
Abstract
Hippophae rhamnoides ssp. sinensis is an ecologically and economically important species that has been widely used as a pioneer plant in China. In this study, we employed both nuclear ISSR and maternal cpSSR markers to survey the genetic diversity and structure of populations of ssp. sinensis representing three different landscapes, the northwestern desert and grassland region, the alpine vegetation region of the Qinghai-Tibetan Plateau, and the northeastern humid forest region. In all, 12 natural populations with a scattered distribution in the area were studied. The genetic diversities of populations were found to be uneven, and the total genetic diversity was low on the basis of both types of marker. Mantel tests based on both individual Euclidean distance matrices and population genetic distance (measured by Φpt) matrices showed that the two marker systems detected similar trends with respect to genetic distances between populations. The analysis of molecular variance (AMOVA) revealed significant differentiation among populations and among regions for both types of marker. Although the detected pattern of isolation-by-distance among all sampled populations confirmed the earlier colonization pathway, the low level of gene flow and the lack of isolation by distance within each region suggested the presence of an additional dispersal barrier. UPGMA dendrograms and PCA plots also revealed clear clustering and significant regional differentiation. Our results indicate that the genetic structure of ssp. sinensis has been affected by habitat fragmentation and restricted population sizes. We propose that the biology of reproduction and ecology have played determinant roles in the development of the regional structure of populations. The genetic information obtained will help to establish conservation strategies and programs for sustainable management of H. rhamnoides ssp. sinensis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hippophae L. is a small genus of Elaeagnaceae. Several scholars have recognized only one species in Hippophae (Servettaz 1909; Avdeyev 1983). The “one species” classification was denied by some authors based on the highly variable morphology. However, the systematic treatment and the relationships between taxa in this genus remain controversial, the number of species and subspecies being the focus of this dispute. Rousi (1971) divided the genus Hippophae into three species with nine subspecies in the species H. rhamnoides. After the revision of Rousi (1971), Liu and He (1978) described a new species, H. neurocarpa S. W. Liu and T. N. He, from the Qinghai Plateau. Lian (1988) established two sections and increased the number of taxa to seven species and nine subspecies on the basis of extensive investigations and systemic studies (Lian and Chen 1996; Lian et al. 1997, 1998). However, the taxonomical treatment that divided the genus into two sections was not supported by Sun et al. (2002) on the basis of ITS sequences. According to the latest systematic treatments of the genus Hippophae L. (Bartish et al. 2002; Swenson and Bartish 2003), the genus comprises seven species, and the species H. rhamnoides circumscribes eight subspecies. All species are diploid (2n = 24), wind-pollinated, and dioecious, and are restricted to the Qinghai-Tibetan Plateau and adjacent areas, with the exception of the species H. rhamnoides L. that occurs widely but sporadically in Asia and Europe (Rousi 1965, 1971; Lu 1997; Lian et al. 2000; Bartish et al. 2000, 2002). Ssp. sinensis and ssp. yunnanensis from H. rhamnoides are endemic to China. Ssp. sinensis is found on the eastern edges of the distribution of H. rhamnoides and is most widely distributed at China at altitudes ranging from 400 to 3,900 m. Some plants belonging to ssp. sinensis also occur at altitudes below 400 m.
Because of its strong ecological adaptability and high tolerance of environmental stresses, especially to extreme conditions, including temperature, drought, high altitude, salinity, alkalinity, and inundation (Lu 1992; Ruan and Li 2002; Sheng et al. 2006), sea buckthorn (Hippophae) is a major pioneer species in arid and semiarid zones and can provide a protective windbreak. Sea buckthorn also has enormous nutritional, medical and ornamental value. It is a multi-purpose plant which has been a target of plant breeding programs, mainly in China, Russia, Central Asia and Europe (Yao and Tigerstedt 1993; Aitzetmuller and Xin 1999; Tang and Tigerstedt 2001). However, the accelerated and uncontrolled use of this important resource has led to deforestation in some parts of its natural distribution, with consequent adverse impact on the ecosystem (Tian et al. 2004a). Knowledge of its genetics, especially of the genetic variation and structure of natural populations, is urgently needed to guide the exploitation of this valuable germplasm and breeding programs.
H. rhamnoides ssp. sinensis is an ecologically and economically important species that has been widely used as a pioneer plant in China. The major regions of northern China within the Loess Plateau areas, where ssp. sinensis occurs, are characterized by orographically diverse landscapes and an array of ridges and ravines. It mainly occurs on sandy soils by river banks or dry river beds, mountain slopes, and valleys in the transition zone of the northwestern desert and grassland region. Plants belonging to ssp. sinensis also grow in the marginal areas of the alpine vegetation of the Qinghai-Tibet Plateau and the northeastern humid forest region, where they mix with other trees and form patchy forests. The discontinuous but extensive distribution in diverse environmental conditions, together with the dioecious breeding system and competition with other species, may have contributed to the genetic structure of this subspecies during its long-term evolution. Substantial morphological variations in the fruits, leaves, and chemical components have been detected (Rousi 1971; Zhao et al. 1991; Lian et al. 2000). Several studies based on molecular markers have been conducted on the genetic diversity and population genetic structure of ssp. sinensis (Yao and Tigerstedt 1993; Sun et al. 2006). However, previous studies were performed on the basis of a single type of molecular marker representing either the nuclear or cytoplastic genome. Comparisons of population diversity and structure represented in different genomes, and patterns of gene flow by seed and pollen in different environments would certainly be an advance compared with previous genetic studies in ssp. sinensis.
