
Abstract
Parsimony-based phylogenetic analyses of the genus Guizotia were undertaken based on DNA sequence data from the following chloroplast DNA (cpDNA) regions: trnT-trnL, trnL-trnF, trnY-rpoB, trnC-petN, psbM-trnD and rps16-trnQ intergenic spacers, trnL, rps16 and matK-5′trnK introns and matK gene. Out of the 26 primers used in this study, 14 were newly designed. The study was conducted to determine (1) the closest relative of Guizotia abyssinica, (2) the taxonomic status of some Guizotia taxa and (3) the subtribal placement of Guizotia in the tribe Heliantheae. The analyses of the sequence data showed that G. abyssinica, G. scabra ssp. scabra, G. scabra ssp. schimperi and G. villosa are phylogenetically closely related. However, G. scabra ssp. schimperi appeared as the most closely related taxon to G. abyssinica. Based on this phylogenetic analysis, we suggest that the two subspecies of G. scabra are better treated as separate species. The analysis also clearly demonstrated that “Chelelu” and “Ketcha” are distinct Guizotia species. The trnT-trnL and trnL-trnF intergenic spacer-based phylogenetic analysis of various subtribes of the tribe Heliantheae strongly supports the placement of the genus Guizotia within the subtribe Milleriinae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Guizotia Cass. is a small but economically important genus that belongs to the family Asteraceae, tribe Heliantheae. Baagøe (1974) circumscribed the genus to six species viz.: G. abyssinica (L. f.) Cass., G. arborescens I. Friis, G. jacksonii (S. Moore) J. Baagøe, G. scabra (Vis.) Chiov. ssp. scabra, G. scabra (Vis.) Chiov. ssp. schimperi (Sch. Bip. in Walp.) J. Baagøe, G. villosa Sch. Bip. in Walp and G. zavattarii Lanza in Chiov. & al. However, the taxonomic status of G. scabra ssp. scabra and G. scabra ssp. schimperi has been questioned based on the existing evidence (Hiremath and Murthy 1992; Hiremath et al. 1992; Dagne 1995; Geleta et al. 2007). After Baagøe’s (1974) taxonomic revision, two new populations of Guizotia, named “Chelelu” and “Ketcha,” were discovered in Ethiopia by Dagne (1995). These populations are morphologically distinct from each other and do not exactly match any of the recognized taxa of the genus Guizotia (Dagne 1995, 2001). Their taxonomic status has not been determined, although we treated them as separate “taxa” for the sake of simplicity.
Guizotia is a diploid Afromontane endemic genus with 2n = 30 chromosomes (e.g., Dagne 1995), which is native to tropical Africa with most of the taxa restricted to East Africa, and with the highest concentration of species in Ethiopia (Baagøe 1974). Guizotia species show narrow endemism, except Guizotia scabra ssp. scabra, which extends from East Africa to Cameroon and the Nigerian highlands with a distributional gap in the Congolian rainforest. This narrow endemism is shown by G. arborescens (southwest of Ethiopia and around the borders of Sudan and Uganda), G. scabra ssp. schimperi (native to the Ethiopian highlands), G. jacksonii (Aberdares, Mt. Kenya and Mt. Elgon in Kenya and Uganda), G. villosa (northern and northwestern part of the Ethiopian highlands) and G. zavattarii (southern Ethiopia and northern Kenya). The genus is comprised of erect and creeping, annual and perennial, herbaceous and shrubby members; all species are wild and/or weedy except G. abyssinica, which is cultivated mainly for its edible oil. Evidence suggests that G. abyssinica might have originated from G. scabra ssp. schimperi through selection and further cultivation (Baagøe 1974; Hiremath and Murthy 1988; Murthy et al. 1993; Dagne 1994, 1995, 2001). However, a firm exclusion of G. scabra ssp. scabra and G. villosa from being an ancestor to G. abyssinica still demands more data (Bekele et al. 2007).
Guizotia has been placed under different subtribes of the tribe Heliantheae by different authors. Consequently, the distinguishing morphological characteristics of the genus are not clear. Bentham (1873) placed the genus under the subtribe Coreopsidinae by suggesting its resemblance to some of the African forms of Coreopsis, without elaborating. After a century Baagøe (1974) suggested transferring the genus to the subtribe Verbesininae mainly due to laterally compressed achenes as opposed to dorsal compression. However, Stuessy (1977), after revising the tribe Heliantheae, maintained the genus within Coreopsidinae based on characteristics such as deeply divided leaves with opposite arrangement, scarious-margined outer phyllaries and orange-brown striae in several floral structures. Later, Robinson (1981) stated that the terete, striate achenes, the ornamented seed coats and the glanduliferous anther appendages are evidence against the placement of the genus within the Coreopsidinae. Consequently, he placed the genus under the subtribe Milleriinae based on close approximation of technical characters despite differences in habit and flower color. The placement of the genus under subtribe Milleriinae was also asserted by Karis (1993), who suggested that delimitation of Milleriinae has to be amended in order to clarify the limit between the Milleriinae and Melampodiinae. Schulz (1990) transferred an African Sigesbeckia species (S. somalensis S. Moore), a member of Milleriinae (Humbles 1972), into Guizotia, suggesting a close resemblance between Guizotia and Milleriinae.
