). Augmentation with DHEA (Strous 2005) or agonists at glycine site (Tsai et al. 2006; Heresco-Levy et al. 1999) has been reported to improve negative symptoms in schizophrenic patients maintained on antipsychotic drugs, excluding clozapine (Lane et al. 2006), which has glutamatergic activity (Advokat 2005; Spurney et al. 1999; Evins et al. 1997). These data suggest that clinical effectiveness may require specific and fine-tuning adjustments of glutamate system.
Transcription and neurotrophic factors in SSRI–antipsychotic combined treatment
Transcription and neurotrophic factors have multiple effects on intracellular signaling pathways and they are key factors in the modulation of neuronal plasticity and synaptic activity (Colombo 2004; Nikitin 2007; Tischmeyer and Grimm 1999; Murer et al. 1999; Wetmore et al. 1990; Linnarsson et al. 2000; Pezet et al. 2002; Pillai 2008; Roberts et al. 2006; Spedding and Gressens 2008; Dono 2003; Otto and Unsicker 1990). Preliminary data suggest that they may have a role in the mechanism of action of the combined treatment.
Subchronic (7 days) fluoxetine–olanzapine treatment suppressed the induction of phospho-cyclic adenosine monophosphate response element-binding protein (pCREB) and the transcription factor Fos in rat frontal cortex and hippocampus (Horowitz et al. 2003), while a single injection had no effect in the hippocampus and striatum (Maragnoli et al. 2004). Studies in brains of naive rats reported modified levels of Fos alone in its complex with Jun (AP-1 complex) following chronic antipsychotic treatment (Cochran et al. 2002; Cohen and Wan 1996; Kontkanen et al. 2002). Moreover, it was suggested that typical and atypical drugs have differential effects on Fos expression (Deutch and Duman 1996; Semba et al. 1999; Wan et al. 1995; Werme et al. 2000) and CREB phosphorylation (Pozzi et al. 2003), which may lead to different neural activity and therapeutic outcome. In addition, postmortem studies in schizophrenia patients reported changes in pCREB (Kyosseva et al. 2000) and Fos (Kyosseva 2004) levels in the thalamus and cerebellar vermis.
The neurotrophic factors, brain-derived neurotrophic factor (BDNF) and fibroblast growth factor (FGF-2) have been implicated in the pathophysiology and the treatment response of schizophrenia (Buckley et al. 2007; Fumagalli et al. 2004; Huang and Lee 2006; Lu and Martinowich 2008; Pillai 2008; Tan et al. 2005). Chronic administration of quetiapine combined with venlafaxine in rats prevented the decrease in hippocampal cell proliferation and BDNF expression induced by chronic restrain stress in a synergistic and dose-dependent manner (Xu et al. 2006). Repeated treatment with the fluoxetine–olanzapine combination induced FGF-2 levels in the striatum and the hippocampus, in addition to the prefrontal cortex in which FGF-2 was upregulated after a single dose only (Maragnoli et al. 2004). In addition, there are indications for distinct effects of typical and atypical antipsychotic drugs on BDNF and FGF-2 (Balu et al. 2008; Chlan-Fourney et al. 2002; Parikh et al. 2004; Pillai et al. 2006; Riva et al. 1999). Future work needs to clarify the significance of these alterations and whether they contribute to the specific effects of the combined drug action or represent a general neuroleptic action.
Calcium signaling pathway
It has been suggested that dysfunction of calcium (Ca) signaling may explain many of the morphological and functional alterations observed in schizophrenia (Lidow 2003). Binding of glutamate to the NMDA receptor, implicated in schizophrenia (Tsai and Coyle 2002), causes an influx of Ca, which triggers the phosphorylation of the postsynaptic brain-specific protein neurogranin by PKC (Rodriguez-Sanchez et al. 1997) and the release of calmodulin from neurogranin (Chakravarthy et al. 1999). The “free” calmodulin complex with Ca and activates Ca2+/calmodulin-dependent kinase II (CaMKII) that plays an important role in synaptic plasticity, learning and memory (Huang et al. 2004; Pak et al. 2000).
Several components of the Ca signaling system have been reported to be abnormal in schizophrenia. Recent studies indicted that neurogranin gene is associated with schizophrenia (Ruano et al. 2008) and that neurogranin and calmodulin protein expression are altered in the prefrontal cortex of schizophrenic patients (Broadbelt and Jones 2008). Loss of neurogranin may lead to changes in long-term potentiation and spatial learning (Pak et al. 2000). There is also evidence that CaMKII is altered in schizophrenia (Novak et al. 2006) and following haloperidol treatment (Fumagalli et al. 2008). Regulators of G-protein signaling (RGS) proteins, RGS4 and RGS2, modulate NMDA receptor activity through serotonergic and noradrenergic receptors (Gu et al. 2007; Liu et al. 2006) and thus affect intracellular Ca levels. Several studies indicate that RGS4, RGS2, RGS5, RGS9 and RGS10 are involved in the pathophysiology of schizophrenia and its drug treatments (Campbell et al. 2008; Erdely et al. 2006; Fatemi et al. 2006; Hishimoto et al. 2004; Mirnics et al. 2001; Seeman et al. 2007). PKCβII is involved in calcium signaling through its interaction with the intracellular scaffold protein RACK1 (Patterson et al. 2004). Interestingly, we have found changes in PKCβII levels in PFC of naive rats following chronic fluvoxamine–haloperidol treatment (Chertkow et al. 2006). There is preliminary clinical evidence that combined SSRI–antipsychotic treatment may have selective effects on components of the calcium cascade (neurogranin, RGS family and Rack1) in PMC from schizophrenia patients (Chertkow et al. 2007b).
Cytokines
Growing evidence demonstrates that the nervous system interacts with the brain and peripheral immune and endocrine systems through neurotransmitters, hormones and cytokines (Kronfol and Remick 2000). In line with this notion, data from the recent years indicated that schizophrenia pathology might involve impairments in cytokine profile, including balance in helper T cells (Th1/Th2), and in the levels of interleukin (IL)-2, IL-6, IL-8 and IL-10 (Potvin et al. 2008; Schwartz and Silver 2000; Zhang et al. 2005). Specifically, it was suggested that elevated cortisol levels are associated with negative symptoms and high IL-2 concentration with positive symptoms (Zhang et al. 2005). The effects of antipsychotic treatment on cytokine network have been previously examined and reviewed (Drzyzga et al. 2006; Pollmacher et al. 1996; Zhang et al. 2005). The most robust data focus on tumor necrosis factor α (TNF-α), IL-2, IL-10 and IL-6 (Drzyzga et al. 2006), but it is as yet unclear whether a characteristic cytokine profile is associated with therapeutic response (Drzyzga et al. 2006).
Clinical support for involvement of cytokines in mechanism of SSRI augmentation comes from a study showing time-dependent changes in the expression levels of IL-8 receptor and chemokine (C–C motif) receptor 1 (CCR1) in PMC of schizophrenia patients following addition of fluvoxamine to ongoing antipsychotic treatment (Chertkow et al. 2007b). Importantly, the molecular changes paralleled reduction in negative symptoms (Chertkow et al. 2007b). IL-8, essential for the directional migration of leukocytes, is increased in the serum of unmedicated chronic schizophrenia patients (Erbagci et al. 2001; Maes et al. 2002; Zhang et al. 2002). Thus, it is plausible that augmentation therapy of SSRI and antipsychotic drug acts to equilibrate the pathological increase in IL-8 receptor concentration.