In this study, we used nuclear ISSR and maternal cpSSR markers to investigate the genetic diversity and structure of populations of ssp. sinensis representing three different landscapes. Our primary purpose was to detect the level and pattern of genetic variation of the subspecies, to compare population structures represented in different genomes, and to estimate gene flow in natural populations via different types of propagules. Knowledge of regional differentiation in two marker systems may provide an important insight into the population genetics of ssp. sinensis and the effect of eco-environmental and climatic factors on the genetic variation. This knowledge could be useful for conservation and breeding programs.
Materials and methods
Plant material
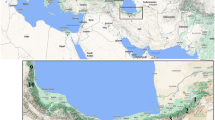
Ssp. sinensis has a zonal distribution pattern controlled by rainfall and heat. Its geographical range is roughly located in the transitional zone of three vegetation regions in China, i.e. the northwestern desert and grassland region, the alpine vegetation region of the Qinghai-Tibetan Plateau, and the eastern humid forest region (Lian and Chen 1992). Twelve populations of ssp. sinensis which occur in various habitats with different climates and topographies within the major distribution area were selected for the study (Table 1; Fig. 1). The 12 populations were divided into three groups according to their geographical regions and the zonal distribution pattern controlled by heat and rainfall. Region 1 consisted of five populations (IMO, HBF, SAXH, SAXW, and SXJ), collected from the northwestern desert and grassland region. Region 2 comprised four populations (SCJ, SCB, SCM, and SCW) from the alpine vegetation region of the Qinghai-Tibetan Plateau. Three populations (LNF, HLJS, and HLJH) from the northeastern humid forest region were included in region 3. All populations were sampled from natural forests, and sampled stands were 150–3,000 m away from the closest neighboring populations. Population sizes varied widely, from about 200 m2 to hundreds hm2, as estimated by the eye.

The locations of the 12 populations of Hippophae rhamnoides ssp. sinensis in China. Different gray lines stand for the territorial boundaries of local provinces of selected populations
All plant material was raised from seeds collected in natural populations with two exceptions:
-
1
In population SCM, sampled material was collected from a population with approx. 120 individuals grown in the Maoxian Mountain Ecosystem Research Station of the Chinese Academy of Science, introduced from their original location in the Jiuzaigou Nature Reserve, China;
-
2
In populations SAXW, SAXH, and IMO, sampled material was collected from a ssp. sinensis nursery representing populations with their original location in the Wuqi county and Huanglong county in the Shaanxi province, and in Ordos in the Inner Mongolia Autonomous Region, respectively.
Seed was generally collected from 16 to 20 widely separated mother plants in each population. The sampled mother plants were separated by a minimum distance of 10 m to prevent collection of ramets from a single individual. Thus, each analyzed individual had a different mother plant.
DNA extraction
DNA was isolated from the fresh leaves of seedlings by the CTAB-based method (Chen et al. 2008). Leaf materials were ground to fine powder in liquid nitrogen and then transferred to 2.0 ml Eppendorf tubes and mixed with 900 μl preheated 2× CTAB extraction buffer containing 1% (v/v) β-mercaptoethanol and 1% (w/v) PVP 40000. DNA quality and quantity were checked by a DNA–protein instrument (Bio-Rad).
ISSR analysis
Based on the clarity and reproducibility of the band patterns, nine primers out of 45 ISSR primers, i.e. 807, 808, 809, 811, 826, 840, 848, 881, and 887, were selected from UBC set #9 of ISSR primers (http://www.biotech.ubc.ca/). The PCRs were performed in a volume of 12.5 μl containing 1.3 μl 10× reaction buffer (TaKaRa, Dalian), 300 μM dNTP (Promega), 0.25 μM primer, 0.5 U Taq polymerase (TaKaRa, Dalian), and 20 ng genomic DNA. For each primer, amplifications were carried out in 96-well plates using the program: initial denaturation step 3 min at 95°C, followed by 40 cycles of 1 min at 95°C, 1 min at 53–57°C, and 1.5 min at 72°C, and a final extension step of 10 min at 72°C. The PCR products were separated on 1.8% agarose gels and stained with 0.1% ethidium bromide. The molecular weights were estimated using the GeneRuler™ 100 bp DNA Ladder Plus (Fermentas). The gel images were recorded and the band sizes were quantified using a Gel Doc 2000 system (Bio-Rad). All of the plant-primer combinations were run more than once to ensure reproducibility. The fragments amplified by ISSR primers were scored for each individual as present (1) or absent (0) on the basis of size comparison with external standards (GeneRuler™ 100 bp DNA Ladder Plus).
