
Abstract
Recent molecular studies have suggested the monophyly of Bolusiella, a small orchid genus comprising five species and one subspecies from Continental Africa, but sampling has been limited. Using the species delimitation presented in the recent taxonomic revision of the genus, this study aimed to confirm the monophyly of Bolusiella and assess the interspecific relationships using a comprehensive sampling and various analytical methods. DNA sequences of one nuclear spacer region (ITS-1) and five plastid regions (matK, rps16, trnL–trnF, trnC–petN, and ycf1) from 20 specimens representing all five species of the genus were analyzed using static homology, dynamic homology, and Bayesian methods. The monophyly of both the genus Bolusiella and each of its five species was confirmed, corroborating the previously published taxonomic revision. The use of dynamic homology methods was not conclusive for this particular group. The results of the total evidence analysis (combining all six sequence regions) using the dynamic homology approach yielded a slightly different hypothesis regarding interspecific relationships (namely the exchange of B. talbotii and Bolusiella iridifolia as the earliest diverging lineage), probably because the nodes in question are supported by a small subset of conflicting characters, compared to the hypotheses resulting from the static homology and Bayesian methods, which are congruent with the results of previous studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The orchid genus Bolusiella Schltr. was established in 1918 and is dedicated to the South African botanist Harry Bolus. It is well delimited both morphologically and geographically. Its six currently recognized taxa (five species and one subspecies) are known only from continental Africa and are easily recognizable by their equitant and fleshy leaves and dense inflorescence. While most of the taxa are widespread in tropical Africa, B. zenkeri (Kraenzl.) Schltr. is only known from the Upper and Lower Guinea domains, and Bolusiella iridifolia subsp. picea P.J.Cribb is confined to the Eastern Afromontane region.
The genus was the subject of preliminary phylogenetic analyses based on molecular data in two studies by Carlsward et al. (2006a, b), which suggested that Bolusiella was monophyletic, but sampling in the genus was limited. These two studies were based on both Bayesian inference and maximum parsimony and yielded identical phylogenetic hypotheses for the evolution of the clade, but due to sampling limitations (only three species: B. iridifolia (Rolfe) Schltr., B. zenkeri, and B. maudiae (Bolus) Schltr. were included), phylogenetic relationships among all the Bolusiella species could not be assessed, nor could the monophyly of its members be tested, even though all the species are morphologically well circumscribed (Verlynde et al. 2013). For example, B. iridifolia has leaves that are deeply sulcate, while those of all other species are not. Similarly, a spur is absent in B. fractiflexa Droissart, Stévart & Verlynde, but present in all other species, where it may either be found in the same plane as the lip (as in B. iridifolia subsp. picea) or completely curled under the lip (as in B. maudiae) (Online Resource 1).
The recent taxonomic revision of Bolusiella (Verlynde et al. 2013) helps to provide greater confidence in the species circumscriptions, allowing us to focus more specifically on the assessment of phylogenetic relationships among these species. The aim of the present study, therefore, was to use various analytical approaches, static homology, direct optimization (using POY version 5.1.2, Varon et al. 2010; Wheeler et al. 2014), and Bayesian inference to confirm the monophyly of Bolusiella and to assess phylogenetic relationships among its species.
Materials and methods
Taxonomic sampling
DNA was obtained from leaf- or floral-tissue samples taken from fertile specimens collected in the wild in Cameroon, Gabon, Guinea-Conakry, and Rwanda. Twenty Bolusiella accessions were sampled for DNA sequencing of one nuclear spacer region (ITS-1) and five plastid sequences (matK, rps16, trnL–trnF, trnC–petN and ycf1) (Online Resource 2). These twenty accessions represent all five species of the genus, but two accessions from the East African B. iridifolia could not be identified to the subspecies level because the voucher specimens lacked flowers.
To assess the monophyly of Bolusiella, sufficient outgroup sampling is necessary (see Darlu and Tassy 1993; Barriel and Tassy 1998). In this study, outgroups included Ancistrorhynchus clandestinus (Lindl.) Schltr., considered to be the sister group of Bolusiella in recent studies (Simo-Droissart et al. 2016), along with specimens of Angraecum bancoense Burg and Angraecum distichum Lindl., which are also included in subtribe Angraecinae, and finally a specimen of Polystachya albescens subsp. imbricata (Rolfe) Summerh., included in the same tribe as the Angraecinae (viz. Vandeae) but in a different subtribe (Polystachyinae). These outgroup taxa were sampled for all six regions.
DNA purification, PCR amplification, and DNA sequencing
The protocol adopted here followed Simo-Droissart et al. (2013), which focused on the phylogenetic study of Angraecum section Pectinaria. The following primers were used for amplification and sequencing of each individual plastid region: Tab-C and Tab-D for the trnL intron and Tab-E and Tab-F for the trnL–trnF intergeneric spacer (Taberlet et al. 1991), rps16-1F and rps16-2R for the rps16 intron (Oxelman et al. 1997), 19F (Molvray et al. 2000), 1326R (Cuénoud et al. 2002), 390F (Cuénoud et al. 2002) and trnK-2R (Johnson and Soltis 1994) for matK, trnC and petN-1R for the trnC–petN intergenic spacer (Lee and Wen 2003), and 3720F, IntR, IntF, and 5500R for ycf1 (Neubig et al. 2009). The nuclear marker ITS-1 was amplified using ITS-A, ITS-B, ITS-C, and ITS-D, designed for angiosperms by Blattner (1999).