Chloroplast DNA (cpDNA) sequence variation is widely used for systematics and phylogenetic inference at different taxonomic levels (e.g., Taberlet et al. 1991; Johnson and Soltis 1994; Liang and Hilu 1996; Hilu and Liang 1997; Bayer et al. 2002; Shaw and Small 2005; Crawford and Mort 2005). Taberlet et al. (1991) reported that the trnT-trnL and trnL-trnF intergenic spacers and trnL intron are useful for evolutionary studies at low taxonomic levels and, since then, these regions have been used extensively for phylogenetic studies. The matK gene has been widely used for infrafamilial phylogenetic inference, sometimes together with the matK-5′trnK and the 3′trnK-matK portion of the trnK intron (e.g., Johnson and Soltis 1994; Padgett et al. 1999; Wagstaff and Breitwieser 2004; Shaw et al. 2005). Based on pairwise sequence divergence analysis between Lactuca and Helianthus, Timme et al. (2007) identified the trnY-rpoB intergenic spacer as one of fast evolving cpDNA regions in Asteraceae. Similarly, trnC-trnD has been reported as one of fast evolving regions of cpDNA (e.g., Lee and Wen 2004; Shaw et al. 2005). The utility of this region, which includes trnC-petN and psbM-trnD intergenic spacers, has been demonstrated for phylogenetic studies at low taxonomic levels in flowering plants (Lee and Wen 2004). The intron of chloroplast gene rps16 has been used for phylogenetic studies in different families of flowering plants (e.g., Oxelman et al. 1997; Baker et al. 2000; Lee and Hymowitz 2001), although its infrageneric resolution was reported to be weak (Baker et al. 2000). The rps16-trnQ intergenic spacer has also been used for phylogenetic studies at a low taxonomic level (Pan et al. 2007).
The present study aimed to determine the phylogenetic relationship between various Guizotia species based on sequence data from the aforementioned cpDNA regions and thereby (1) to assess the validity of the previous suggestion about the origin of G. abyssinica and (2) to determine the taxonomic status of Chelelu, Ketcha and the two subspecies of G. scabra. Phylogenetic analysis of various Heliantheae species was another objective of this study in order to comment on the subtribal placement of the genus Guizotia within the tribe Heliantheae.
Materials and methods
Plant material and DNA extraction
Five Guizotia species, out of a total of six (Baagøe 1974), and two yet taxonomically unclassified Guizotia populations (Dagne 1995) were used in this study. The seed samples and voucher specimens of these taxa were collected from various regions in Ethiopia (Table 1). Voucher specimens are being described for the purpose of taxonomic revision of the genus at the Swedish University of Agricultural Sciences (SLU, Sweden) and will be submitted to Addis Ababa University Herbarium (Ethiopia). In this study, each taxon was represented by two to four samples. Seeds were grown in a greenhouse, and fresh leaves from 15-day-old plants were used for genomic DNA extraction. DNA was extracted by a modified CTAB procedure as described in Assefa et al. (2003). DNA quality and concentration were determined using the NanoDrop® ND-1000 spectrophotometer (Saveen Werner, Sweden).
PCR and sequencing
Target DNA regions (Table 2) were amplified using a GeneAMP PCR system 9700 thermocycler with the following temperature profiles: initial 3 min denaturing at 94°C and final 7 min extension at 72°C with 30 intervening cycles of 1 min denaturing at 94°C, 1 min primer annealing at 48°C and 2 min primer extension at 72°C. The whole trnK intron including the matK gene was amplified using primers MG1 and MG15 (Liang and Hilu 1996), while the trnT-trnL intergenic spacer was amplified using primers a (B48557) and b (A4929; Taberlet et al. 1991). The trnL intron and the trnL-trnF intergenic spacer were amplified as a single fragment using primers c (B49317) and f (A50272) (Taberlet et al. 1991; Table 2). The matK gene was sequenced from the 5′ end to near the 3′ end (83%) using sequencing primers 1110R, 1240R, 1408F, 1541R and 1694F, respectively (Bayer et al. 2002; Table 2). Primer 1110R sequenced about 41% of the matK-5′trnK portion of the trnK intron and the 5′most portion of the matK gene. A complete sequence of the trnT-trnF intergenic spacer was obtained by sequencing both strands using primer a (B48557) and primer b (A4929). The whole length of the trnL intron and the trnL-trnF intergenic spacer was sequenced using primers c (B49317) and e (B49873), respectively (Table 2).