cpSSR analysis
To date, no chloroplast microsatellite sequences have been reported for Hippophae rhamnoides. In this study, ten conserved primer pairs developed for dicotyledonous angiosperms (Weising and Gardner 1999), six universal primer pairs developed from Arabidopsis thaliana (Cheng et al. 2006), and three primer pairs developed on the basis of chloroplast genomes of Solanaceous plants (Bryan et al. 1999) were used to detect cpDNA microsatellite loci. In addition, seven primer pairs denoted CCMP2, CCMP3, CCMP4, CCMP6, CCMP7, ARCP4, and NTCP9, which produced fragments unambiguously scored across all samples, were taken into consideration for further analysis. The amplification reactions were performed in a volume of 10 μl containing 1.0 μl 10× reaction buffer (TaKaRa, Dalian), 300 mM dNTPs (Promega), 0.2 mM of both primers specified for each microsatellite locus, 0.5 U Taq polymerase (TaKaRa), and 10–20 ng DNA. PCR amplifications were performed using the profile: 3 min of denaturation at 94°C; 30 cycles of denaturation (45 s at 94°C), annealing (30 s at 48–55°C), and extension (45 s at 72°C), followed by a final extension step for 7 min at 72°C. The amplified PCR products were separated on a LI-COR 4200 DNA Analyzer Sequencer using 6% denaturing polyacrylamide gels (KB-plus solution, LI-COR). The electrophoresed gels were silver-stained, and the fingerprints were visualized and scored using Genescan™ Analysis 3.1 software (Applied Biosystems). Fragment sizes were estimated using a standard molecular weight marker pBR322 DNA/HaeIII. In all cases, PCR reactions were performed at least twice in order to ensure the reproducibility of the reactions. Because the chloroplast genome is haploid and does not undergo recombination, it can be viewed as a single locus, and the size scores for the fourteen fragments included in the analysis were combined in order to derive the chloroplast haplotype of each individual. Nevertheless, we use the term locus to refer to a cpSSR site, and allele to refer to a size variant at a given cpSSR site.
Data analysis
The genetic diversity statistics based on ISSR markers, including the percentage of polymorphic loci per population (PLP), Nei’s gene diversity (H Ene), the effective number of alleles (N e), the total genetic diversity (H Tne), and genetic diversity within populations (H Sne) (Nei 1978), were generated by use of the software Popgen 1.31 (Yeh et al. 1997) to describe genetic variation at intra and inter-population levels. The Arlequin ver 3.11 software package (Excoffier et al. 2007) was used to compute variation data for the chloroplast haplotype within and among populations, including the number of haplotypes (n h), the effective number of haplotypes (n E), the unbiased haplotypic diversity (H Ecp), the average diversity over populations (H Scp), and the total gene diversity (H Tcp) (Nei 1987).
Pairwise genetic distances were calculated between both individuals and populations. Calculation of pairwise Euclidean distances between individuals for binary and haploid data followed the method of Huff et al. (1993). The degree of relatedness between the genetic distance matrices generated by the two types of marker was measured using the Mantel matrix-correspondence test (Mantel 1967). To facilitate comparisons between patterns of molecular variance of cpSSR and binary ISSR markers, the degree of genetic differentiation based on both marker types was estimated using the fixation indices at three hierarchical levels: among populations (Φpt), among populations within regions (Φpr), and among regions (Φrt) by analysis of molecular variance (AMOVA) in GenAlEx software ver 6 (Peakall and Smouse 2006). Significance values were computed by a permutation test from 1,000 permutations. The effective number of migrants (N m) was indirectly estimated using Wright’s (1969) formula N m = 0.25 (1 − F st)/F st, where F st was substituted by Φpt, because Φpt is analogous to F st when the data are haploid or binary (Peakall et al. 1995). To visualize similarities between populations, modified Roger’s distances (Wright 1978) were calculated to construct dendrograms using UPGMA (unweighted pair-group arithmetic mean-method) for the 12 populations. The bootstrap values were obtained by use of TFPGA software (Miller 2000), with 1,000 replications to evaluate the internal support for the tree. Furthermore, in order to combine the information provided by different loci, a multivariate approach based on principal-components analysis (PCA) was performed with the informative ISSR loci allele frequencies and the transformed haplotype frequencies from the cpSSR data set. The possible relationship between genetic and geographical distances was assessed by a Mantel test in GenAlEx software ver 6, which tested isolation by distance on the basis of the modified Roger’s distances between populations against the natural logarithm of geographical distances between populations.
Results
Genetic diversity
The ISSR patterns obtained using the nine selected primers out of 45 tested ISSR primers were found to be highly reproducible and polymorphic in all populations. A total of 252 reproducible bands were scored, of which 237 bands were polymorphic among the 12 populations. Of the total of 252 bands scored, four bands were population-specific, present in single populations (two in population IMO and two in population SCW). In individual populations, the percentage of polymorphic loci (PLP) ranged from 38.5 to 61.1%. Nei’s gene diversities (H Ene) and effective numbers of alleles per locus (N e) varied from 0.106 to 0.187 and from 1.21 to 1.30, respectively (Table 2). Among the 12 populations investigated, population IMO had the highest variability and population HLJH had the lowest. The average Nei’s gene diversity (H Sne) calculated across populations equaled 0.144. For the entire sample set, the total genetic diversity (H Tne) and N e value were 0.276 and 1.45, respectively. At the region level, genetic diversity was lowest in region 2, with H Ene and N e values of 0.201 and 1.31.