Leaf or floral tissue was dried in silica gel for DNA extraction (Chase and Hills 1991). Total DNA was extracted from fresh (1 g) or dried material (0.3 g) using one of two methods. The first method used 1 g of fresh leaves in a modified 2× CTAB protocol (Doyle and Doyle 1987). Proteins were removed with SEVAG (chloroform/isoamyl alcohol 24:1), and DNA was precipitated with ethanol (− 20 °C). At the end of the extraction protocol, turbid or colored DNA extracts were purified further on Macherey–Nagel columns. For some samples, an alternative extraction method used 0.3 g of dried material with the NucleoSpin® plant kit from Macherey–Nagel, following the manufacturer’s protocol.
PCR amplifications were carried out using Biometra TProfessional thermocyclers (PTC–100 or PTC–200; Bio-Rad Laboratories, Inc.) in total volumes of 25 μL, each reaction containing 1–2 μL of template DNA (of unknown concentration), 0.12 μL (5 U/μL) of Taq polymerase (Qiagen), 2.5 μL PCR buffer, 1 μL MgCl2 (25 mM), 0.5 μL dNTPs (10 μM), 0.25 μL of each primer (10 μM), and 18.37–19.37 μL of H2O. The PCR amplification profiles used for the trnL–trnF region, trnC–petN, the rps16 intron, and ITS-1 consisted of an initial denaturation at 94 °C for 3 min followed by 30 cycles of 30 s at 94 °C, 30 s at 52 °C, and 1 min at 72 °C, with a final extension at 72 °C for 10 min. Amplification of matK (19F–1326R and 390F–trnK2R) and ycf1 (3720F–intR and intF–5500R) involved an initial denaturation at 94 °C for 3 min followed by 30 cycles of 30 s at 94 °C, 30 s at 52 °C, and 1 min 30 s at 72 °C, with a final extension at 72 °C for 10 min. PCR products were purified by enzymatic digestion using ExoSAP (Qiagen).
Cycle sequencing was carried out using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Inc., ABI, Lennik, the Netherlands) with the same primers used for PCR amplification: 1.5 μL of sequencing buffer, 1 μL of BigDye terminator with 0.2 μL of 10 μM primer, 1–3 μL of amplified product (of unknown concentration), and 4.3–6.3 μL of H2O, for a total reaction volume of 10 μL. Cycle sequencing conditions were as follows: a premelt of 1 min (96 °C), 25 cycles each with 10 s of denaturation (96 °C), 5 s of annealing (52 °C), and 4 min of elongation (60 °C). Cycle sequencing products were purified through ethanol precipitation. Sequences were generated on an ABI 3100 following the manufacturer’s protocols (ABI). Both strands were sequenced to assure accurate base calling.
Sequence editing
Complementary and overlapping sequences were assembled using CodonCode Aligner (version 4.2.7, CodonCode Corporation). Each individual base position was examined for agreement between the two to four contigs from both strands. Consensus sequences were edited manually.
Phylogenetic analyses
We used the molecular phylogenetic study of Angraecum section Pectinaria as a template for Bayesian and static homology methodology (Simo-Droissart et al. 2013). For each of the following analytical approaches, phylogenetic analyses were conducted first for each region separately (i.e., ITS-1, matK, rps16, trnC–petN, trnL–trnF, and ycf1). Following this, a single concatenated dataset for the five plastid regions was analyzed (five-marker plastid dataset), and then all six regions (plastid and nuclear) regions were analyzed in a single combined dataset (six-marker combined dataset). We therefore used eight different datasets (six individual markers and two combined datasets).
In addition, given its recent history, Bolusiella was considered as a useful model for testing the utility of various analytical approaches, static homology, direct optimization (using POY version 5.1.2, Varon et al. 2010; Wheeler et al. 2014) as well as Bayesian inference. In Simo-Droissart et al. (2013), the choice was made to code indels as missing data. In this study, indels were coded both as binary presence/absence characters and as missing data in the parsimony analyses. The genus has a relatively small number of species, allowing easy manipulation of datasets, and previous molecular studies provide preliminary evidence of its monophyly.
Finally, concerning the influence of gaps, transitions, and transversions, dynamic homology analyses performed under equal weighting (indel:Tv:Ts=1:1:1) minimized character conflict and yielded the lowest ILD values (Table 1). Therefore, results from this weighting scheme are reported below for parsimony and direct optimization analyses.