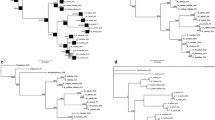
Fourteen new primers were designed to amplify and sequence the rps16 intron and the rps16-trnQ, trnY-rpoB, trnC-petN and psbM-trnD intergenic spacers (Fig. 1; Table 2) using the Primer3 primer designing program (Rozen and Skaletsky 2000). These regions were selected based on published reports and the degree of sequence divergence between G. abyssinica and Helianthus annuus (Genbank accession numbers EU549769 and DQ383815, respectively). The primers were designed to the conserved regions of the two sequences and named based on their 5′ position (forward primers) and 3′ position (reverse primers) in the G. abyssinica cpDNA. About 58% of the rps16 intron and 89% of the rps16-trnQ intergenic spacer were amplified in three segments with a combination of six primers (Fig. 1a; Table 2). Primers 5700R, 5985F and 6538F were used to sequence these regions. The whole trnC-petN intergenic spacer was amplified and sequenced by primers 9351F and 10175R (Fig. 1b; Table 2). Similarly, primers 10771F and 11495R were used to amplify the entire psbM-trnD intergenic spacer. Primer 10771F was used to sequence this spacer. The trnY-rpoB intergenic spacer was amplified in two segments with a combination of four primers (Fig. 1c; Table 2). Three of the four primers (11680F, 12141F and 12141R) were used to sequence this region. The alignment of DNA sequences from G. abyssinica, H. annuus and Lactuca sativa L. (Genbank accession number AP007232) showed that the primer annealing sites for 9 of the 14 newly designed primers are conserved in L. sativa as well. The sites for the remaining five primers are different at 1 or 2 nucleotide positions (see Table 2).

Schematic representation of the relative positions of the newly designed primers (arrows) used in this study within the rps16-trnQ, trnC-trnD and trnY-rpoB regions of cpDNA (a–c). The size of coding regions is represented proportionally (note: rpoB gene is represented partially). Except for the trnY-rpoB intergenic spacer, the size of the noncoding regions are shortened (broken lines) and thus are not proportional to the size of the coding regions and trnY-rpoB intergenic spacer
The PCR products were purified with a QIAquick PCR purification kit (Qiagen GmbH, Germany) using a microcentrifuge according to the manufacturer’s instructions. We employed cycle sequencing using the ABI PRISM® BigDye® Terminator v3.1 cycle sequencing kit (Applied Biosystems) for trnT-trnL, trnL-trnF intergenic spacers, trnL and matK-5′trnK introns and matK gene. Cycle sequencing was performed in a final volume of 10 μl containing 1× BigDye sequencing buffer, 2 μ1 of BigDye RR-100 cycle sequencing mix, 5 pmol of sequencing primer and 50–100 ng of purified double-stranded PCR product. Cycle sequencing reactions were carried out at 95°C for 30 s, 50°C for 15 s and 60°C for 4 min for 25 cycles. The product of the sequencing reaction was precipitated using a mixture of 29 μl of 96% ethanol and 1 μl of 3 M sodium acetate (pH 5.2) and centrifuged at 13,200 rpm for 30 min. The precipitate was washed with 150 μl of 70% ethanol, air dried and submitted to BM labbet (http://www.BMlabbet.se) for sequencing with an ABI PRISM® 3100 genetic analyzer (Applied Biosystems). In the case of the rps16 intron, rps16-trnQ, trnC-petN, psbM-trnD and trnY-rpoB intergenic spacers, 8 μl of purified PCR product (50–100 ng) was mixed with 2 μl of 5 μM sequencing primers and sent to the sequencing facility at the University of Oslo (http://www.bio.uio.no/ABI-lab/), where DNA sequencing for these regions was done. The nucleotide sequences of the ten cpDNA regions from representative samples of each Guizotia taxon were submitted to the nucleotide sequence database, and their Genbank accession numbers are given in Table 3.
Outgroup and additional ingroup taxa selection
Two data matrices were analyzed in this study. The first data matrix contains all ten cpDNA regions sequenced. In this data matrix, Helianthus annuus (subtribe Helianthinae) was used as an outgroup to analyze the phylogenetic relationship between Guizotia species. The second data matrix, which contains sequences from Guizotia and 29 additional Asteraceae species, was used to analyze the phylogenetic relationship between the genus Guizotia and various genera in the tribe Heliantheae and other closely related tribes (Table 3; Fig. 3). These species were selected based on their taxonomic position within Asteraceae, availability of their sequences in the nucleotide data base and the extent of DNA sequence divergence between them and Guizotia. Except for Eupatorium cannabinum (tribe Eupatorieae), Stevia rebaudiana (tribe Eupatorieae), Inula Britannica (tribe Inuleae) and Hieracium pilosella (Cichorioideae; tribe Cichorieae), the remaining 25 species belong to tribe Heliantheae (Table 3; Fig. 3). The subtribal nomenclature of the Heliantheae species is based on the treatment of the tribe by Robinson (1981). All non-Guizotia DNA sequences were retrieved from the National Center for Biotechnology Information (NCBI) database. The Genbank accession numbers of these sequences are given in Table 4. In the later analysis, where 28 of the 29 species were used as additional ingroup taxa and H. pilosella was used as an outgroup, only sequence data from the trnT-trnL and trnL-trnF intergenic spacers were used.