Summary and future perspective
Negative symptoms in schizophrenia, like other “treatment resistant” symptoms of psychiatric illnesses, continue to be a therapeutic challenge. New molecular targets are needed to develop novel and more effective pharmacotherapeutic compounds. Given the complexity of the symptoms, it is unlikely that “single-action” drugs, given alone, will be effective and a multifunctional approach, as in augmentation treatment, may be needed. The studies reviewed here encourage the view that combined SSRI–antipsychotic treatment results in unique molecular changes in the brain which differ from those of the individual drugs (Fig. 1). We have highlighted some plausible molecular meeting points for actions of pharmacologically distinct treatments which may underlie their convergent clinical effects. These are localized anatomically, and involve systems at various cellular levels, downstream from the initial impact of the drugs on membrane receptors or transporters (Fig. 2). They modify widespread pathways and inter-cellular processes such as, rate of neurotransmission, connectivity, plasticity and proliferation.

A schematic illustration of some signaling pathways potentially involve in the synergistic therapeutic effect of SSRI–antipsychotic combination. The diagram depict the connection between some of the molecular targets discuss in the text, which may underlie the synergistic therapeutic effect of SSRI–antipsychotic combination. Intracellular signaling cascades and neural action are modified through G-protein coupled receptors activated or inhibited by natural ligands (5HT, IL-8, etc.) or medications. Enhanced cortical DA/NE release following SSRI–antipsychotic combination involves the action on 5HT-1A and 5HT-2 receptors and the input of tyrosine hydroxylase (TH), catalyzing DA synthesis. Receptor activity is also modulated intracellularly by RGS family proteins. Conductance of a Cl− through GABA-A receptor channel is regulated by PKCβ acting on β3 subunit and by the natural ligand GABA, whose level is modified by GAD67. GABA-A activity is also modulated by negative coupling (for example with D2-family receptor antagonists) with subtypes of Gi and/or Go, and the consequent un-inhibition of adenylyl cyclase (AC) and phospholipase C (PLC). Higher levels of cAMP enhance cAMP-dependent protein kinase activity (PKA) and GABA-A phosphorylation. In addition, activation of the cAMP pathway increases catecholamine synthesis via increases phosphorylation of TH. The relation of Rack1 and neurogranin, which are discussed in the text, is also depicted in this diagram. PKC protein kinase C, PKA protein kinase A, PIP 2 phosphatidylinositol-4,5-bisphosphate, DAG diacylglycerol, IP 3 inositol trisphosphate, 5HT serotonin, RGS regulator of G-proteins, GAD glutamate decarboxylase
In current psychiatric practice, a decision to add a second drug is usually made after monotherapy fails to achieve a satisfactory response (Ostroff and Nelson 1999; Rutherford et al. 2007; Silver et al. 1996; Tani et al. 2004; Zink 2005). This imposes both an interval between drug administration and a treatment-specific intake order (i.e. in schizophrenia antipsychotics are given first, while in depression and OCD, SSRIs initiate the treatment). It is unclear if and how the interval between the onsets of administration of the two drugs and their order influences response. For example, in depression co-administration of risperidone and fluvoxamine, from the beginning of therapy is efficacious (Hirose and Ashby 2002), while in treatment of negative symptoms augmentation typically begins several weeks into antipsychotic treatment. These parameters have implications for both the understanding of the mechanism of combined antipsychotic–antidepressant therapy and for deciding on the appropriate treatment regimen.
Clearly, the available data are limited, and much research is needed to identify and validate the molecular targets of SSRI–antipsychotic and other augmentation treatments. Conceptually, the method of isolating molecular effects of pharmacologically distinct but clinically convergent drugs provides a useful “window” for understanding of the mechanisms of multifunctional treatments and development of new more effective drugs for the treatment of schizophrenia.
References
Adson DE, Kushner MG, Eiben KM, Schulz SC (2004) Preliminary experience with adjunctive quetiapine in patients receiving selective serotonin reuptake inhibitors. Depress Anxiety 19:121–126
Advokat C (2005) Differential effects of clozapine versus other antipsychotics on clinical outcome and dopamine release in the brain. Essent Psychopharmacol 6:73–90
Ago Y, Nakamura S, Baba A, Matsuda T (2005) Sulpiride in combination with fluvoxamine increases in vivo dopamine release selectively in rat prefrontal cortex. Neuropsychopharmacology 30:43–51
Alex KD, Pehek EA (2007) Pharmacologic mechanisms of serotonergic regulation of dopamine neurotransmission. Pharmacol Ther 113:296–320
Amsterdam JD, Shults J (2005) Comparison of fluoxetine, olanzapine, and combined fluoxetine plus olanzapine initial therapy of bipolar type I and type II major depression—lack of manic induction. J Affect Disord 87:121–130
Andreasen NC, Nopoulos P, Schultz S, Miller D, Gupta S, Swayze V, Flaum M (1994) Positive and negative symptoms of schizophrenia: past, present, and future. Acta Psychiatr Scand Suppl 384:51–59
Avenoso A, Spina E, Campo G, Facciola G, Ferlito M, Zuccaro P, Perucca E, Caputi AP (1997) Interaction between fluoxetine and haloperidol: pharmacokinetic and clinical implications. Pharmacol Res 35:335–339
Baldessarini RJ, Cohen BM, Teicher MH (1988) Significance of neuroleptic dose and plasma level in the pharmacological treatment of psychoses. Arch Gen Psychiatry 45:79–91
Balu DT, Hoshaw BA, Malberg JE, Rosenzweig-Lipson S, Schechter LE, Lucki I (2008) Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res 1211:37–43
Barbee JG, Conrad EJ, Jamhour NJ (2004) The effectiveness of olanzapine, risperidone, quetiapine, and ziprasidone as augmentation agents in treatment-resistant major depressive disorder. J Clin Psychiatry 65:975–981
Benes FM, Vincent SL, Marie A, Khan Y (1996) Up-regulation of GABAA receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience 75:1021–1031
Bergemann N, Frick A, Parzer P, Kopitz J (2004) Olanzapine plasma concentration, average daily dose, and interaction with co-medication in schizophrenic patients. Pharmacopsychiatry 37:63–68
Berman RM, Narasimhan M, Charney DS (1997) Treatment-refractory depression: definitions and characteristics. Depress Anxiety 5:154–164