Dynamic homology approach
Direct optimization (Wheeler 1996; Gladstein and Wheeler 1996; D’Haese 2004) is a method of phylogenetic analysis that does not rely on a priori multiple alignments. Instead, it is based on a dynamic approach (hence, dynamic homology) in which both nucleotide substitutions and insertion–deletion events are considered evolutionary events to be optimized simultaneously to derive the best trees, without reference to a preexisting alignment (Wheeler et al. 2006). Despite the potential for this method, its application has been somewhat limited, but has included several animal phyla, such as Arthropoda (e.g., D’Haese 2002, 2003; Giribet et al. 2001; Arango and Wheeler 2007) and Annelida (Worsaae et al. 2005), and in the orders Squamata and Chiroptera (Frost et al. 2001; Giannini 2003, respectively). Dynamic homology has not been widely used in angiosperms. We only found ten studies, of which three involved dicots (Gottlieb et al. 2005; Weese and Johnson 2005; Pedraza-Peñalosa 2010), five monocots, mainly Poaceae (Lehtonen and Myllys 2008; Souto et al. 2006; Cialdella et al. 2007, 2010; Petersen et al. 2011) and two broader studies including both dicots and monocots (Aagesen 2005; Catalano et al. 2009). To date, dynamic homology has not been used on any dataset concerning the Orchidaceae family.
Phylogenetic analyses using the dynamic homology framework (Wheeler 1996) were performed using the parsimony criterion as implemented in POY version 5.1.2 (Varon et al. 2010; Wheeler et al. 2014). Each analysis was run for nine different transformation-cost regimes (Sankoff matrices for indel, transversion, and transition costs) to test the stability of the results. The influence of gap/transversion and transition/transversion costs was explored through sensitivity analysis (Wheeler 1996) to avoid an arbitrary choice of parameters. Three indel/transversion cost ratios (1, 2 and 4) and three transversion/transition cost ratios (1, 2 and 4) resulted in nine individual analyses for each combination (matK, ycf1, rps16, trnL–trnF, trnC–petN, ITS-1, plastid dataset, and combined dataset). For each of eight datasets combined with nine Sankoff matrices, the analytical procedure followed a three-step strategy: In step 1, a starting pool of 1000 Wagner trees was generated through random addition sequence (RAS). Each replicate was explored by a combination of TBR and SPR branch swapping and then subjected to parsimony ratcheting (Nixon 1999). The resulting topologies were then explored by tree fusing (Goloboff 1999). A final, more thorough branch swapping was performed by retaining trees up to 10% longer than the optimal ones, then retaining only the optimal trees as the result of this step. In step 2, for each of the eight datasets, trees resulting from the nine different transformation-cost regimes were concatenated for a supplementary tree-fusing step. Finally, in step 3, an additional round of tree fusing using iterative pass optimization (Wheeler 2003) for final refinement based on trees obtained for the previous two steps of the analyses.
Character congruence was used as an optimality criterion to choose the parameter set that maximizes congruence among loci. Congruence was measured by the incongruence length difference (ILD) metrics (Mickevich and Farris 1981). This value is calculated by dividing the difference between the overall tree length and the sum of its data components: (length combined–length individual sets)/length combined. The tree from the analysis that minimizes character conflict among all data is taken as the best overall explanation of character variation, and thus the best estimate of the phylogeny.
Approach based on aligned sequences
Consensus sequences were aligned with the CLUSTAL plugin (Larkin et al. 2007) implemented within Geneious (version 4.8.5, Biomatters), with default settings (Online Resource 3).
Static homology—Maximum parsimony (MP) analyses were performed using POY, under the equal weighting scheme (indel:Tv:Ts=1:1:1). Indels were coded in two different ways, as binary characters (present/absent) and as missing data. Heuristic searches were performed using tree bisection-reconnection (TBR) branch swapping, with 1000 replicates of random-taxon addition sequence, holding ten trees at each step. In a second round of analysis, we used all trees found in the tree-limited analysis as starting trees, with a limit of 10,000 trees, which were then swapped to completion.
Bayesian analysis—Bayesian analyses were performed using the MrBayes (Ronquist and Huelsenbeck 2003; Ronquist et al. 2012) module implemented within Geneious version 4.8.5 (Kearse et al. 2012) on the combined matrix, with one partition per gene (six partitions in total). Analyses were run for 2,000,000 generations with four chains (default temperatures) using a model-jumping approach that allows sampling across the entire general time reversible (GTR) model space (i.e., no best-fitting models were defined a priori; Huelsenbeck et al. 2004) and with model parameters unlinked between partitions. We followed the protocol of Simo-Droissart et al. (2013) where trees were sampled every 500 generations, resulting in a total of 4001 trees per run from which the first 500 (12.5%) were discarded as the burn-in phase. In order to avoid any bias, we checked empirically, within the MrBayes module in Geneious, that the plateau was reached at 2,000,000 generations by running the analyses with 10,000,000 generations. In the same fashion, we checked the length of the burn-in phase and adapted the amount of trees to be discarded to avoid discarding too many alternative solutions.
Clade support values—Levels of internal support were estimated for both the dynamic and static homology approaches, using two methods, the bootstrap protocol (Efron 1979; Felsenstein 1985) with 1000 replicates and the decay index (Bremer 1988; Donoghue et al. 1992), both of which were calculated from the aligned data (homology hypothesis implied for the optimal topology) using POY, building, and swapping 1000 trees from the optimal tree.