Sequence alignment and data analysis
Sequences were edited using BIOEDIT version 7.0.5 (Hall 2005), and the quality of the sequences was visually inspected using SEQUENCE SCANNER version 1.0 (Applied Biosystems). Sequences were aligned using CLUSTAL X version 1.81 (Thompson et al. 1997), followed by manual adjustment. This phylogenetic analysis was carried out using PAUP* 4.0 Beta 10 (Swofford 2000). Gaps created during sequence alignment were treated as missing data, but to exploit the utility of indel positions, parsimony informative indels (PII) were scored with the simple indel coding method of Simmons and Ochoterena (2000). Phylogenetic analyses were conducted based on both unweighted and weighted characters. In the latter case, the first codon positions of the matK gene weighed twice the weight of all other characters in the data set and transversions cost twice transitions (see, for example, Sankoff et al. 1976; Bofkin and Goldman 2007). Trees were constructed using the maximum parsimony optimality criterion. Heuristic searches were performed through random sequence addition with 1,000 replicates using various branch swapping and branch length optimization options. Both strict and bootstrap 50% majority rule consensus trees were constructed and clade support was estimated using bootstrap values (1,000 bootstrap replicates with 100 random additions).
Results
Sequence data description
In this study, full sequence length was obtained for the trnT-trnL, trnL-trnF, trnC-petN and psbM-trnD intergenic spacers and the trnL intron. More than 80% of the matK gene and rps16-trnQ intergenic spacer sequences were also obtained (Table 5). Significant length variations between Guizotia species were detected in the trnT-trnL intergenic spacer, ranging from 582 (G. zavattarii) to 634 (Ketcha) nucleotides. The sequence length variation was mainly due to the number of tandem repeats of “TATAGAAGATGAAAGAAGATAGA,” which were four, three and two for Ketcha, G. arborescens and the rest of the taxa, respectively. In this spacer, gaps accounted for 7.8% of the total aligned length. Sequence length variation was less than 10 nucleotides in the other regions (Table 5). Gaps in the rps16 intron and the trnC-petN intergenic spacer were mainly due to length variation in mononucleotide repeats (“C”s, “A”s and “T”s). Microsatellites of 10–17 “C”s in the rps16 intron and 9–16 “A”s in the trnC-petN intergenic spacer are especially interesting in that they differentiate most of the Guizotia species. A six-nucleotide-long indel unique to G. abyssinica was also obtained in the rps16 intron. Similarly, seven- and eight-nucleotide-long unique indels were obtained in Ketcha and Chelelu, respectively, in the trnY-rpoB intergenic spacer. No indels were evident in the trnL and matK-5′trnK introns, the matK gene and the psbM-trnD spacer aligned sequences. Four variable sites were revealed among the Guizotia taxa within the matK gene, two resulting in synonymous and two in non-synonymous amino acid substitutions. In the case of non-synonymous substitutions, exchanges of asparagine with lysine and valine with isoleucine were observed.
The relative utility of the cpDNA regions for phylogenetic studies within Asteraceae
The relative utility of the ten cpDNA loci used in this study was assessed by comparing the number and percentage of parsimony informative characters from each region. The comparison was made based on the aligned sequences of G. abyssinica, H. annuus and L. sativa (Table 5). The rps16-trnQ intergenic spacer provided the highest number of parsimony informative characters (33) followed by trnC-petN (31). The percent parsimony informative characters (PPIC) ranged from 0.009 (trnL intron) to 0.039 (trnC-petN intergenic spacer). Thus, trnC-petN and rps16-trnQ are seem to be evolving faster than the other regions. The least parsimony informative characters were obtained from trnL and matK-5′trnK introns. Generally, introns provided fewer parsimony informative characters than the intergenic spacers. The degree of informativeness of variable characters was estimated based on the percent parsimony informative variable sites (PPIVC; Table 5). The highest and lowest PPIVC were obtained for rps16-trnQ (27%) and trnY-rpoB (9.7%) intergenic spacers, respectively. Generally, the less commonly used cpDNA regions (trnY-rpoB, rps16-trnQ, trnC-petN and psbM-trnD intergenic spacers) provided more parsimony informative characters than the more commonly used regions (trnT-trnL-trnF regions and matK gene) and therefore should be preferred for phylogenetic studies at a low taxonomic level, at least within Asteraceae.
Phylogenetic inference of the genus Guizotia
A total of 6,722 aligned length of nucleotides (including the outgroup species; H. annuus) were used for analysis of phylogenetic relationship between Guizotia species. Out of the 408 variable characters, 46 characters were parsimony informative, excluding parsimony informative indels. Twenty-four parsimony informative indels were recorded in this data set. The phylogenetic analysis of this sequence data (unweighted characters; without including scored PII) using a tree-bisection-reconnection (TBR) branch swapping algorithm resulted in 438 equally parsimonious trees [tree length = 438; consistency index (CI) = 0.94; homoplasy index (HI) = 0.05; retention index (RI) = 0.89]. Phylogenetic analyses conducted based on unweighted and weighted characters resulted in identical tree topology.
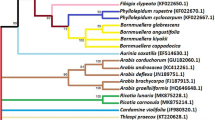
The inclusion of the scored parsimony informative indels (PII) into the analysis did not change the tree topology, although it causes a slight change in clade support. The bootstrap 50% majority rule consensus tree (1,000 bootstrap replicates with 100 random additions; MaxTrees = 500), which is similar to a strict consensus tree, presented in Fig. 2 was constructed based on weighted characters without including scored PII. The phylogenetic analysis of the combined data from trnT-trnL and trnL-trnF intergenic spacers was based on the total aligned length of 888 nucleotides. Out of the 301 variable characters in this sequence, 122 characters were parsimony informative. The phylogenetic analysis of the sequence data (weighted characters; excluding PII) using a TBR branch swapping algorithm resulted in 780 equally parsimonious trees (tree length = 780; CI = 0.75; HI = 0.25; RI = 0.64). The strict consensus tree of 10,000 trees was similar to the bootstrap 50% majority rule consensus tree (1,000 bootstrap replicates with 100 random additions; MaxTrees = 10,000; data not shown). Strict consensus trees were constructed both with and without scored PII. The former offers greater resolution within Guizotia; both results, however, are congruent in their placement of Guizotia within Milleriinae. The strict consensus tree of 324 trees constructed by including scored PII is given in Fig. 3 with clade support from the bootstrap 50% majority rule consensus tree given above the branches.