Bloch MH, Landeros-Weisenberger A, Kelmendi B, Coric V, Bracken MB, Leckman JF (2006) A systematic review: antipsychotic augmentation with treatment refractory obsessive-compulsive disorder. Mol Psychiatry 11:622–632
Blum BP, Mann JJ (2002) The GABAergic system in schizophrenia. Int J Neuropsychopharmacol 5:159–179
Bodkin JA, Siris SG, Bermanzohn PC, Hennen J, Cole JO (2005) Double-blind, placebo-controlled, multicenter trial of selegiline augmentation of antipsychotic medication to treat negative symptoms in outpatients with schizophrenia. Am J Psychiatry 162:388–390
Bogan AM, Koran LM, Chuong HW, Vapnik T, Bystritsky A (2005) Quetiapine augmentation in obsessive-compulsive disorder resistant to serotonin reuptake inhibitors: an open-label study. J Clin Psychiatry 66:73–79
Bonaccorso S, Meltzer HY, Li Z, Dai J, Alboszta AR, Ichikawa J (2002) SR46349-B, a 5-HT(2A/2C) receptor antagonist, potentiates haloperidol-induced dopamine release in rat medial prefrontal cortex and nucleus accumbens. Neuropsychopharmacology 27:430–441
Broadbelt K, Jones LB (2008) Evidence of altered calmodulin immunoreactivity in areas 9 and 32 of schizophrenic prefrontal cortex. J Psychiatr Res 42:612–621
Brosen K (1998) Differences in interactions of SSRIs. Int Clin Psychopharmacol 13(Suppl 5):S45–S47
Brown EB, McElroy SL, Keck PE Jr, Deldar A, Adams DH, Tohen M, Williamson DJ (2006) A 7-week, randomized, double-blind trial of olanzapine/fluoxetine combination versus lamotrigine in the treatment of bipolar I depression. J Clin Psychiatry 67:1025–1033
Buckley PF, Mahadik S, Pillai A, Terry A Jr (2007) Neurotrophins and schizophrenia. Schizophr Res 94:1–11
Burton S (2006) Symptom domains of schizophrenia: the role of atypical antipsychotic agents. J Psychopharmacol 20:6–19
Bystritsky A, Ackerman DL, Rosen RM, Vapnik T, Gorbis E, Maidment KM, Saxena S (2004) Augmentation of serotonin reuptake inhibitors in refractory obsessive-compulsive disorder using adjunctive olanzapine: a placebo-controlled trial. J Clin Psychiatry 65:565–568
Campbell DB, Lange LA, Skelly T, Lieberman J, Levitt P, Sullivan PF (2008) Association of RGS2 and RGS5 variants with schizophrenia symptom severity. Schizophr Res 101:67–75
Carey PD, Vythilingum B, Seedat S, Muller JE, van Ameringen M, Stein DJ (2005) Quetiapine augmentation of SRIs in treatment refractory obsessive-compulsive disorder: a double-blind, randomised, placebo-controlled study [ISRCTN83050762]. BMC Psychiatry 5:5
Chaichan W (2004) Olanzapine plus fluvoxamine and olanzapine alone for the treatment of an acute exacerbation of schizophrenia. Psychiatry Clin Neurosci 58:364–368
Chakravarthy B, Morley P, Whitfield J (1999) Ca2+–calmodulin and protein kinase Cs: a hypothetical synthesis of their conflicting convergences on shared substrate domains. Trends Neurosci 22:12–16
Chertkow Y, Weinreb O, Youdim MB, Silver H (2006) The effect of chronic co-administration of fluvoxamine and haloperidol compared to clozapine on the GABA system in the rat frontal cortex. Int J Neuropsychopharmacol 9:287–296
Chertkow Y, Weinreb O, Youdim MB, Silver H (2007a) Dopamine and serotonin metabolism in response to chronic administration of fluvoxamine and haloperidol combined treatment. J Neural Transm 114(11):1443–1454
Chertkow Y, Weinreb O, Youdim MB, Silver H (2007b) Gene expression changes in peripheral mononuclear cells from schizophrenic patients treated with a combination of antipsychotic with fluvoxamine. Prog Neuropsychopharmacol Biol Psychiatry 31:1356–1362
Chiu CC, Lane HY, Huang MC, Liu HC, Jann MW, Hon YY, Chang WH, Lu ML (2004) Dose-dependent alternations in the pharmacokinetics of olanzapine during coadministration of fluvoxamine in patients with schizophrenia. J Clin Pharmacol 44:1385–1390
Chlan-Fourney J, Ashe P, Nylen K, Juorio AV, Li XM (2002) Differential regulation of hippocampal BDNF mRNA by typical and atypical antipsychotic administration. Brain Res 954:11–20
Citrome L, Jaffe A, Levine J, Lindenmayer JP (2005) Dosing of quetiapine in schizophrenia: how clinical practice differs from registration studies. J Clin Psychiatry 66:1512–1516
Cochran SM, McKerchar CE, Morris BJ, Pratt JA (2002) Induction of differential patterns of local cerebral glucose metabolism and immediate-early genes by acute clozapine and haloperidol. Neuropharmacology 43:394–407
Cohen BM, Wan W (1996) The thalamus as a site of action of antipsychotic drugs. Am J Psychiatry 153:104–106
Colombo PJ (2004) Learning-induced activation of transcription factors among multiple memory systems. Neurobiol Learn Mem 82:268–277
Corey-Lisle PK, Birnbaum H, Greenberg P, Marynchenko M, Dube S (2003) Economic impact of olanzapine plus fluoxetine combination therapy among patients treated for depression: a pilot study. Psychopharmacol Bull 37:90–98
Corya SA, Williamson D, Sanger TM, Briggs SD, Case M, Tollefson G (2006) A randomized, double-blind comparison of olanzapine/fluoxetine combination, olanzapine, fluoxetine, and venlafaxine in treatment-resistant depression. Depress Anxiety 23:364–372
Cremers TI, Rea K, Bosker FJ, Wikstrom HV, Hogg S, Mork A, Westerink BH (2007) Augmentation of SSRI effects on serotonin by 5-HT2C antagonists: mechanistic studies. Neuropsychopharmacology 32:1550–1557
D’Amico G, Cedro C, Muscatello MR, Pandolfo G, Di Rosa AE, Zoccali R, La Torre D, D’Arrigo C, Spina E (2003) Olanzapine augmentation of paroxetine-refractory obsessive-compulsive disorder. Prog Neuropsychopharmacol Biol Psychiatry 27:619–623
de Oliveira IR, Dardennes RM, Amorim ES, Diquet B, de Sena EP, Moreira EC, de Castro-e-Silva EJ, Payan C, Fermanian J, Marcilio C et al (1995) Is there a relationship between antipsychotic blood levels and their clinical efficacy? An analysis of studies design and methodology. Fundam Clin Pharmacol 9:488–502
Dell’Osso B, Altamura AC, Mundo E, Marazziti D, Hollander E (2007) Diagnosis and treatment of obsessive-compulsive disorder and related disorders. Int J Clin Pract 61:98–104
Deng C, Huang XF (2006) Increased density of GABAA receptors in the superior temporal gyrus in schizophrenia. Exp Brain Res 168:587–590
Denys D, de Geus F, van Megen HJ, Westenberg HG (2004a) A double-blind, randomized, placebo-controlled trial of quetiapine addition in patients with obsessive-compulsive disorder refractory to serotonin reuptake inhibitors. J Clin Psychiatry 65:1040–1048
Denys D, Klompmakers AA, Westenberg HG (2004b) Synergistic dopamine increase in the rat prefrontal cortex with the combination of quetiapine and fluvoxamine. Psychopharmacology (Berl) 176:195–203