Results
Monophyly of the genus and species
When markers were analyzed individually, Bolusiella was found to be monophyletic in all of the resulting topologies except for ycf1 (when analyzed under static homology with indels coded as binary characters and dynamic homology approaches, respectively, Online Resource 4e–f), trnL–trnF (when analyzed under dynamic homology approaches, Online Resource 4i), and for rps16 sequences (analyzed using dynamic homology, Online Resource 4q).
Species were also generally found to be monophyletic, except for B. maudiae when ITS-1 sequences were analyzed under static homology approach, for B. iridifolia with ycf1 sequences, and B. talbotii with rps16 sequences analyzed under static homology with indels coded as binary characters and dynamic homology approaches (Table 2; Online Resource 4a–t). However, resolution and branch support among these species are clearly insufficient to recover interspecific relationships within Bolusiella based on any individual plastid dataset, or even when only the five plastid sequences are combined (Fig. 1; Online Resource 4a–c, e–g, i–k, m–o, and q–s). Yet, when all sequences are combined, Bolusiella along with its species is found to be monophyletic, and analyses yielded well-resolved trees with high branch support (Fig. 2).

Phylogenetic trees resulting from nuclear marker ITS-1 dataset search: a dynamic homology analysis (strict consensus tree with bootstrap percentages shown above or below branches); b static Homology analysis (maximum parsimony), with indels treated as characters (strict consensus tree with bootstrap percentages shown above or below branches); c static homology analysis (maximum parsimony), with indels treated as missing data (strict consensus tree with bootstrap percentages shown above or below branches); d Bayesian analysis (strict consensus tree with posterior probability values shown above or below branches)

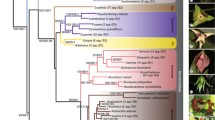
Phylogenetic trees resulting from six-marker combined dataset (ITS-1, matK, rps16, trnL–F, trnC–petN, and ycf1) search: a dynamic homology analysis (strict consensus tree with bootstrap percentages shown above or below branches); b static homology analysis (maximum parsimony), with indels treated as characters (strict consensus tree with bootstrap percentages shown above or below branches); c static homology analysis (maximum parsimony), with indels treated as missing data (strict consensus tree with bootstrap percentages shown above or below branches); d Bayesian analysis (strict consensus tree with posterior probability values shown above or below branches)
Phylogenetic relationships
When individual markers were analyzed separately using dynamic homology, there was insufficient resolution to recover the monophyly of Bolusiella or to assess interspecific relationships. Three of the six analyses of single regions (trnL–trnF, rps16 and ycf1) placed outgroup taxa among Bolusiella (Online Resource 4e–l&q). Analyses for rps16 show insufficient differences between sequences to resolve relationships among all angraecoid specimens (Online Resource 4q). However, in combination, these markers yielded well-resolved trees with high support values (bootstrap (BS) = 81–100%). Within the Bolusiella clade, B. talbotii represented the earliest diverging lineage, sister to a clade comprising all other species (BS = 99%). Inside this latter clade, B. fractiflexa + B. maudiae (BS = 100%) are sister to a clade uniting B. iridifolia and B. zenkeri (BS = 99%) (Fig. 2a).
In the results from the parsimony analyses, differences occur depending on whether indels were coded as missing or binary (presence/absence) characters. In the parsimony trees, B. iridifolia appears as the earliest diverging lineage, sister to the B. talbotii and B. zenkeri group, and to B. maudiae and B. fractiflexa group (Fig. 2c). The topology of the tree resulting from the six-marker combined dataset analysis where gaps are coded as binary characters is not the same as the topology of the tree resulting from the analyses of the six-marker combined dataset where gaps were coded as missing data. However, giving the support levels in the former, the two trees are actually very similar, the latter resolving B. talbotii as the sister group to B. zenkeri while the relationships between B. talbotii, B. zenkeri, and the B. fractiflexa + B. maudiae group are not resolved in the former. While the bootstrap values of the internal nodes of the genus are rather low, support values for the monophyly of individual taxa are still well supported (BS = 99–100%) (Fig. 2b).
The six-marker combined dataset Bayesian analysis yielded a well-resolved tree with high posterior probabilities (PP = 1). Within the genus, B. iridifolia is the earliest diverging lineage, sister to a clade uniting B. talbotii and B. zenkeri, and sister to B. fractiflexa and B. maudiae. The topology of this tree is identical to the one obtained using the nuclear marker (ITS-1) alone (Figs. 1, 2d).
Discussion
For five of the eight datasets used in our study, the implied alignment generated by POY resulted in shorter trees than the analyses based on CLUSTAL alignments (with the exception of ycf1, the five-marker plastid dataset, and the six-marker combined dataset). This result stands in contrast to what Weese and Johnson (2005) described for their study of the genus Saltugilia (V.E. Grant & A.G. Day) L.A. Johnson (Polemoniaceae).