The bootstrap 50% majority rule consensus tree generated from a phylogenetic analysis of DNA sequence data from ten cpDNA regions (scored parsimony informative indels excluded). Bootstrap values greater than 50 are given above the branches

Strict consensus of 324 trees from the analysis of trnT-trnL and trnL-trnF intergenic spacer sequences of Guizotia, an additional 25 species of tribe Heliantheae, 2 species of tribe Eupatorieae and 1 species of tribe Inuleae (scored parsimony informative indels were included). Hieracium pilosella was used as an outgroup. Numbers above the branches are bootstrap values from a bootstrap 50% majority rule consensus tree
Discussion
Chloroplast DNA loci, which are often assumed to be uniparentally inherited and non-recombining, have been extensively used for systematics and phylogenetics. However, the rate of evolution of the cpDNA genome is slower than that of the nuclear genome. Correspondingly, the cpDNA regions that have been used for phylogenetic studies are less variable than the most extensively used nuclear loci, internal transcribed spacers of nuclear ribosomal DNA (ITS) (e.g., Small et al. 2004; Mort et al. 2007). It is often difficult to obtain adequate resolution of any phylogeny of closely related taxa using few cpDNA loci due to the low number of phylogenetically informative characters. Hence, the practice of acquiring sequence data from several loci is a proven means of acquiring a better resolved phylogeny (Mort et al. 2007).
In this study, we investigated ten cpDNA regions to resolve the phylogeny of the genus Guizotia. Five of the ten loci were amplified and sequenced using newly designed primers. The sequences of most of these primers are conserved in Guizotia, Helianthus and Lactuca, and thus can be used in a wide range of Asteraceae species. Some primers may need a modification at 1–2 nucleotide positions in order to be used in species distantly related to Guizotia (see Table 2). Generally, there is a trend for some cpDNA regions to be phylogenetically more informative than others (Shaw et al. 2005). However, it is also likely that each family or major lineage has a different degree of variability in different cpDNA regions (Timme et al. 2007). For example, Shaw et al. (2005) examined sequence variations of 21 cpDNA noncoding regions and reported that the trnT-trnL intergenic spacer provides a higher number of potentially informative characters than most of the regions they examined, which includes the psbM-trnD intergenic spacer. However, in this study the psbM-trnD intergenic spacer provided more parsimony informative characters than the trnT-trnL intergenic spacer, and therefore the psbM-trnD intergenic spacer should be preferred over the trnT-trnL intergenic spacer, at least in Asteraceae. Out of the ten cpDNA regions used in this study, the rps16-trnQ and trnC-petN intergenic spacers are the top two in terms of their phylogenetic utilities in Asteraceae and can provide better phylogenetic structure at low taxonomic levels. The phylogenetic utility of the rps16-trnQ spacer at a low taxonomic level has also been commented upon recently in the family Apiaceae (Downie et al. 2008).
Phylogenetic relationship between Guizotia species
The phylogenetic analysis of 6,722-bp-long aligned sequences from ten cpDNA regions for eight Guizotia taxa generated four major clades with a moderate to high bootstrap support (Fig. 2). The resulting parsimony trees were well resolved and comparable to those generated using nuclear ITS sequences (Bekele et al. 2007). The first clade (clade I) comprises G. abyssinica, G. scabra ssp. scabra, G. scabra ssp. schimperi and G. villosa. This clade was also observed during phylogenetic analyses of several subsets of the entire data set, although within the clade resolution was low. It is interesting to note that these taxa share complete sequence similarity in the matK gene, but they differ at least at two of the four parsimony informative characters from the other Guizotia species. The result suggests a close phylogenetic relationship between these taxa. However, two subclades (subclades A and B) were formed under clade I, which answered the two major questions of this study: the closest relative of G. abyssinica and the taxonomic status of the two subspecies of G. scabra. Subclade A comprises G. abyssinica and G. scabra ssp. schimperi, whereas subclade B contains G. scabra ssp. scabra and G. villosa (Fig. 2). Dagne (1995) obtained the same grouping based on chromosome morphology. Further, these subclades were also recovered during phylogenetic analysis of the genus based on ITS data (Bekele et al. 2007). However, the present result is different in that G. abyssinica appeared as a single separate group closely related to G. scabra ssp. schimperi. The grouping of one of the G. abyssinica samples together with G. villosa and G. scabra ssp. scabra in the ITS-based analysis (Bekele et al. 2007) might be the result of gene flow and genetic recombination in the nuclear ITS region, as the two subspecies of G. scabra, G. villosa and G. abyssinica are cross-compatible.