Deutch AY, Duman RS (1996) The effects of antipsychotic drugs on Fos protein expression in the prefrontal cortex: cellular localization and pharmacological characterization. Neuroscience 70:377–389
Devoto P, Flore G, Vacca G, Pira L, Arca A, Casu MA, Pani L, Gessa GL (2003) Co-release of noradrenaline and dopamine from noradrenergic neurons in the cerebral cortex induced by clozapine, the prototype atypical antipsychotic. Psychopharmacology (Berl) 167:79–84
Dono R (2003) Fibroblast growth factors as regulators of central nervous system development and function. Am J Physiol Regul Integr Comp Physiol 284:R867–R881
Doree JP, Des Rosiers J, Lew V, Gendron A, Elie R, Stip E, Tourjman SV (2007) Quetiapine augmentation of treatment-resistant depression: a comparison with lithium. Curr Med Res Opin 23:333–341
Dremencov E, El Mansari M, Blier P (2007) Noradrenergic augmentation of escitalopram response by risperidone: electrophysiologic studies in the rat brain. Biol Psychiatry 61:671–678
Drzyzga L, Obuchowicz E, Marcinowska A, Herman ZS (2006) Cytokines in schizophrenia and the effects of antipsychotic drugs. Brain Behav Immun 20:532–545
Dube S, Tollefson GD, Thase ME, Briggs SD, Van Campen LE, Case M, Tohen M (2007) Onset of antidepressant effect of olanzapine and olanzapine/fluoxetine combination in bipolar depression. Bipolar Disord 9:618–627
Erbagci AB, Herken H, Koyluoglu O, Yilmaz N, Tarakcioglu M (2001) Serum IL-1beta, sIL-2R, IL-6, IL-8 and TNF-alpha in schizophrenic patients, relation with symptomatology and responsiveness to risperidone treatment. Mediators Inflamm 10:109–115
Erdely HA, Tamminga CA, Roberts RC, Vogel MW (2006) Regional alterations in RGS4 protein in schizophrenia. Synapse 59:472–479
Erzegovesi S, Guglielmo E, Siliprandi F, Bellodi L (2005) Low-dose risperidone augmentation of fluvoxamine treatment in obsessive-compulsive disorder: a double-blind, placebo-controlled study. Eur Neuropsychopharmacol 15:69–74
Evins AE, Amico ET, Shih V, Goff DC (1997) Clozapine treatment increases serum glutamate and aspartate compared to conventional neuroleptics. J Neural Transm 104(6–7):761–766
Fatemi SH, Reutiman TJ, Folsom TD, Bell C, Nos L, Fried P, Pearce DA, Singh S, Siderovski DP, Willard FS, Fukuda M (2006) Chronic olanzapine treatment causes differential expression of genes in frontal cortex of rats as revealed by DNA microarray technique. Neuropsychopharmacology 31:1888–1899
Fava M (2001) Augmentation and combination strategies in treatment-resistant depression. J Clin Psychiatry 62(Suppl 18):4–11
Feng J, Cai X, Zhao J, Yan Z (2001) Serotonin receptors modulate GABA(A) receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. J Neurosci 21:6502–6511
Francobandiera G (2001) Olanzapine augmentation of serotonin uptake inhibitors in obsessive-compulsive disorder: an open study. Can J Psychiatry 46:356–358
Fumagalli F, Molteni R, Bedogni F, Gennarelli M, Perez J, Racagni G, Riva MA (2004) Quetiapine regulates FGF-2 and BDNF expression in the hippocampus of animals treated with MK-801. Neuroreport 15:2109–2112
Fumagalli F, Frasca A, Racagni G, Riva MA (2008) Dynamic regulation of glutamatergic postsynaptic activity in rat prefrontal cortex by repeated administration of antipsychotic drugs. Mol Pharmacol 73:1484–1490
Gobert A, Rivet JM, Cistarelli JM, Millan MJ (1997) Buspirone enhances duloxetine- and fluoxetine-induced increases in dialysate levels of dopamine and noradrenaline, but not serotonin, in the frontal cortex of freely moving rats. J Neurochem 68:1326–1329
Goff DC, Freudenreich O, Evins AE (2001) Augmentation strategies in the treatment of schizophrenia. CNS Spectr 6:904, 907–911
Goforth HW, Carroll BT (2007) Aripiprazole augmentation of tranylcypromine in treatment-resistant major depression. J Clin Psychopharmacol 27:216–217
Gu Z, Jiang Q, Yan Z (2007) RGS4 modulates serotonin signaling in prefrontal cortex and links to serotonin dysfunction in a rat model of schizophrenia. Mol Pharmacol 71:1030–1039
Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA (2008) Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry 165:479–489
Heinz A, Knable MB, Coppola R, Gorey JG, Jones DW, Lee KS, Weinberger DR (1998) Psychomotor slowing, negative symptoms and dopamine receptor availability—an IBZM SPECT study in neuroleptic-treated and drug-free schizophrenic patients. Schizophr Res 31:19–26
Hellerstein DJ (2004) Aripiprazole as an adjunctive treatment for refractory major depression. Prog Neuropsychopharmacol Biol Psychiatry 28:1347–1348
Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Silipo G, Lichtenstein M (1999) Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psychiatry 56:29–36
Hiemke C, Peled A, Jabarin M, Hadjez J, Weigmann H, Hartter S, Modai I, Ritsner M, Silver H (2002) Fluvoxamine augmentation of olanzapine in chronic schizophrenia: pharmacokinetic interactions and clinical effects. J Clin Psychopharmacol 22:502–506
Hirose S, Ashby CR Jr (2002) An open pilot study combining risperidone and a selective serotonin reuptake inhibitor as initial antidepressant therapy. J Clin Psychiatry 63:733–736
Hishimoto A, Shirakawa O, Nishiguchi N, Aoyama S, Ono H, Hashimoto T, Maeda K (2004) Novel missense polymorphism in the regulator of G-protein signaling 10 gene: analysis of association with schizophrenia. Psychiatry Clin Neurosci 58:579–581
Hollander E, Baldini Rossi N, Sood E, Pallanti S (2003) Risperidone augmentation in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. Int J Neuropsychopharmacol 6:397–401
Horowitz JM, Goyal A, Ramdeen N, Hallas BH, Horowitz AT, Torres G (2003) Characterization of fluoxetine plus olanzapine treatment in rats: a behavior, endocrine, and immediate-early gene expression analysis. Synapse 50:353–364
Huang TL, Lee CT (2006) Associations between serum brain-derived neurotrophic factor levels and clinical phenotypes in schizophrenia patients. J Psychiatr Res 40:664–668
Huang KP, Huang FL, Jager T, Li J, Reymann KG, Balschun D (2004) Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci 24:10660–10669
Huang M, Ichiwaka J, Li Z, Dai J, Meltzer HY (2006a) Augmentation by citalopram of risperidone-induced monoamine release in rat prefrontal cortex. Psychopharmacology (Berl) 185:274–281
Huang XF, Han M, Huang X, Zavitsanou K, Deng C (2006b) Olanzapine differentially affects 5-HT2A and 2C receptor mRNA expression in the rat brain. Behav Brain Res 171:355–362
Ichikawa J, Li Z, Dai J, Meltzer HY (2002) Atypical antipsychotic drugs, quetiapine, iloperidone, and melperone, preferentially increase dopamine and acetylcholine release in rat medial prefrontal cortex: role of 5-HT1A receptor agonism. Brain Res 956:349–357