Analyses of ITS-1 resulted in the same topology regardless of the method used. This includes the monophyly of Bolusiella, as well as each of its species (with the exception of B. maudiae using the static homology analyses). The four ITS-1 trees also agree in the placement of B. iridifolia as the earliest diverging lineage in the genus, and the sister-group relationship of two pairs of species, B. talbotii + B. zenkeri and B. maudiae + B. fractiflexa (Fig. 1). Trees resulting from the ITS-1 dataset are also congruent with the trees based on parsimony and Bayesian analyses of the six-marker combined dataset. The agreement between ITS-1 and the six-marker combined dataset suggests that the highest rate of phylogenetically informative characters is derived from ITS-1 and that the plastid markers are much less variable (Álvarez and Wendel 2003). This interpretation agrees with the lack of resolution in the trees based on separate plastid analyses. In the molecular study of continental African species of Angraecum section Pectinaria (Simo-Droissart et al. 2013), the marker with the highest rate of PICs (Phylogenetically Informative Characters, a nucleotide character state shared by two or more taxa) is ITS-1 (19.3%), with nearly twice as many as the plastid markers (an average of 10% for each plastid markers). In our case, the rate of PICs in ITS-1 is 13.56% (99 PICs), whereas the average rate for the plastid markers is 8.16%.
When the ITS-1 dataset is analyzed using dynamic homology, the resulting tree is congruent with the ITS-1 trees obtained using other methods, but not with the trees from the analyses of the six-marker combined dataset (Figs. 1, 2). Moreover, the different analyses of the five-marker plastid dataset do not yield congruent results and do not match the trees obtained using the nuclear marker (Online Resource 4u–x). Despite this, both dynamic and static homologies of the five-marker plastid dataset (coding indels as binary characters) result in identical topologies.
When using the six-marker combined dataset, the earliest diverging lineage of the Bolusiella clade differed in trees based on static homology and Bayesian approaches compared to those derived using dynamic homology. In the Bayesian tree, the lineage leading to B. iridifolia is the earliest diverging one, but in the dynamic homology tree, it is B. talbotii. The topology where B. iridifolia is the earliest diverging lineage was also found in previous molecular phylogenies (Carlsward et al. 2006a, b) based on a much broader sampling of angraecoid orchids (but only three species of Bolusiella). All these observations suggest that the basal placement of the B. iridifolia lineage is the most probable phylogenetic hypothesis.
Indels: valuable or missing data?
Similar to the results of Simo-Droissart et al. (2013), our study shows the same tree topology when analyzing the six-marker combined dataset using both indel treatments, except for the clade uniting Bolusiella talbotii and B. zenkeri, which is not supported when indels were coded as a binary character (Fig. 2b). Furthermore, the lengths of the trees obtained when indels are coded as missing data are logically always shorter than those obtained when indels are coded as discrete characters (Table 3). This can be easily understood because indels are supplementary and “valuable” characters, as measured by levels of homoplasy. However, once the sequences have been aligned, comparisons or homology hypotheses should apply to all positions, some of which may contain bases and gaps (Giribet and Wheeler 1999; Padial et al. 2014). That is, indels have become part of the pattern as much as any other nucleotide or amino acid. The pattern used to code characters for phylogenetic analysis, and consequently the putative recognition of transitions, transversions, and indels in DNA sequences, is the one created by the alignment, not the unaligned pattern that occurs in organisms (Simmons and Ochoterena 2000). This loss of information, when indels are coded as missing data, produces less resolved trees, and interspecific relationships are rarely resolved when plastid markers are analyzed individually (Table 2; Online Resource 4b–c, f–g, j–k, n–o & r–s), a conclusion similar to that of Heath Ogden and Rosenberg (2006). However, in our dataset for Bolusiella, indels did not represent valuable information when analyzing the six-marker combined dataset and therefore should be treated as missing data.
Conclusion
The aims of our study were to test the monophyly of each taxon conisdered and to assess phylogenetic relationships among them using the direct optimization protocol (Wheeler et al. 2014), as well as standard Bayesian and maximum parsimony analyses. While our study confirms the previously published taxonomy of Bolusiella (Verlynde et al. 2013), these results also show a difference in the evolution of plastid and nuclear regions. Results obtained from dynamic homology and those from standard phylogenetic approaches (parsimony and Bayesian approaches using an aligned dataset) allowed us to confirm the monophyly of Bolusiella and its species. The results also confirm that this genus represents five distinct species, notwithstanding the fact that the two subspecies of B. iridifolia were not clearly differentiated. The use of dynamic homology for this dataset was conclusive in that it provided an estimate of the phylogeny, albeit one that is slightly different from the one obtained with probabilistic methods. This method also provided a test of the monophyly of the taxa, but failed to recover interspecific relationships resolved with the other methods, probably because the nodes in question are being supported by a small subset of conflicting characters. Therefore, a broader phylogenetic analysis with more species representing different African angraecoid genera would be helpful to test further the utility of direct optimization in assessing interspecific relationships within the large orchid family.