This study clearly showed that G. scabra ssp. schimperi is the closest relative of G. abyssinica. Evidence regarding the origin and domestication of crop plants can be generated from various sources, such as history, linguistics, archeobotany, comparative morphology, phytogeography, cytogenetics and molecular biology. Although archeobotanical evidence regarding the origin and domestication of G. abyssinica is lacking, based on morphological, phytogeographical and cytological evidence, G. abyssinica was suggested to originate from G. scabra ssp. schimperi through selection and further cultivation (Baagøe 1974; Hiremath and Murthy 1988; Murthy et al. 1993; Dagne 1994, 1995, 2001). Morphologically, G. abyssinica most resembles G. scabra ssp. schimperi (Baagøe 1974). The percentage of crossability and genome homology between these taxa and the mean pollen fertility of their hybrid were higher than that obtained between G. abyssinica and the other Guizotia species (Hiremath and Murthy 1992; Murthy et al. 1993; Dagne 1994). Given this evidence, the absence of the wild form of G. abyssinica, and the result of this particular study, it is safe to conclude that G. scabra ssp. schimperi is the progenitor of G. abyssinica.
Despite their significant morphological differences, Baagøe (1974) united G. scabra and G. schimperi and renamed them as G. scabra ssp. scabra and G. scabra ssp. schimperi, respectively. However, after analyzing their karyotypes, Hiremath and Murthy (1992) and Dagne (1995) suggested that these taxa should be considered as separate species. Similarly, Hiremath et al. (1992) reported that the two subspecies differ distinctly in their genome size and, based on this, advised treating them as independent species. Analysis of genetic relationships between various Guizotia species based on molecular marker data (Geleta et al. 2007) also supports the treatment of these taxa as independent species. In this phylogenetic analysis, G. scabra ssp. scabra and G. scabra ssp. schimperi were under different subclades (Fig. 2). G. scabra ssp. schimperi is more closely related to G. abyssinica than to G. scabra ssp. scabra, and G. scabra ssp. scabra is more closely related to G. villosa than to G. scabra ssp. schimperi. Thus, given the previous evidence and this study, G. scabra ssp. schimperi and G. scabra ssp. scabra should be treated as separate species.
The Chelelu and Ketcha populations were discovered and considered as new Guizotia species in 1995 (see Dagne 1995). The study, based on cross-compatibility and chromosome pairing of the hybrids between Chelelu and other Guizotia species (Dagne 2001), strengthens this consideration. In this phylogenetic analysis, these taxa form their own clade, being nested between known Guizotia species (Clade II; Fig. 2), which confirms the previous results obtained using molecular markers (Geleta et al. 2007), cytogenetics and crossing experiments (Dagne 1995, 2001). Our results also suggest that these taxa are more closely related to each other than to other Guizotia species. However, they form their own separate subclades with high bootstrap support. Chelelu is a riverine perennial plant that can be distinguished from Ketcha and other Guizotia species by its rhizomatous-like vegetative propagation and seed color (Dagne 1995). Generally, the data collected so far—be it morphology, karyotype, crossing experiments, meiotic behavior of hybrids, molecular markers or this particular study—show with certainty that Chelelu and Ketcha belong to the genus Guizotia and that they are distinct enough to be treated as separate species. The taxonomic revision of the genus Guizotia is underway in our department. It will be of interest to see the relationship between Sigesbeckia species, moved to the genus Guizotia by Schulz (1990), and the species excluded from the genus Guizotia by Baagøe (1974) as well as the two subspecies of G. scabra, Chelelu and Ketcha in the revision of the genus by considering as many characters as possible.
The position of the genus Guizotia within the tribe Heliantheae
The genus Guizotia has been placed under different subtribes (Coreopsidinae, Verbesininae and Milleriinae) of the tribe Heliantheae by different authors at different times (Baagøe 1974; Stuessy 1977; Robinson 1981; Karis 1993). One of the objectives of this study was to evaluate the position of the genus Guizotia within Heliantheae by including representative sequences from various subtribes of the tribe Heliantheae into the phylogenetic analysis. According to Stuessy’s systematic review of Heliantheae (Stuessy 1977), Acmella radicans and Verbesina jacksonii belong to subtribe Verbesininae, a subtribe that later was assimilated into Robinson’s subtribe Ecliptinae (Robinson 1981). Coreopsis petrophiloides and Milleria quinqueflora are nomenclatural type species of the tribes Coreopsidinae and Milleriinae, respectively.