Javitt DC (2006) Is the glycine site half saturated or half unsaturated? Effects of glutamatergic drugs in schizophrenia patients. Curr Opin Psychiatry 19:151–157
Kalkman HO, Loetscher E (2003) GAD(67): the link between the GABA-deficit hypothesis and the dopaminergic- and glutamatergic theories of psychosis. J Neural Transm 110:803–812
Kane JM, Khanna S, Rajadhyaksha S, Giller E (2006) Efficacy and tolerability of ziprasidone in patients with treatment-resistant schizophrenia. Int Clin Psychopharmacol 21:21–28
Keck PE Jr, Corya SA, Altshuler LL, Ketter TA, McElroy SL, Case M, Briggs SD, Tohen M (2005) Analyses of treatment-emergent mania with olanzapine/fluoxetine combination in the treatment of bipolar depression. J Clin Psychiatry 66:611–616
Kemp DE, Dago PL, Straus JL, Fleck J, Karaffa M, Gilmer WS (2007) Aripiprazole augmentation for treatment-resistant bipolar depression: sustained remission after 36 months. J Clin Psychopharmacol 27:304–305
Ketter TA, Wang PW, Chandler RA, Culver JL, Alarcon AM (2006) Adjunctive aripiprazole in treatment-resistant bipolar depression. Ann Clin Psychiatry 18:169–172
Koch S, Perry KW, Bymaster FP (2004) Brain region and dose effects of an olanzapine/fluoxetine combination on extracellular monoamine concentrations in the rat. Neuropharmacology 46:232–242
Konradi C, Heckers S (2003) Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther 97(2):153–179
Kontkanen O, Lakso M, Wong G, Castren E (2002) Chronic antipsychotic drug treatment induces long-lasting expression of fos and jun family genes and activator protein 1 complex in the rat prefrontal cortex. Neuropsychopharmacology 27:152–162
Kronfol Z, Remick DG (2000) Cytokines and the brain: implications for clinical psychiatry. Am J Psychiatry 157:683–694
Kuroki T, Meltzer HY, Ichikawa J (1999) Effects of antipsychotic drugs on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens. J Pharmacol Exp Ther 288:774–781
Kyosseva SV (2004) Differential expression of mitogen-activated protein kinases and immediate early genes fos and jun in thalamus in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 28:997–1006
Kyosseva SV, Elbein AD, Hutton TL, Griffin ST, Mrak RE, Sturner WQ, Karson CN (2000) Increased levels of transcription factors Elk-1, cyclic adenosine monophosphate response element-binding protein, and activating transcription factor 2 in the cerebellar vermis of schizophrenic patients. Arch Gen Psychiatry 57:685–691
Lammers CH, Deuschle M, Weigmann H, Hartter S, Hiemke C, Heese C, Heuser I (1999) Coadministration of clozapine and fluvoxamine in psychotic patients—clinical experience. Pharmacopsychiatry 32:76–77
Landen M, Thase ME (2006) A model to explain the therapeutic effects of serotonin reuptake inhibitors: the role of 5-HT2 receptors. Psychopharmacol Bull 39:147–166
Lane HY, Huang CL, Wu PL, Liu YC, Chang YC, Lin PY, Chen PW, Tsai G (2006) Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to clozapine for the treatment of schizophrenia. Biol Psychiatry 60:645–649
Li XM, Perry KW, Wong DT, Bymaster FP (1998) Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology (Berl) 136:153–161
Lidow MS (2003) Calcium signaling dysfunction in schizophrenia: a unifying approach. Brain Res Brain Res Rev 43:70–84
Liegeois JF, Ichikawa J, Meltzer HY (2002) 5-HT(2A) receptor antagonism potentiates haloperidol-induced dopamine release in rat medial prefrontal cortex and inhibits that in the nucleus accumbens in a dose-dependent manner. Brain Res 947:157–165
Linnarsson S, Willson CA, Ernfors P (2000) Cell death in regenerating populations of neurons in BDNF mutant mice. Brain Res Mol Brain Res 75:61–69
Liu W, Yuen EY, Allen PB, Feng J, Greengard P, Yan Z (2006) Adrenergic modulation of NMDA receptors in prefrontal cortex is differentially regulated by RGS proteins and spinophilin. Proc Natl Acad Sci USA 103:18338–18343
Lu B, Martinowich K (2008) Cell biology of BDNF and its relevance to schizophrenia. Novartis Found Symp 289:119–129, discussion 129–135, 193–195
Maes M, Bocchio Chiavetto L, Bignotti S, Battisa Tura GJ, Pioli R, Boin F, Kenis G, Bosmans E, de Jongh R, Altamura CA (2002) Increased serum interleukin-8 and interleukin-10 in schizophrenic patients resistant to treatment with neuroleptics and the stimulatory effects of clozapine on serum leukemia inhibitory factor receptor. Schizophr Res 54:281–291
Maragnoli ME, Fumagalli F, Gennarelli M, Racagni G, Riva MA (2004) Fluoxetine and olanzapine have synergistic effects in the modulation of fibroblast growth factor 2 expression within the rat brain. Biol Psychiatry 55:1095–1102
Marazziti D, Pfanner C, Dell’Osso B, Ciapparelli A, Presta S, Corretti G, Di Nasso E, Mungai F, Dell’Osso L (2005) Augmentation strategy with olanzapine in resistant obsessive compulsive disorder: an Italian long-term open-label study. J Psychopharmacol 19:392–394
Marek GJ, Martin-Ruiz R, Abo A, Artigas F (2005) The selective 5-HT2A receptor antagonist M100907 enhances antidepressant-like behavioral effects of the SSRI fluoxetine. Neuropsychopharmacology 30:2205–2215
McDougle CJ, Epperson CN, Pelton GH, Wasylink S, Price LH (2000) A double-blind, placebo-controlled study of risperidone addition in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder. Arch Gen Psychiatry 57:794–801
McElroy SL, Suppes T, Frye MA, Altshuler LL, Stanford K, Martens B, Leverich GS, Post RM, Keck PE Jr (2007) Open-label aripiprazole in the treatment of acute bipolar depression: a prospective pilot trial. J Affect Disord 101:275–281
Melia KR, Rasmussen K, Terwilliger RZ, Haycock JW, Nestler EJ, Duman RS (1992) Coordinate regulation of the cyclic AMP system with firing rate and expression of tyrosine hydroxylase in the rat locus coeruleus: effects of chronic stress and drug treatments. J Neurochem 58:494–502
Mereu G, Casu M, Gessa GL (1983) (−)-Sulpiride activates the firing rate and tyrosine hydroxylase activity of dopaminergic neurons in unanesthetized rats. Brain Res 264:105–110
Millan MJ, Lejeune F, Gobert A (2000) Reciprocal autoreceptor and heteroreceptor control of serotonergic, dopaminergic and noradrenergic transmission in the frontal cortex: relevance to the actions of antidepressant agents. J Psychopharmacol 14:114–138
Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P (2001) Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry 6:293–301
Moghaddam B, Bunney BS (1990) Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J Neurochem 54:1755–1760