References
Aagesen L (2005) Direct optimization, affine gap costs, and node stability. Molec Phylogen Evol 36:641–653. https://doi.org/10.1016/j.ympev.2005.04.012
Álvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Molec Phylogen Evol 29:417–434. https://doi.org/10.1016/S1055-7903(03)00208-2
Arango CP, Wheeler WC (2007) Phylogeny of the sea spiders (Arthropoda, Pycnogonida) based on direct optimization of six loci and morphology. Cladistics 23:255–293. https://doi.org/10.1111/j.1096-0031.2007.00143.x
Barriel V, Tassy P (1998) Rooting with multiple outgroups: consensus versus parsimony. Cladistics 14:193–200. https://doi.org/10.1111/j.1096-0031.1998.tb00332.x
Blattner FR (1999) Direct amplification of the entire ITS region from poorly preserved plant material using recombinant PCR. Biotechniques 27:1180–1186
Bremer K (1988) The limits of amino acid sequence data in angiosperm phylogenetic reconstruction. Evolution 42:795–803. https://doi.org/10.1111/j.1558-5646.1988.tb02497.x
Carlsward BS, Stern W, Bytebier B (2006a) Comparative vegetative anatomy and systematics of the angraecoids (Vandeae, Orchidaceae) with an emphasis on the leafless habit. Bot J Linn Soc 151:165–218. https://doi.org/10.1111/j.1095-8339.2006.00502.x
Carlsward BS, Whitten WM, Williams NH, Bytebier B (2006b) Molecular phylogenetics of Vandeae (Orchidaceae) and the evolution of leaflessness. Amer J Bot 93:770–786. https://doi.org/10.3732/ajb.93.5.770
Catalano SA, Saidman BO, Vilardi JC (2009) Evolution of small inversions in chloroplast genome: a case study from a recurrent inversion in angiosperms. Cladistics 25:93–104. https://doi.org/10.1111/j.1096-0031.2008.00236.x
Chase MW, Hills HG (1991) Silica gel: an ideal material for field preservation of leaf samples for DNA studies. Taxon 40:215–220. https://doi.org/10.2307/1222975
Cialdella AM, Giussani LM, Aagesen L, Zuloaga FO, Morrone O (2007) A phylogeny of Piptochaetium (Poaceae: Pooideae: Stipeae) and related genera based on a combined analysis including trnL-F, rpl16, and morphology. Syst Bot 32:545–559. https://doi.org/10.1600/036364407782250607
Cialdella AM, Salariato DL, Aagesen L, Giussani LM, Zuloaga FO, Morrone O (2010) Phylogeny of new world stipeae (Poaceae): an evaluation of the monophyly of Aciachne and Amelichloa. Cladistics 26:563–578. https://doi.org/10.1111/j.1096-0031.2010.00310.x
Cuénoud P, Savolainen V, Chatrou LW, Powell M, Grayer RJ, Chase MW (2002) Molecular phylogenetics of Caryophyllales based on nuclear 18S rDNA and plastid rbcL, atpB, and matK DNA sequences. Amer J Bot 89:132–144. https://doi.org/10.3732/ajb.89.1.132
CodonCode Aligner (version 4.2.7) CodonCode Corporation. Available at: http://www.codoncode.com
Darlu P, Tassy P (1993) La reconstruction phylogénétique. Masson, Paris
D’Haese CA (2002) Were the first springtails semi-aquatic? A phylogenetic approach by means of 28S rDNA and optimization alignment. Proc Roy Soc London, Ser B, Biol Sci 269:1143. https://doi.org/10.1098/rspb.2002.1981
D’Haese CA (2003) Morphological appraisal of Collembola phylogeny with special emphasis on Poduromorpha and a test of the aquatic origin hypothesis. Zool Scripta 32:563–586. https://doi.org/10.1046/j.1463-6409.2003.00134.x
D’Haese CA (2004) Les principes de l’Optimisation Directe. Biosystema 22:49–58
Donoghue MJ, Olmstead RG, Smith JF, Palmer JD (1992) Phylogenetic relationships of dipsacales based on rbcL sequences. Ann Missouri Bot Gard 79:333–345. https://doi.org/10.2307/2399772
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry Bulletin 19:11–15
Efron B (1979) Bootstrap methods: another look at the jackknife. Ann Stat 29:1–26
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. https://doi.org/10.2307/2408678
Frost DR, Rodrigues MT, Grant T, Titus TA (2001) Phylogenetics of the lizard genus Tropidurus (Squamata: Tropiduridae: Tropidurinae): direct optimization, descriptive efficiency, and sensitivity analysis of congruence between molecular data and morphology. Molec Phylogen Evol 21:352–371. https://doi.org/10.1006/mpev.2001.1015
Giannini N (2003) A phylogeny of megachiropteran bats (Mammalia: Chiroptera: Pteropodidae) based on direct optimization analysis of one nuclear and four mitochondrial genes. Cladistics 19:496–511. https://doi.org/10.1016/j.cladistics.2003.09.002
Giribet G, Wheeler W (1999) On gaps. Molec Phylogen Evol 13:132–143. https://doi.org/10.1006/mpev.1999.0643
Giribet G, Edgecombe GD, Wheeler WC (2001) Arthropod phylogeny based on eight molecular loci and morphology. Nature 413:157–161. https://doi.org/10.1038/35093097
Gladstein D, Wheeler WC (1996) POY: phylogeny reconstruction via direct optimization. American Museum of Natural History, New York