The analyses proved the high reproducibility of cpSSR fingerprints. Six loci (CCMP2, CCMP3, CCMP4, CCMP6, ARCP4, and NTCP9) out of the nineteen chloroplast microsatellites screened were polymorphic, with a total of 14 alleles. Among these, two alleles (ARCP4-1 and CCMP2-2) were found only in population SCW. The 14 alleles produced 15 different haplotypes, of which 13 haplotypes were common to all populations but two haplotypes were found only in population SCW (Table 2). Nei’s unbiased haplotypic diversity (H Ecp) and the number of haplotypes (n h) were highest in populations HLJH and SXJ, and lowest in populations HLJS and SCM. The level of polymorphism obtained with the cpSSR was very low for all the populations, with the total gene diversity (H Tcp) and the average diversity (H Scp) equalling 0.207 and 0.091 (Table 2). At the region level, region 3 had the highest level of haplotypic diversity, with H Ecp and n E values of 0.214 and 1.37, respectively, and genetic variability was lowest in region 1.
Genetic differentiation
Pairwise Euclidean distances between individuals were computed on the basis of ISSR and cpSSR data. A standardized Mantel statistic of r = 0.177 (P = 0.001) was obtained for the relationship between pairwise individual genetic distances computed with the two types of marker. Mantel’s test showed that two individual genetic distance matrices based on ISSR and cpSSR markers were weakly but significantly correlated. The two marker systems therefore detected similar trends with regard to genetic distances between populations.
Pairwise genetic distances between populations were estimated on the basis of ISSR and cpSSR data using the Φpt estimator. Based on ISSR data, the values ranged from 0.019 to 0.822, populations HLJS and LNF being the most differentiated populations (Table 3). On the basis of cpSSR data, the Φpt values ranged from 0.000 to 0.867 (Table 3). Large genetic distance estimated by Φpt and significant genetic structure were found for most pairwise population comparisons within regions 2 and 3. Low Φpt values based on cpSSR data were found between populations within region 1. The Mantel’s test showed that there was a weak but significant correlation between the ISSR and cpSSR divergence matrices based on the Φpt estimator (r = 0.185 and P = 0.05).
At the region level, the Φpt values based on ISSR markers were 0.425, 0.309, and 0.476 for regions 1, 2, and 3, respectively (Table 4). The overall gene flow among the 12 populations was 0.282, and the gene flow among populations within the three regions was 0.338, 0.559 and 0.276, respectively. For the chloroplast genome, region 1 was least differentiated with the Φpt value equal to 0.072, and the Φpt values of regions 2 and 3 equal to 0.489 and 0.586, respectively, showing significant differentiation (Table 4). Consequently, based on cpSSR data, the gene flow among populations within the three regions was 3.222, 0.261, and 0.176, respectively, and the overall gene flow among the 12 populations was 0.165.
Analysis of molecular variance
The apportionment of genetic variation among populations and among regions was measured by fixation indices. For ISSR markers, almost a half of the genetic variation was found among populations and the other half within populations. Among hierarchical levels, differentiation among populations and among populations within regions was higher (Φpt = 0.470, P = 0.001; Φpr = 0.431, P = 0.001) than that among the three regions (Φrt = 0.067, P = 0.001) (Table 5).
For cpSSR markers, analysis of molecular variance showed that approximately 40.1% genetic variation was found within populations, and 30.9 and 29% among the three regions and among populations within regions, respectively (Table 5). Among hierarchical levels, differentiation among populations, among populations within regions, and among regions equaled 0.602 (P = 0.001), 0.423 (P = 0.001), and 0.309 (P = 0.001), respectively (Table 5).
Relationships among populations
The UPGMA dendrograms based on modified Roger’s distances between populations revealed clear clustering with above 60% bootstrap value support, except for a few clusters with very low bootstrap values (<50%) (Fig. 2). The two phenetic trees based on the two types of marker showed relatively similar clustering patterns, except for a few populations. For instance, populations IMO, HBF, and LNF, SAXW and HLJH, and SCJ, SCJ, and SCM congruently formed three sub-clusters according to the phenetic trees (Fig. 2). The main difference between the two trees concerned population SCW, which clustered together with other populations for ISSR data but differed substantially from the others according to cpSSR data. The second difference between the two trees concerned populations SAXW and HLJH, which differed substantially from the others according to ISSR markers but clustered together with other populations according to cpSSR data.

UPGMA dendrogram obtained for the 12 populations of Hippophae rhamnoides ssp. sinensis by nuclear ISSR and cpSSR analysis using the modified Roger index (Wright 1978). Bootstrap values above 50% obtained from 1,000 replicate analyses are shown
The relationships among populations were further illustrated by the results from principal-components analysis (PCA) based on the ISSR and cpSSR matrices. All populations could be clearly distinguished by both principal-components analyses (Fig. 3), and the percentages of the total variation explained by the first two coordinates were 57.7 and 88.9%, respectively. The plots showed different types of distribution among populations, the ISSR points being more evenly dispersed than those of cpSSR data. For example, in the case of cpSSRs, populations from regions 1 and 2 demonstrated regional differentiation between populations sampled from these two areas. Populations SCW and HLJS were far from each other, which was probably responsible for most among-population divergence in cpSSR.