In this phylogenetic analysis, M. quinqueflora appeared as the closest species to the genus Guizotia. The other two species of the subtribe Milleriinae (Rumfordia penninervis and Sigesbeckia blakei) were also positioned within the clade that contains the genus Guizotia, next to M. quinqueflora (Fig. 3). In other words, all three Milleriinae species included in this phylogenetic analysis were closely grouped with the genus Guizotia with a moderate level of bootstrap support (Fig. 3). On the other hand, Acmella radicans, Verbesina jacksonii and Coreopsis petrophiloides were revealed to be distantly related to Guizotia (Fig. 3). Thus, our data support the placement of the genus Guizotia under the subtribe Milleriinae (Robinson 1981; Bergqvist et al. 1992; Karis 1993). Smallanthus microcephalus was revealed to be the closest to the Milleriinae clade. In Robinson’s (1981) comprehensive taxonomic treatment of the tribe Heliantheae, S. microcephalus was placed under the subtribe Melampodiinae. However, Panero et al. (1999), in their phylogenetic analysis of the subtribe Ecliptinae, based on chloroplast restriction site data, indicated that the genus Smallanthus is closely related to the genus Rumfordia and collectively to other Milleriinae genera. These authors advised the transfer of this genus from Robinson’s subtribe Melampodiinae to the subtribe Milleriinae, which further strengthens the placement of the genus Guizotia under the subtribe Milleriinae.
Although discussing the phylogenetic relationships between various genera and subtribes of tribe Heliantheae is beyond the scope of this paper, it is worth mentioning some interesting points. The phylogenetic analysis revealed that Ecliptinae is a highly diversified subtribe that appeared to be polyphyletic, as species under this subtribe were placed in different major clades (Fig. 3). This result is in agreement with Panero et al. (1999), who reported the polyphyletic nature of this subtribe and the distribution of its genera in four different lineages. The amorphous nature of the subtribe Ecliptinae was already noted by Robinson (1981), though he regarded it in his taxonomic revision as natural by comparing it with other subtribes such as Helianthinae. Inula britannica, which belongs to the tribe Inuleae, was closely grouped with Verbesina jacksonii (Fig. 3), which still reflects the highly diverse nature of the Robinson’s subtribe Ecliptinae. The result may also reflect the close phylogenetic relationship between the tribes Heliantheae and Inuleae. In this analysis, Enhydra sessilis (Enhydrinae) and Palafoxia arida (Chaenactidinae) form their own separate clade with high bootstrap support. Such a close relationship between these genera was not indicated in Robinson’s review of the tribe Heliantheae. Clibadium alatum (Clibadiinae) and Wollastonia biflora (Ecliptinae) were grouped together, which is in agreement with the result obtained by Panero et al. (1999), who suggested the inclusion of the genus Clibadium in the subtribe Ecliptinae.
References
Assefa K, Merker A, Hailu T (2003) Inter simple sequence repeat (ISSR) analysis of genetic diversity in tef [Eragrostis tef (Zucc.) Trotter]. Hereditas 139:174–183
Baagøe J (1974) The genus Guizotia (Compositae). A taxonomic revision. Bot Tidsskr 69:1–39
Baker WJ, Hedderson TA, Dransfield J (2000) Molecular phylogenetics of subfamily Calamoideae (Palmae) based on nrDNA ITS and cpDNA rps16 intron sequence data. Mol Phyl Evol 14:195–217
Bayer RJ, Greber DG, Bagnall NH (2002) Phylogeny of Australian Gnaphalieae (Asteraceae) based on chloroplast and nuclear sequences, the trnL Intron, trnL/trnF intergenic spacer, matK, and ETS. Syst Bot 27:801–814
Bekele E, Geleta M, Dagne K et al (2007) Molecular phylogeny of genus Guizotia (Asteraceae) using DNA sequences derived from ITS. Genet Resour Crop Evol 54:1419–1427
Bentham G (1873) Notes on the classification, history, and geographical distribution of Compositeae. J Linn Soc Bot 13:335–577
Bergqvist G, Bremer B, Bremer K (1992) Chloroplast DNA restriction site variation and phylogenetic interrelationships of some genera of the Heliantheae sensu lato (Asteraceae). Nord J Bot 12:149–154
Bofkin L, Goldman N (2007) Variation in evolutionary processes at different codon positions. Mol Biol Evol 24:513–521
Crawford DJ, Mort ME (2005) Phylogeny of eastern North American coreopsis (Asteraceae-Coreopsideae): insights from nuclear and plastid sequences and comments on character evolution. Am J Bot 92:330–336
Dagne K (1994) Meiosis in interspecific hybrids and genomic interrelationships in Guizotia Cass. (Compositae). Hereditas 121:119–129
Dagne K (1995) Karyotypes, C-banding and nucleolar numbers in Guizotia (Compositae). Pl Syst Evol 195:121–135
Dagne K (2001) Cytogenetics of new Guizotia Cass. (Compositae), interspecific hybrids pertaining to genomic and phylogenetic affinities. Pl Syst Evol 230:1–11
Downie SR, Katz-Downie DS, Sun F-J, Lee C-S (2008) Phylogeny and biogeography of Apiaceae tribe Oenantheae inferred from nuclear rDNA ITS and cpDNA psbI-5′trnK(UUU) sequences, with emphasis on the North American Endemics clade. Can J Bot 86:1039–1064