Goloboff PA (1999) Analyzing large data sets in reasonable times: solutions for composite optima. Cladistics 15:415–428. https://doi.org/10.1111/j.1096-0031.1999.tb00278.x
Gottlieb AM, Giberti GC, Poggio L (2005) Molecular analyses of the genus Ilex (Aquifoliaceae) in southern South America, evidence from AFLP and ITS sequence data. Amer J Bot 92:352–369. https://doi.org/10.3732/ajb.92.2.352
Geneious (version 4.8.5) Biomatters. Available at: http://www.geneious.com/
Heath Ogden T, Rosenberg MS (2006) How should gaps be treated in parsimony? A comparison of approaches using simulation. Molec Phylogen Evol 42:817–826. https://doi.org/10.1016/j.ympev.2006.07.021
Huelsenbeck JP, Larget B, Alfaro ME (2004) Bayesian phylogenetic model selection using reversible jump Markov chain Monte Carlo. Molec Biol Evol 21:1123–1133. https://doi.org/10.1093/molbev/msh123
Johnson LA, Soltis DE (1994) matK DNA sequences and phylogenetic reconstruction in Saxifragaceae s. str. Syst Bot 19:143–156. https://doi.org/10.2307/2419718
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Lee C, Wen J (2003) Phylogeny of Panax using chloroplast trnC–trnD intergenic region and the utility of trnC–trnD in interspecific studies of plants. Molec Phylogen Evol 31:894–903. https://doi.org/10.1016/j.ympev.2003.10.009
Lehtonen S, Myllys L (2008) Cladistic analysis of Echinodorus (Alismataceae): simultaneous analysis of molecular and morphological data. Cladistics 24:218–239. https://doi.org/10.1111/j.1096-0031.2007.00177.x
Mickevich MF, Farris JS (1981) The implications of congruence in Menidia. Syst Zool 30:351–370. https://doi.org/10.2307/2413255
Molvray MP, Kores J, Chase MW (2000) Polyphyly of mycoheterotrophic orchids and functional influences on floral and molecular characters. In: Wilson KL, Morrison DA (eds) Monocots: systematics and evolution. CSIRO Publishing, Collingwood, pp 441–448
Neubig KM, Whitten WM, Carlsward BS, Blanco MA, Endara L, Williams NH, Moore M (2009) Phylogenetic utility of ycf1 in orchids: a plastid gene more variable than matK. Pl Syst Evol 277:75–84. https://doi.org/10.1007/s00606-008-0105-0
Nixon KC (1999) The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics 15:407–414. https://doi.org/10.1111/j.1096-0031.1999.tb00277.x
Oxelman B, Lidén M, Berglund D (1997) Chloroplast rps16 intron phylogeny of the tribe Sileneae (Caryophyllaceae). Pl Syst Evol 206:393–410. https://doi.org/10.1007/BF00987959
Padial JM, Grant T, Frost DR (2014) Molecular systematics of terraranas (Anura: Brachycephaloidea) with an assessment of the effects of alignment and optimality criteria. Zootaxa 3825:1–132. https://doi.org/10.11646/zootaxa.3825.1.1
Pedraza-Peñalosa P (2010) Insensitive blueberries: a total evidence analysis of Disterigma s.l. (Ericaceae) exploring transformation costs. Cladistics 26:388–407. https://doi.org/10.1111/j.1096-0031.2009.00293.x
Petersen G, Aagesen L, Seberg O, Larsen IH (2011) When is enough, enough in phylogenetics? A case in point from Hordeum (Poaceae). Cladistics 27:428–446. https://doi.org/10.1111/j.1096-0031.2011.00347.x
Ronquist F, Huelsenbeck JP (2003) MrBayes: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1972–1974. https://doi.org/10.1093/bioinformatics/btg180
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Syst Biol 49:369–381. https://doi.org/10.1080/10635159950173889
Simo-Droissart M, Micheneau C, Sonké B, Droissart V, Plunkett GM, Lowry P, Hardy OJ, Stévart T (2013) Morphometrics and molecular phylogenetics of the continental African species of Angraecum section Pectinaria (Orchidaceae). Pl Syst Evol 146:295–309. https://doi.org/10.5091/plecevo.2013.900
Simo-Droissart M, Sonké B, Droissart V, Micheneau C, Lowry P, Hardy OJ, Plunkett GM, Stévart T (2016) Morphometrics and molecular phylogenetics of Angraecum section Dolabrifolia (Orchidaceae, Angraecinae). Pl Syst Evol 302:1027–1045. https://doi.org/10.1007/s00606-016-1315-5
Souto DF, Catalano SA, Tosto D, Bernasconi P, Sala A, Wagner M, Corach D (2006) Phylogenetic relationships of Deschampsia antarctica (Poaceae): insights from nuclear ribosomal ITS. Pl Syst Evol 261:1–9. https://doi.org/10.1007/s00606-006-0425-x
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Pl Molec Biol 17:1105–1109. https://doi.org/10.1007/BF00037152
Varon A, Vinh LS, Wheeler WC (2010) POY version 4: phylogenetic analysis using dynamic homologies. Cladistics 26:72–85. https://doi.org/10.1111/j.1096-0031.2009.00282.x