The PCA plot of coordinates via a covariance matrix with data standardization based on pairwise Nei’s genetic distances. PCA plot of coordinates: a based on ISSR markers; b based on cpSSR markers
To explore how much genetic differentiation could be explained by geographical distances between pairs of populations, modified Roger’s distance values were plotted against the natural logarithm of geographical distances between populations. Although the overall Mantel test revealed a significant correlation between genetic and geographical distances based on both ISSR (r = 0.269, P = 0.043) and cpSSR (r = 0.386, P = 0.006) markers, the genetic distances between pairs of populations within regions 1 and 2, on the basis of ISSR and cpSSR markers, were not positively correlated with geographical distances (P > 0.28 and P > 0.15, respectively).
Discussion
Habitat fragmentation is expected to have a detrimental impact on the genetic diversity of plant species (O’Connell et al. 2006; Isagi et al. 2007; Lu et al. 2009), but several studies have revealed that population responses to fragmentation are variable (Jump and Penuelas 2006; Kettle et al. 2007). In this study, the genetic diversity (H Sne = 0.144) based on ISSR markers was within the general range previously detected for ssp. sinensis and other subspecies belonging to H. rhamnoides using RAPD and ISSR markers (H e = 0.117–0.204) (Bartish et al. 1999, 2000; Tian et al. 2004a, b; Sun et al. 2006; Chen et al. 2010), but lower than the observed diversity (H e = 0.249) for H. rhamnoides based on the same type of marker (ISSR) in the Wolong Nature Reserve of China (Chen et al. 2008). The observed genetic diversity in ssp. sinensis on the basis of cpSSRs was lower than cpSSR diversities found in natural populations of other angiosperms, for example Dipteronia Oliv (Yang et al. 2008). Unfortunately, as far as we are aware, there is lack of available diversity data for the chloroplast genome of Hippophae, and no comparison of cpSSR diversity can be performed here.
H. rhamnoides ssp. sinensis forests, which have endured habitat fragmentation for several centuries, occur in the hilly-gully region of the Loess Plateau and the desert-grassland region in northwestern China, the transition zones of the mountain-plain of the northeastern humid forest region, and the alpine vegetation region of the Qinghai-Tibetan Plateau. The different landscapes and rugged topography have divided ssp. sinensis forests into fragmented and small populations. Decreasing migration between and within populations and the effects of genetic drift may be responsible for the low level of genetic diversity present in ssp. sinensis and reduced ability to withstand variable climatic conditions during early successional stages. In addition, the high degree of vegetative reproduction may be a reason for the relatively low genetic diversity within populations of ssp. sinensis.
The amount of genetic diversity varied among populations of ssp. sinensis and between landscapes. Fluctuations in population size, and different landscape and variable climatic conditions in fragmented habitats may account for the uneven distribution of genetic diversity. For example, population SCW had a high level of genetic diversity when compared with populations from the same region, which may be related to a larger population size, high density, and ecological optima. Population SCW is located in the middle altitudinal zone in the Wolong Nature Reserve of China, where favorable ecological conditions (e.g. abundant rainfall and optimum temperature; Table 1) enable its continuous distribution covering the zone from 2,000 to 2,600 m above sea level. Evidence for the optimum zone for H. rhamnoides occurring in the Wolong Natural Reserve has also been documented by Li et al. (2007). In general, genetic diversity is known to be positively correlated with population size (Frankham 1996). Limited gene exchange and competition with other species may contribute to the observed lower genetic diversity of populations SCM, SCJ, and SCB, originally located on the fringe of the forest, where plants belonging to ssp. sinensis mix with other trees and form patchy forests. Several mountains, for example the Hengduan Mountains and Saluli Mountain, and Jinsha River cross this area. Mountains, river, and tall trees form barriers and are important in preventing gene exchange, which consequently leads to isolation, genetic drift effects, and low genetic diversity in populations of ssp. sinensis in this region.
In populations from the northeastern humid forest region, the different landform features between populations and variable hydrothermal conditions may account for the difference in the level of genetic variation. Population HLJH is located in the plain region, which is a landform that enables effective gene exchange by seed dispersal. Populations HLJS and LNF occur in the transition zone of the mountain-plain area, where the rugged topography of the transition zone isolates populations and hampers gene exchange. In populations from the northwestern desert and grassland region, different population sizes and levels of gene flow may contribute to the observed varying levels of genetic variability among populations. There is an array of ridges and ravines in most areas of the Loess Plateau with the altitude range between 1,000 and 2,000. Steep slopes and ravines divide the ssp. sinensis forest into discrete fragmented populations with different sizes and block gene exchange. Hence variable levels of genetic diversity occur.