Morishita S, Arita S (2003) Suitable dose and duration of fluvoxamine administration to treat depression. Psychiatry Clin Neurosci 57:177–181
Murer MG, Boissiere F, Yan Q, Hunot S, Villares J, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R (1999) An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer’s disease. Neuroscience 88:1015–1032
Nemeroff CB (1997) Dosing the antipsychotic medication olanzapine. J Clin Psychiatry 58(Suppl 10):45–49
Nierenberg AA (2007) Combined olanzapine plus fluoxetine modestly improves symptoms of acute bipolar I depression compared to lamotrigine. Evid Based Ment Health 10:12
Nikitin VP (2007) A new mechanism of synapse-specific neuronal plasticity. Neurosci Behav Physiol 37:559–570
Nomikos GG, Iurlo M, Andersson JL, Kimura K, Svensson TH (1994) Systemic administration of amperozide, a new atypical antipsychotic drug, preferentially increases dopamine release in the rat medial prefrontal cortex. Psychopharmacology (Berl) 115:147–156
Novak G, Seeman P, Tallerico T (2006) Increased expression of calcium/calmodulin-dependent protein kinase IIbeta in frontal cortex in schizophrenia and depression. Synapse 59:61–68
O’Connor M, Silver H (1998) Adding risperidone to selective serotonin reuptake inhibitor improves chronic depression. J Clin Psychopharmacol 18:89–91
Ordway GA, Szebeni K (2004) Effect of repeated treatment with olanzapine or olanzapine plus fluoxetine on tyrosine hydroxylase in the rat locus coeruleus. Int J Neuropsychopharmacol 7:321–327
Ostroff RB, Nelson JC (1999) Risperidone augmentation of selective serotonin reuptake inhibitors in major depression. J Clin Psychiatry 60:256–259
Otto D, Unsicker K (1990) Basic FGF reverses chemical and morphological deficits in the nigrostriatal system of MPTP-treated mice. J Neurosci 10:1912–1921
Owen RT (2006) Olanzapine/fluoxetine combination for bipolar depression and other mood disorders: a review. Drugs Today (Barc) 42:185–192
Pak JH, Huang FL, Li J, Balschun D, Reymann KG, Chiang C, Westphal H, Huang KP (2000) Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc Natl Acad Sci USA 97:11232–11237
Papakostas GI, Petersen TJ, Kinrys G, Burns AM, Worthington JJ, Alpert JE, Fava M, Nierenberg AA (2005) Aripiprazole augmentation of selective serotonin reuptake inhibitors for treatment-resistant major depressive disorder. J Clin Psychiatry 66:1326–1330
Parikh V, Khan MM, Mahadik SP (2004) Olanzapine counteracts reduction of brain-derived neurotrophic factor and TrkB receptors in rat hippocampus produced by haloperidol. Neurosci Lett 356:135–139
Patkar AA, Peindl K, Mago R, Mannelli P, Masand PS (2006) An open-label, rater-blinded, augmentation study of aripiprazole in treatment-resistant depression. Prim Care Companion J Clin Psychiatry 8:82–87
Patterson RL, van Rossum DB, Barrow RK, Snyder SH (2004) RACK1 binds to inositol 1,4,5-triphosphate receptors and mediates Ca2+ release. Proc Natl Acad Sci USA 101:2328–2332
Pehek EA, Nocjar C, Roth BL, Byrd TA, Mabrouk OS (2006) Evidence for the preferential involvement of 5-HT2A serotonin receptors in stress- and drug-induced dopamine release in the rat medial prefrontal cortex. Neuropsychopharmacology 31:265–277
Pezet S, Cunningham J, Patel J, Grist J, Gavazzi I, Lever IJ, Malcangio M (2002) BDNF modulates sensory neuron synaptic activity by a facilitation of GABA transmission in the dorsal horn. Mol Cell Neurosci 21:51–62
Pillai A (2008) Brain-derived neurotropic factor/TrkB signaling in the pathogenesis and novel pharmacotherapy of schizophrenia. Neurosignals 16:183–193
Pillai A, Terry AV Jr, Mahadik SP (2006) Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BDNF levels in rat striatum and hippocampus. Schizophr Res 82:95–106
Pinna G, Costa E, Guidotti A (2006) Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake. Psychopharmacology (Berl) 186:362–372
Pollmacher T, Hinze-Selch D, Mullington J (1996) Effects of clozapine on plasma cytokine and soluble cytokine receptor levels. J Clin Psychopharmacol 16:403–409
Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E (2008) Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry 63:801–808
Poyurovsky M, Kurs R, Weizman A (2003) Olanzapine–sertraline combination in schizophrenia with obsessive-compulsive disorder. J Clin Psychiatry 64:611
Pozzi L, Hakansson K, Usiello A, Borgkvist A, Lindskog M, Greengard P, Fisone G (2003) Opposite regulation by typical and atypical anti-psychotics of ERK1/2, CREB and Elk-1 phosphorylation in mouse dorsal striatum. J Neurochem 86:451–459
Rapaport MH, Gharabawi GM, Canuso CM, Mahmoud RA, Keller MB, Bossie CA, Turkoz I, Lasser RA, Loescher A, Bouhours P, Dunbar F, Nemeroff CB (2006) Effects of risperidone augmentation in patients with treatment-resistant depression: results of open-label treatment followed by double-blind continuation. Neuropsychopharmacology 31:2505–2513
Rasmussen K (2006) Creating more effective antidepressants: clues from the clinic. Drug Discov Today 11:623–631
Reynolds GP, Zhang ZJ, Beasley CL (2001) Neurochemical correlates of cortical GABAergic deficits in schizophrenia: selective losses of calcium binding protein immunoreactivity. Brain Res Bull 55:579–584
Riva MA, Molteni R, Tascedda F, Massironi A, Racagni G (1999) Selective modulation of fibroblast growth factor-2 expression in the rat brain by the atypical antipsychotic clozapine. Neuropharmacology 38:1075–1082
Roberts DS, Hu Y, Lund IV, Brooks-Kayal AR, Russek SJ (2006) Brain-derived neurotrophic factor (BDNF)-induced synthesis of early growth response factor 3 (Egr3) controls the levels of type A GABA receptor alpha 4 subunits in hippocampal neurons. J Biol Chem 281:29431–29435
Rodriguez-Sanchez P, Tejero-Diez P, Diez-Guerra FJ (1997) Glutamate stimulates neurogranin phosphorylation in cultured rat hippocampal neurons. Neurosci Lett 221:137–140
Rollema H, Lu Y, Schmidt AW, Sprouse JS, Zorn SH (2000) 5-HT(1A) receptor activation contributes to ziprasidone-induced dopamine release in the rat prefrontal cortex. Biol Psychiatry 48:229–237
Ruano D, Aulchenko YS, Macedo A, Soares MJ, Valente J, Azevedo MH, Hutz MH, Gama CS, Lobato MI, Belmonte-de-Abreu P, Goodman AB, Pato C, Heutink P, Palha JA (2008) Association of the gene encoding neurogranin with schizophrenia in males. J Psychiatr Res 42:125–133
Rummel C, Kissling W, Leucht S (2006) Antidepressants for the negative symptoms of schizophrenia. Cochrane Database Syst Rev 3:CD005581
Rutherford B, Sneed J, Miyazaki M, Eisenstadt R, Devanand D, Sackeim H, Roose S (2007) An open trial of aripiprazole augmentation for SSRI non-remitters with late-life depression. Int J Geriatr Psychiatry 22(10):986–991