Geleta M, Bryngelsson T, Bekele E et al (2007) Comparative analysis of genetic relationship and diagnostic markers of several taxa of Guizotia Cass. (Asteraceae) as revealed by AFLPs and RAPDs. Pl Syst Evol 265:221–239
Hall T (2005) BioEdit v. 7.0.5. Biological sequence alignment editor for Windows. Ibis Therapeutics a division of Isis pharmaceuticals. http://www.mbio.ncsu.edu/BioEdit/bioedit.html. Accessed 06 Dec 2008
Hilu KW, Liang H (1997) The matK Gene: sequence variation and application in plant systematics. Am J Bot 84:830–839
Hiremath SC, Murthy HN (1988) Domestication of niger (Guizotia abyssinica). Euphytica 37:225–228
Hiremath SC, Murthy HN (1992) Cytological studies in Guizotia (Asteraceae). Caryologia 45:69–82
Hiremath SC, Murthy HN, Salimath SS (1992) Quantitative nuclear DNA differences associated with genome evolution in Guizotia (Compositae). Genetica 85:241–247
Humbles JE (1972) Observations of genus Sigesbeckia L. Ciencia Y Naturaleza 13:2–19
Johnson LA, Soltis E (1994) matK DNA sequences and phylogenetic reconstruction in Saxifragaceae s.str. Syst Bot 19:143–156
Karis PO (1993) Heliantheae sensu lato (Asteraceae) Clades and classification. Plant Syst Evol 188:139–195
Lee J, Hymowitz T (2001) A molecular phylogenetic study of the subtribe Glycininae (Leguminosae) derived from the chloroplast DNA Rps16 intron sequences. Am J Bot 88:2064–2073
Lee C, Wen J (2004) Phylogeny of Panax using chloroplast trnC–trnD intergenic region and the utility of trnC–trnD in interspecific studies of plants. Mol Phyl Evol 31:894–903
Liang H, Hilu KW (1996) Application of the matK gene sequences to grass systematics. Can J Bot 74:125–134
Mort ME, Archibald JK, Randle CP et al (2007) Inferring phylogeny at low taxonomic levels: Utility of rapidly evolving cpDNA and nuclear ITS Loci. Am J Bot 94:173–183
Murthy HN, Hiremath SC, Salimath SS (1993) Origin, evolution and genome differentiation in Guizotia abyssinica and its wild species. Theor Appl Genet 87:587–592
Oxelman B, Liden M, Berglund D (1997) Chloroplast rps 16 intron phylogeny of the tribe Sileneae (Caryophyllaceae). Plant Syst Evol 206:393–410
Padgett DJ, Les DH, Crow GE (1999) Phylogenetic Relationships in Nuphar (Nymphaeaceae): evidence from morphology, chloroplast DNA, and nuclear ribosomal DNA. Am J Bot 86:1316–1324
Pan J, Zhang D, Sang T (2007) Molecular phylogenetic evidence for the origin of a diploid hybrid of Paeonia (Paeoniaceae). Am J Bot 94:400–408
Panero JL, Jansen RK, Jennifer A et al (1999) Phylogenetic relationships of subtribe Ecliptinae (Asteraceae: Heliantheae) based on chloroplast DNA restriction site data. Am J Bot 86:413–427
Robinson H (1981) A revision of the tribal and subtribal limits of the Heliantheae (Asteraceae). Smithsonian Contrib Bot 51:1–102
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386
Sankoff D, Cedergren RJ, Lapalme G (1976) Frequency of insertion-deletion, transversion, and transition in the evolution of 5S ribosomal RNA. J Mol Evol 7:133–149
Schulz DL (1990) Zur kenntnis der Gattung Sigesbeckia L. in Africa. Gleditschia 18:211–218
Shaw J, Small RL (2005) Chloroplast DNA phylogeny and phylogeography of the north Americans plums (Prunus subgenus Prunus section prunocerasus, Rosaceae). Am J Bot 92:2011–2030
Shaw J, Lickey EB, Beck JT et al (2005) The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot 92:142–166
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Syst Biol 49:369–381
Small RL, Cronn RC, Wendel JF (2004) Use of nuclear genes for phylogeny construction in plants. Aust Syst Bot 17:145–170
Stuessy TF (1977) Heliantheae-systematic review. In: Heywood VH, Harborne JB, Turner BL (eds) The biology and chemistry of the Compositae, vol 1. Academic Press, London, pp 1106–1118
Swofford DL (2000) PAUP*: Phylogenetic analysis using parsimony, version 4.0, beta. Sinauer Associates Inc., Sunderland
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Thompson JD, Gibson TJ, Plewniak F et al (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 25:4876–4882
Timme RE, Kuehl JV, Jeffrey L, Boore JL, Jansen RK (2007) A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am J Bot 94:302–312
Wagstaff SJ, Breitwieser I (2004) Phylogeny and classification of Brachyglottis (Senecioneae, Asteraceae): an example of a rapid species radiation in New Zealand. Syst Bot 29:1003–1010
Acknowledgments
We thank Dr. Anders Carlsson and Dr. Peter Olsson for valuable discussions on the data analyses and Ms. Therése Bengtsson for her assistance with the laboratory work. We would also like to thank Professor Bengt Oxelman for his critical reading and highly valuable comments and suggestions on this manuscript. This work was financed by the Swedish International Development Agency (SIDA/SAREC) through the International Science Program (ISP).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Geleta, M., Bekele, E., Dagne, K. et al. Phylogenetics and taxonomic delimitation of the genus Guizotia (Asteraceae) based on sequences derived from various chloroplast DNA regions. Plant Syst Evol 289, 77–89 (2010). https://doi.org/10.1007/s00606-010-0334-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-010-0334-x