Genetic analyses revealed a high level of genetic differentiation among populations of ssp. sinensis based on ISSR (Φpt = 0.470). The estimate is close to the values reported in earlier RAPD and ISSR analyses in ssp. sinensis (F st = 0.418, Tian et al. 2004b) and other subspecies belonging to H. rhamnoides (G st = 0.459, Chen et al. 2010). Comparably, the cpSSR markers revealed high genetic differentiation for regions (Φrt = 0.309) and for populations (Φpt = 0.602). The existence of disjunct regions with distinct nuclear DNA band patterns and cpDNA haplotypes (e.g. IMO and SCW) suggests that large areas of unsuitable habitat (>100 km) impose significant barriers to pollen and seed dispersal. It is therefore likely that the genetic structure observed has been affected by habitat fragmentation. According to Heuertz et al. (2003), gene flow mediated by pollen and seed play determinant roles in the establishment of genetic structure. Previous research shows that the distance of pollen dispersal in H. rhamnoides is less than 20 m with 60–90% falling within 12 m (Tian et al. 1993; Wang et al. 1989). Long-distance dispersal by birds between isolated populations is expected to be rare, in view of the high mountains between populations of ssp. sinensis. For neutral genes, N m values below unity indicate that genetic drift is a major factor affecting population structure (Hartl and Clark 1989). The indirect estimates of gene flow based on both types of marker in ssp. sinensis suggest that levels of gene flow are not high enough to counteract differentiation due to genetic drift.
Isolation-by-distance was observed in the investigated populations of ssp. sinensis but there was no extreme barrier to dispersal throughout the area of our study. Given the low dispersal ability and gene flow observed, some breakdown in dispersal has occurred. The lack of isolation by distance within regions 1 and 2 further suggests some additional breakdown in dispersal within each region. At present, we are unable to explain this puzzle and offer two possible explanations, which are not mutually exclusive. First, our results confirmed the pathway of the earlier colonization events in ssp. sinensis. According to Lian and Chen (1992), this subspecies originated in the eastern Himalayas and subsequently dispersed to the southwestern region of the Greater Khingan Mountains along the eastern and northeastern marginal zone of the Qinghai-Tibetan Plateau and Loess plateau.
Again, chance events during expansion after earlier colonizations of ssp. sinensis and habitat fragmentation may explain the lack of isolation by distance within regions. The lack of correlation between geographical and genetic distances within the alpine vegetation region of the Qinghai-Tibetan Plateau could be because of multiple independent colonization events or sub-differentiation during expansion. The gene flow within the northwestern desert and grassland region is mainly because of seed dispersal, which may in part explain the lack of isolation by distance. During cold winters, when snow covers most other plants, fruits of spp. sinensis become the main food source for many birds in the northwestern desert and grassland region. Birds feeding on ssp. sinensis are likely to move irregularly among the isolated populations in the landscape, as seed resources on any one site become quickly exhausted, and consequent gene flow effects on the genetic structure are expected. Data collected from fruit foraging patterns of frugivorous birds also indicate that seed dispersal via birds’ digestion processes plays a vital role in renewal and maintenance of the community of ssp. sinensis (Lu et al. 2006).
Regional differentiation between the northwestern desert and grassland region and the alpine vegetation region of the Qinghai-Tibetan Plateau was observed. This result may be explained largely by the ecology and biology of reproduction of ssp. sinensis. The northwestern region of China belongs to the Loess plateau, where topography and climate conditions are more uniform than those in the northeastern humid forest region and the alpine vegetation region of the Qinghai-Tibetan Plateau. Uniform landscape and climate conditions tend to reduce genetic differentiation among populations. The northeastern humid forest region and the alpine vegetation region of the Qinghai-Tibetan Plateau have extremely complex topography and variable climatic conditions, which consequently, leads to genetic difference among populations. Additionally, morphological traits provide some support to the clustering pattern. As a result of adaptation to dry and barren habitats, plants occurring in the northwestern region are usually shrublike and have small, prickly fruits. Given the more suitable conditions (heat and rainfall), plants of ssp. sinensis in the alpine vegetation region and the northeastern humid forest region are trees and/or hemi-arbor and have big, succulent fruits. Therefore, the adaptation of populations of ssp. sinensis to the local environment in the three regions, as Lian et al. (2000) suggested, may account for the observed population structures and relationships, given that the habitats of ssp. sinensis vary greatly across its distribution.
The overall conclusions of this study are that populations of ssp. sinensis have a relatively low level of genetic diversity and significant genetic differentiation exists among populations and regions. Complex topography and heterogeneous climatic conditions throughout the range of ssp. sinensis could explain habitat fragmentation and restricted size of populations. Reduced gene flow between populations and increased genetic drift within populations were probably the main factors responsible for the genetic structure revealed in our samples. Our results have a number of implications for the management and conservation of ssp. sinensis. Conservation measurements should be focused on the conservation of regional populations in or ex situ. In a situation of limited management capacity of germplasm conservation in situ, the advised approach is to establish seed orchards of ssp. sinensis based on seed harvesting and collection of vegetative tissue. Using populations with high genetic variation and uniqueness, on the basis of both nuclear and chloroplast markers, as seed reservoirs is of great benefit to maintaining a wide genetic base. This would guarantee the maintenance of most of the subspecies’ genetic variation, including uncommon and unique alleles. Further advanced research on ssp. sinensis, especially investigations on eco-physiological, demographic, and genetic characteristics, will facilitate future programs of sustainable management and conservation of H. rhamnoides ssp. sinensis.