Schmitt A, May B, Muller B, Jatzko A, Petroianu G, Braus DF, Henn FA (2003) Effects of chronic haloperidol and clozapine treatment on AMPA and kainate receptor binding in rat brain. Pharmacopsychiatry 36:292–296
Schwartz M, Silver H (2000) Lymphocytes, autoantibodies and psychosis—coincidence versus etiological factor: an update. Isr J Psychiatry Relat Sci 37:32–36
Seager MA, Huff KD, Barth VN, Phebus LA, Rasmussen K (2004) Fluoxetine administration potentiates the effect of olanzapine on locus coeruleus neuronal activity. Biol Psychiatry 55:1103–1109
Seager MA, Barth VN, Phebus LA, Rasmussen K (2005) Chronic coadministration of olanzapine and fluoxetine activates locus coeruleus neurons in rats: implications for bipolar disorder. Psychopharmacology (Berl) 181:126–133
Seeman P, Ko F, Jack E, Greenstein R, Dean B (2007) Consistent with dopamine supersensitivity, RGS9 expression is diminished in the amphetamine-treated animal model of schizophrenia and in postmortem schizophrenia brain. Synapse 61:303–309
Semba J, Sakai MW, Suhara T, Akanuma N (1999) Differential effects of acute and chronic treatment with typical and atypical neuroleptics on c-fos mRNA expression in rat forebrain regions using non-radioactive in situ hybridization. Neurochem Int 34:269–277
Sevincok L, Topuz A (2003) Lack of efficacy of low doses of quetiapine addition in refractory obsessive-compulsive disorder. J Clin Psychopharmacol 23:448–450
Shapira NA, Ward HE, Mandoki M, Murphy TK, Yang MC, Blier P, Goodman WK (2004) A double-blind, placebo-controlled trial of olanzapine addition in fluoxetine-refractory obsessive-compulsive disorder. Biol Psychiatry 55:553–555
Shelton RC (2003) The combination of olanzapine and fluoxetine in mood disorders. Expert Opin Pharmacother 4:1175–1183
Shelton RC (2006) Olanzapine/fluoxetine combination for bipolar depression. Expert Rev Neurother 6:33–39
Shelton RC, Williamson DJ, Corya SA, Sanger TM, Van Campen LE, Case M, Briggs SD, Tollefson GD (2005) Olanzapine/fluoxetine combination for treatment-resistant depression: a controlled study of SSRI and nortriptyline resistance. J Clin Psychiatry 66:1289–1297
Shi L, Namjoshi MA, Swindle R, Yu X, Risser R, Baker RW, Tohen M (2004) Effects of olanzapine alone and olanzapine/fluoxetine combination on health-related quality of life in patients with bipolar depression: secondary analyses of a double-blind, placebo-controlled, randomized clinical trial. Clin Ther 26:125–134
Shim SS, Hammonds MD, Kee BS (2008) Potentiation of the NMDA receptor in the treatment of schizophrenia: focused on the glycine site. Eur Arch Psychiatry Clin Neurosci 258:16–27
Silver H (2001) Fluvoxamine as an adjunctive agent in schizophrenia. CNS Drug Rev 7:283–304
Silver H, Nassar A (1992) Fluvoxamine improves negative symptoms in treated chronic schizophrenia: an add-on double-blind, placebo-controlled study. Biol Psychiatry 31:698–704
Silver H, Shmugliakov N (1998) Augmentation with fluvoxamine but not maprotiline improves negative symptoms in treated schizophrenia: evidence for a specific serotonergic effect from a double-blind study. J Clin Psychopharmacol 18:208–211
Silver H, Kaplan A, Jahjah N (1995) Fluvoxamine augmentation for clozapine-resistant schizophrenia. Am J Psychiatry 152:1098
Silver H, Kushnir M, Kaplan A (1996) Fluvoxamine augmentation in clozapine-resistant schizophrenia: an open pilot study. Biol Psychiatry 40:671–674
Silver H, Barash I, Aharon N, Kaplan A, Poyurovsky M (2000) Fluvoxamine augmentation of antipsychotics improves negative symptoms in psychotic chronic schizophrenic patients: a placebo-controlled study. Int Clin Psychopharmacol 15:257–261
Silver H, Knoll G, Isakov V, Goodman C, Finkelstein Y (2005) Blood DHEAS concentrations correlate with cognitive function in chronic schizophrenia patients: a pilot study. J Psychiatr Res 39:569–575
Simon JS, Nemeroff CB (2005) Aripiprazole augmentation of antidepressants for the treatment of partially responding and nonresponding patients with major depressive disorder. J Clin Psychiatry 66:1216–1220
Simpson GM, Glick ID, Weiden PJ, Romano SJ, Siu CO (2004) Randomized, controlled, double-blind multicenter comparison of the efficacy and tolerability of ziprasidone and olanzapine in acutely ill inpatients with schizophrenia or schizoaffective disorder. Am J Psychiatry 161:1837–1847
Siris SG (1993) Adjunctive medication in the maintenance treatment of schizophrenia and its conceptual implications. Br J Psychiatry 22(Suppl):66–78
Sokolski KN (2007) Adjunctive aripiprazole in bipolar I depression. Ann Pharmacother 41:35–40
Sokolski KN, Denson TF (2003) Adjunctive quetiapine in bipolar patients partially responsive to lithium or valproate. Prog Neuropsychopharmacol Biol Psychiatry 27:863–866
Spedding M, Gressens P (2008) Neurotrophins and cytokines in neuronal plasticity. Novartis Found Symp 289:222–233, discussion 233–240
Spina E, de Leon J (2007) Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol 100:4–22
Sproule BA, Otton SV, Cheung SW, Zhong XH, Romach MK, Sellers EM (1997) CYP2D6 inhibition in patients treated with sertraline. J Clin Psychopharmacol 17:102–106
Spurney CF, Baca SM, Murray AM, Jaskiw GE, Kleinman JE, Hyde TM (1999) Differential effects of haloperidol and clozapine on ionotropic glutamate receptors in rats. Synapse 34(4):266–276
Stoll AL, Haura G (2000) Tranylcypromine plus risperidone for treatment-refractory major depression. J Clin Psychopharmacol 20:495–496
Strous RD (2005) Dehydroepiandrosterone (DHEA) augmentation in the management of schizophrenia symptomatology. Essent Psychopharmacol 6:141–147
Szabo ST, Blier P (2002) Effects of serotonin (5-hydroxytryptamine, 5-HT) reuptake inhibition plus 5-HT(2A) receptor antagonism on the firing activity of norepinephrine neurons. J Pharmacol Exp Ther 302:983–991
Szegedi A, Anghelescu I, Wiesner J, Schlegel S, Weigmann H, Hartter S, Hiemke C, Wetzel H (1999) Addition of low-dose fluvoxamine to low-dose clozapine monotherapy in schizophrenia: drug monitoring and tolerability data from a prospective clinical trial. Pharmacopsychiatry 32:148–153
Tan YL, Zhou DF, Cao LY, Zou YZ, Zhang XY (2005) Decreased BDNF in serum of patients with chronic schizophrenia on long-term treatment with antipsychotics. Neurosci Lett 382:27–32
Tani K, Takei N, Kawai M, Suzuki K, Sekine Y, Toyoda T, Minabe Y, Mori N (2004) Augmentation of milnacipran by risperidone in treatment for major depression. Int J Neuropsychopharmacol 7:55–58
Tarazi FI, Baldessarini RJ, Kula NS, Zhang K (2003) Long-term effects of olanzapine, risperidone, and quetiapine on ionotropic glutamate receptor types: implications for antipsychotic drug treatment. J Pharmacol Exp Ther 306:1145–1151