Verlynde S, Dubuisson JY, Stévart T, Simo M, Geerinck D, Sonké B, Cawoy V, Descourvières P, Droissart V (2013) Taxonomic revision of the genus Bolusiella (Orchidaceae, Angraecinae) with a new species from Cameroon, Burundi and Rwanda. Phytotaxa 114:1–22. https://doi.org/10.11646/phytotaxa.114.1.1
Weese TL, Johnson LA (2005) Utility of NADP-dependent isocitrate dehydrogenase for species-level evolutionary inference in angiosperm phylogeny: a case study in Saltugilia. Molec Phylogen Evol 36:24–41. https://doi.org/10.1016/j.ympev.2005.03.004
Wheeler WC (1996) Optimization alignment: the end of multiple sequence alignment in phylogenetics? Cladistics 12:1–9. https://doi.org/10.1111/j.1096-0031.1996.tb00189.x
Wheeler WC (2003) Iterative pass optimization of sequence data. Cladistics 19:254–260. https://doi.org/10.1111/j.1096-0031.2003.tb00368.x
Wheeler WC, Aagensen L, Arango CP, Faivovich J, Grant T, D’Haese CA, Janies D, Smith WL, Varon A, Giribet G (2006) Dynamic homology and phylogenetic systematics: a unified approach using POY. American Museum of Natural History, New York
Wheeler WC, Lucaroni N, Hong L, Crowley LM, Varón A (2014) POY version 5: phylogenetic analysis using dynamic homologies under multiple optimality criteria. Cladistics 31:189–196. https://doi.org/10.1111/cla.12083
Worsaae K, Nygren A, Rouse GW, Giribet G, Persson J, Sundberg P, Pleijel F (2005) Phylogenetic position of Nerillidae and Aberranta (Polychaeta, Annelida), analysed by direct optimization of combined molecular and morphological data. Zool Scripta 34:313–328. https://doi.org/10.1111/j.1463-6409.2005.00190.x
Acknowledgements
We want to thank Clément Schneider and Julio Pedraza for their assistance in the use of the Museum National d’Histoire Naturelle’s parallel mainframe cluster (PCIA, UMS2700) in the early stages of this project. The authors are grateful to the people growing orchids in the Yaoundé shadehouse (Cameroon), in the Sibang shadehouse (Gabon) and in the Nimba shadehouse (Republic of Guinea) for the collection of specimens and leaf samples. We would also like to thank Gilbert Delepierre, Jean-Paul Lebel, and Eberhard Fischer for the collection of leaf samples in Rwanda and Kenya. We are grateful to the American Orchid Society for financial support to M. Simo-Droissart, V. Droissart and T. Stévart. This research was supported by the US National Science Foundation (1051547, T. Stévart as PI, G. Plunkett as co-PI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors of this paper “Molecular phylogeny of the genus Bolusiella (Orchidaceae, Angraecinae)” declare that they have no conflict of interest.
Additional information
Handling editor: Mark Mort.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
Morphological differences among Bolusiella species: Diagnostic characters (in bold) distinguishing the six recognized species of Bolusiella. (PDF 77 kb)
Online Resource 2
Taxonomic sample for the molecular study: Missing data for concerned regions are indicated with “–” and available data with “+.” (PDF 78 kb)
Online Resource 3
Nucleotide sequence alignment matrix of 6 combined marker dataset (ITS-1, matK, rps16, trnL–F, trnC–petN and ycf1). (NEX 159 kb)
Online Resource 4
Additional phylogenetic trees resulting of each 5 chloroplast marker and the 5 chloroplast combined dataset obtained with Dynamic Homology analysis, Static Homology analysis (Maximum parsimony), with indels treated as characters, Static Homology analysis (Maximum parsimony), with indels treated as missing data and Bayesian analysis. (PDF 406 kb)
Information on Electronic Supplementary Material
Information on Electronic Supplementary Material
Online Resource 1. Morphological differences among Bolusiella species: Diagnostic characters (in bold) distinguishing the six recognized species of Bolusiella.
Online Resource 2. Taxonomic sample for the molecular study: Missing data for concerned regions are indicated with “–” and available data with “+.”
Online Resource 3. Nucleotide sequence alignment matrix of 6 combined marker dataset (ITS-1, matK, rps16, trnL–F, trnC–petN and ycf1).
Online Resource 4. Additional phylogenetic trees resulting of each 5 chloroplast marker and the 5 chloroplast combined dataset obtained with Dynamic Homology analysis, Static Homology analysis (Maximum parsimony), with indels treated as characters, Static Homology analysis (Maximum parsimony), with indels treated as missing data and Bayesian analysis.
Rights and permissions
About this article

Cite this article
Verlynde, S., D’Haese, C.A., Plunkett, G.M. et al. Molecular phylogeny of the genus Bolusiella (Orchidaceae, Angraecinae). Plant Syst Evol 304, 269–279 (2018). https://doi.org/10.1007/s00606-017-1474-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-017-1474-z