References
Aitzetmuller K, Xin Y (1999) Sea buckthorn and sea buckthorn oils—recent developments in China and central Asia. Nahrung 43:228–232
Avdeyev VI (1983) Novaya taksonomiya roda oblepikha: Hippophae L. Izv. Akad. Nauk. Tadzh. SSR. Biol. Nauk. 4:11–17
Bartish IV, Jeppsson N, Nybom H (1999) Population genetic structure in the dioecious pioneer plant species Hippophae rhamnoides investigated by random amplified polymorphic DNA (RAPD) markers. Mol Ecol 8:791–802
Bartish IV, Jeppsson N, Bartish GI, Lu R, Nybom H (2000) Inter- and intraspecific genetic variation in Hippophae (Elaeagnaceae) investigated by RAPD markers. Plant Syst Evol 225:85–101
Bartish IV, Jeppsson N, Nybom H, Swenson U (2002) Phylogeny of Hippophae (Elaeagnaceae) inferred from parsimony analysis of chloroplast DNA and morphology. Syst Bot 27:41–54
Bryan GJ, McNicoll J, Ramsay G, Meyer RC, De Jong WS (1999) Polymorphic simple sequence repeat markers in chloroplast genomes of Solanaceous plants. Theor Appl Genet 99:859–867
Chen G, Wang Y, Zhao C, Korpelainen H, Li C (2008) Genetic diversity of Hippophae rhamnoides populations at varying altitudes in the Wolong Natural Reserve of China as revealed by ISSR markers. Silvae Genet 57:29–36
Chen W, Su X, Zhang H, Sun K, Ma RJ, Chen XL (2010) High genetic differentiation of Hippophae rhamnoides ssp. yunnanensis (Elaeagnaceae), a plant endemic to the Qinghai-Tibet Plateau. Biochem Genet 48:565–576
Cheng YJ, Meng HJ, Guo WW, Deng XX (2006) Universal chloroplast primer pairs for simple sequence repeat analysis in diverse genera of fruit crops. J Hortic Sci Biotechnol 81:132–138
Excoffier L, Guillaume L, Schneider S (2007) ARLEQUIN (version 3.11): an integrated software package for population genetics data analysis. Computational and Molecular Population Genetics Lab (CMPG). Zoological Institute, University of Berne, Switzerland
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Hartl DL, Clark AG (1989) Principles of population genetics. Sinauer, Sunderland
Heuertz M, Vekemans X, Hausman JF, Palada M, Hardy OJ (2003) Estimating seed vs. pollen dispersal from spatial genetic structure in the common ash. Mol Ecol 12:2483–2495
Huff DR, Peakall R, Smouse PE (1993) RAPD variation within and among natural populations of outcrossing buffalo grass Buchloe dactyloides (Nutt) Engelm. Theor Appl Genet 86:927–934
Isagi Y, Tateno R, Matsuki Y, Hirao A, Watanabe S, Shibata M (2007) Genetic and reproductive consequences of forest fragmentation for populations of Magnolia obovata. Ecol Res 22:382–389
Jump AS, Penuelas J (2006) Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc Natl Acad Sci USA 103:8096–8100
Kettle CJ, Hollingsworth PM, Jaffre T, Moran B, Ennos RA (2007) Identifying the early genetic consequences of habitat degradation in a highly threatened tropical conifer, Araucaria nemorosa Laubenfels. Mol Ecol 16:3581–3591
Li C, Xu G, Zang R, Korpelainen H, Berninger F (2007) Sex-related differences in leaf morphological and physiological responses in Hippophae rhamnoides along an altitudinal gradient. Tree Physiol 27:399–406
Lian YS (1988) New discoveries of the genus Hippophae L. (Elaeagnaceae). Acta Phytotax Sin 26:235–237 in Chinese
Lian YS, Chen XL (1992) The ecogeographical distribution of Hippophae rhamnoides subsp. sinensis and its phytogeographical significance. Acta Phytotax Sin 30:349–355 in Chinese
Lian YS, Chen XL (1996) The systematic classification of the genus Hippophae. Hippophae 9:15–24
Lian YS, Chen XL, Wang F (1997) New discoveries of the genus Hippophae L. II. In: Lu S, Li M, Hu J, Liu S (eds) Worldwide research and development of sea buckthorn. China Science and Technology Press, Beijing, pp 60–65
Lian Y, Chen X, Lian H (1998) Systematic classification of the genus Hippophae L. Seabuckthorn Res 1:13–23
Lian YS, Lu SG, Xue SK, Chen XL (2000) Biology and chemistry of the genus Hippophae. Gansu Science and Technology Press, Lanzhou, pp 1–226 (in Chinese)
Liu SW, He TN (1978) The genus Hippophae from Qing-Zang plateau. Acta Phytotax Sin 16:106–108 (in Chinese)
Lu R (1992) Sea buckthorn—a multipurpose plant species for fragile mountains. ICIMOD Occasional Paper No. 20. Kathmandu, Nepal
Lu R (1997) Eco-geographical distribution of sea buckthorn and prospects of International cooperation. In: Lu S, Li M, Hu J, Liu S (eds) Worldwide research and development of sea buckthorn. China Science and Technology Press, Beijing, pp 123–129
Lu XW, Sun K, Ma RJ, Zhang H, Su X, Wang ML (2006) Fruits foraging patterns and seed dispersal effect of frugivorous birds on Hippophae rhamnoides sinensis. Front Biol China 3:318–322