Tischmeyer W, Grimm R (1999) Activation of immediate early genes and memory formation. Cell Mol Life Sci 55:564–574
Tohen M, Vieta E, Calabrese J, Ketter TA, Sachs G, Bowden C, Mitchell PB, Centorrino F, Risser R, Baker RW, Evans AR, Beymer K, Dube S, Tollefson GD, Breier A (2003) Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 60:1079–1088
Tsai G, Coyle JT (2002) Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol 42:165–179
Tsai GE, Yang P, Chang YC, Chong MY (2006) d-Alanine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 59:230–234
Verma V, Lim EP, Han SP, Nagarajah R, Dawe GS (2007) Chronic high-dose haloperidol has qualitatively similar effects to risperidone and clozapine on immediate-early gene and tyrosine hydroxylase expression in the rat locus coeruleus but not medial prefrontal cortex. Neurosci Res 57:17–28
Volavka J, Cooper TB, Czobor P, Lindenmayer JP, Citrome LL, Mohr P, Bark N (2000) High-dose treatment with haloperidol: the effect of dose reduction. J Clin Psychopharmacol 20:252–256
Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2000) Decreased glutamic acid decarboxylase 67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry 57:237–245
Wan W, Ennulat DJ, Cohen BM (1995) Acute administration of typical and atypical antipsychotic drugs induces distinctive patterns of Fos expression in the rat forebrain. Brain Res 688:95–104
Weigmann H, Gerek S, Zeisig A, Muller M, Hartter S, Hiemke C (2001) Fluvoxamine but not sertraline inhibits the metabolism of olanzapine: evidence from a therapeutic drug monitoring service. Ther Drug Monit 23:410–413
Weinberger DR, Berman KF (1996) Prefrontal function in schizophrenia: confounds and controversies. Philos Trans R Soc Lond B Biol Sci 351:1495–1503
Weiss EL, Potenza MN, McDougle CJ, Epperson CN (1999) Olanzapine addition in obsessive-compulsive disorder refractory to selective serotonin reuptake inhibitors: an open-label case series. J Clin Psychiatry 60:524–527
Werme M, Ringholm A, Olson L, Brene S (2000) Differential patterns of induction of NGFI-B, Nor1 and c-fos mRNAs in striatal subregions by haloperidol and clozapine. Brain Res 863:112–119
Westerink BH, Kawahara Y, De Boer P, Geels C, De Vries JB, Wikstrom HV, Van Kalkeren A, Van Vliet B, Kruse CG, Long SK (2001) Antipsychotic drugs classified by their effects on the release of dopamine and noradrenaline in the prefrontal cortex and striatum. Eur J Pharmacol 412:127–138
Wetmore C, Ernfors P, Persson H, Olson L (1990) Localization of brain-derived neurotrophic factor mRNA to neurons in the brain by in situ hybridization. Exp Neurol 109:141–152
Wetzel H, Anghelescu I, Szegedi A, Wiesner J, Weigmann H, Harter S, Hiemke C (1998) Pharmacokinetic interactions of clozapine with selective serotonin reuptake inhibitors: differential effects of fluvoxamine and paroxetine in a prospective study. J Clin Psychopharmacol 18:2–9
Williams R (2001) Optimal dosing with risperidone: updated recommendations. J Clin Psychiatry 62:282–289
Worthington JJ 3rd, Kinrys G, Wygant LE, Pollack MH (2005) Aripiprazole as an augmentor of selective serotonin reuptake inhibitors in depression and anxiety disorder patients. Int Clin Psychopharmacol 20:9–11
Xu H, Chen Z, He J, Haimanot S, Li X, Dyck L, Li XM (2006) Synergetic effects of quetiapine and venlafaxine in preventing the chronic restraint stress-induced decrease in cell proliferation and BDNF expression in rat hippocampus. Hippocampus 16:551–559
Yamamoto BK, Cooperman MA (1994) Differential effects of chronic antipsychotic drug treatment on extracellular glutamate and dopamine concentrations. J Neurosci 14:4159–4166
Yasui-Furukori N, Kondo T, Mihara K, Inoue Y, Kaneko S (2004) Fluvoxamine dose-dependent interaction with haloperidol and the effects on negative symptoms in schizophrenia. Psychopharmacology (Berl) 171:223–227
Yoshimura R, Kaneko S, Shinkai K, Nakamura J (2006) Successful treatment for obsessive-compulsive disorder with addition of low-dose risperidone to fluvoxamine: implications for plasma levels of catecholamine metabolites and serum brain-derived neurotrophic factor levels. Psychiatry Clin Neurosci 60:389–393
Yoshino T, Nisijima K, Shioda K, Yui K, Katoh S (2004) Perospirone, a novel atypical antipsychotic drug, potentiates fluoxetine-induced increases in dopamine levels via multireceptor actions in the rat medial prefrontal cortex. Neurosci Lett 364:16–21
Youngren KD, Inglis FM, Pivirotto PJ, Jedema HP, Bradberry CW, Goldman-Rakic PS, Roth RH, Moghaddam B (1999) Clozapine preferentially increases dopamine release in the rhesus monkey prefrontal cortex compared with the caudate nucleus. Neuropsychopharmacology 20:403–412
Zhang W, Perry KW, Wong DT, Potts BD, Bao J, Tollefson GD, Bymaster FP (2000) Synergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology 23:250–262
Zhang XY, Zhou DF, Zhang PY, Wu GY, Cao LY, Shen YC (2002) Elevated interleukin-2, interleukin-6 and interleukin-8 serum levels in neuroleptic-free schizophrenia: association with psychopathology. Schizophr Res 57:247–258
Zhang XY, Zhou DF, Cao LY, Wu GY, Shen YC (2005) Cortisol and cytokines in chronic and treatment-resistant patients with schizophrenia: association with psychopathology and response to antipsychotics. Neuropsychopharmacology 30:1532–1538
Zhang HX, Zhao JP, Lv LX, Li WQ, Xu L, Ouyang X, Yuan ZQ, Huang JS (2007) Explorative study on the expression of neuregulin-1 gene in peripheral blood of schizophrenia. Neurosci Lett 438(1):1–5
Zink M (2005) Augmentation of olanzapine in treatment-resistant schizophrenia. J Psychiatry Neurosci 30:409–415
Zink M, Schmitt A, May B, Muller B, Braus DF, Henn FA (2004a) Differential effects of long-term treatment with clozapine or haloperidol on GABA transporter expression. Pharmacopsychiatry 37:171–174
Zink M, Schmitt A, May B, Muller B, Demirakca T, Braus DF, Henn FA (2004b) Differential effects of long-term treatment with clozapine or haloperidol on GABAA receptor binding and GAD67 expression. Schizophr Res 66:151–157
Acknowledgment
The authors gratefully acknowledge the generous assistance of Prof. Hiemke and co-workers at Mainz, Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chertkow, Y., Weinreb, O., Youdim, M.B.H. et al. Molecular mechanisms underlying synergistic effects of SSRI–antipsychotic augmentation in treatment of negative symptoms in schizophrenia. J Neural Transm 116, 1529–1541 (2009). https://doi.org/10.1007/s00702-009-0255-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-009-0255-4