Lu Z, Wang Y, Zhang X, Korpelainen H, Li C (2009) Genetic variation of isolated Picea balfouriana populations from the southeast of the Qinghai-Tibet Plateau. Ann For Sci 66:607 (p1–10)
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Miller M (2000) TFPGA (version 1.3): tools for population genetic analysis, a windows program for the analysis of allozyme and molecular population genetic data. Department of Biological Sciences, Northern Arizona University
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
O’Connell LM, Mosseler A, Rajora OP (2006) Impacts of forest fragmentation on the mating system and genetic diversity of white spruce (Picea glauca) at the landscape level. Heredity 97:418–426
Peakall R, Smouse PE (2006) GENALEX 6: Genetic Analysis in Excel. Population genetic software for teaching and research. Australian National University, Canberra
Peakall R, Smouse PE, Huff DR (1995) Evolutionary implications of allozyme and RAPD variation in diploid populations of dioecious buffalograss Buchloe dactyloides. Mol Ecol 4:135–147
Rousi A (1965) Observations on the cytology and variation of European and Asiatic populations of Hippophae rhamnoides. Ann Bot Fennici 2:1–18
Rousi A (1971) The genus Hippophae L. A taxonomic study. Ann Bot Fennici 8:177–227
Ruan CJ, Li DQ (2002) Analysis on the community characteristics of Hippophae rhamnoides L. plantation and water and nutrition of woodland in Loess Hilly Region. J Appl Ecol 13:1061–1064 (in Chinese)
Servettaz C (1909) Monographie des Eleagnacees. Beih Bot Centralbl 25:1–40
Sheng HM, An LZ, Chen T, Xu SJ, Liu GX, Zheng XL, Pu LL, Liu YJ, Lian YS (2006) Analysis of the genetic diversity and relationships among and within species of Hippophae (Elaeagnaceae) based on RAPD markers. Plant Syst Evol 260:25–37
Sun K, Chen X, Ma R, Li C, Wang Q, Ge S (2002) Molecular phylogenetics of Hippophae L. (Elaeagnaceae) based on the internal transcribed spacer (ITS) sequences of nrDNA. Plant Syst Evol 235:121–134
Sun K, Chen W, Ma RJ, Chen XL, Li A, Ge S (2006) Genetic variation in Hippophae rhamnoides ssp. sinensis (Elaeagnaceae) revealed by RAPD markers. Biochem Genet 44:186–197
Swenson U, Bartish IV (2003) Taxonomic synopsis of Hippophae (Elaeagnaceae). Nordic J Bot 22:369–374
Tang X, Tigerstedt PMA (2001) Variation of physical and chemical characters within an elite sea buckthorn (Hippophae rhamnoides L.) breeding population. Sci Horitic 88:203–214
Tian LC, Guan FL, Zhang MY (1993) The preliminary study of flowering features in Hippophae rhamnoides L. ssp. sinensis Rousi and artificial hybridization. Hippophae 6:29–33
Tian C, Lei Y, Shi S, Nan P, Chen J, Zhong Y (2004a) Genetic diversity of sea buckthorn (Hippophae rhamnoides) populations in northeastern and northwestern China as revealed by ISSR markers. New For 27:229–237
Tian C, Nan P, Shi S, Chen J, Zhong Y (2004b) Molecular genetic variation in Chinese populations of three subspecies of Hippophae rhamnoides. Biochem Genet 42:259–267
Wang ZK, Guo BP, Yan LY (1989) Study on features of pollen dispersal of sea buckthorn in mountains. In: Proceedings of International Symposium on Sea Buckthorn, Beijing, pp 186–189
Weising K, Gardner RC (1999) A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome 42:9–19
Wright S (1969) Evolution and genetics of populations, vol 2. The theory of gene frequencies. University of Chicago Press, Chicago
Wright S (1978) Evolution and genetics of populations, vol 4. Variability within and among populations. University of Chicago Press, Chicago
Yang Y, Yao Y, He H (2008) Influence of ambient and enhanced ultraviolet-B radiation on the plant growth and physiological properties in two contrasting populations of Hippophae rhamnoides. J Plant Res 121:377–385
Yao Y, Tigerstedt PMA (1993) Isozyme studies of genetic diversity and evolution in Hippophae. Genet Resour Crop Evol 40:153–164
Yeh FC, Yang RC, Boyle T (1997) POPGENE (version 1.32): software Microsoft Windows-based freeware for population genetic analysis. University of Alberta, Canada
Zhao H, Zhu C, Gao C, Li H, Liu Z, Sun W (1991) Geographical variation of fruit traits of the Chinese sea buckthorn and selection of provenances for fruit use. Hippophae 4:15–18
Acknowledgments
The research was supported by the Program of “Knowledge Innovation Engineering” of the Chinese Academy of Sciences (no. KSCX2-YW-Z-1019) and the Program of “the Light Foundation” of the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Y., Jiang, H., Peng, S. et al. Genetic structure in fragmented populations of Hippophae rhamnoides ssp. sinensis in China investigated by ISSR and cpSSR markers. Plant Syst Evol 295, 97–107 (2011). https://doi.org/10.1007/s00606-011-0466-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-011-0466-7