
Abstract
In-depth study of cellular heterogeneity of rare cells (e.g. circulating tumour cells (CTCs) and circulating foetal cells (CFCs)) is greatly needed in disease management but has never been completely explored due to the current technological limitations. We have developed a retrieval method for single-cell detection using a static droplet array (SDA) device through liquid segmentation with almost no sample loss. We explored the potential of using SDA for low sample input and retrieving the cells of interest using everyday laboratory equipment for downstream molecular analysis. This single-cell isolation and retrieval method is low-cost, rapid and provides a solution to the remaining challenge for single rare cell detection. The entire process takes less than 15 min, is easy to fabricate and allows for on-chip analysis of cells in nanolitre droplets and retrieval of desired droplets. To validate the applicability of our device and method, we mimicked detection of single CTCs by isolating and retrieving single cells and perform real-time PCR on their mRNA contents.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The study of cell populations at single-cell resolution and understanding the cellular heterogeneity among them can reveal important information regarding different cell types, functionality, conditions and circuits via genomic, transcriptomic, proteomic and metabolomic analysis [1, 2]. Single-cell analysis can provide insights on healthy and diseased behaviours of individual cells and can play a crucial role in precision diagnostics and therapeutics [3]. Among different analysis types, single-cell genomic and transcriptomic analysis has drawn the most attention in the past decade due to its capabilities in discovering cellular information [4]. Typically, single-cell genomic and transcriptomic analysis involves isolation and lysis of single cells to extract, amplify and barcode the DNA/RNA of each cell individually. For RNA analysis, an additional step of reverse transcription is needed to create complementary DNA (cDNA) due to the unstable nature of RNAs [5]. Conventionally, single-cell isolation and analysis were possible through limited dilution [6], micromanipulation [7], fluorescence-activated cell sorting (FACS) [8], laser-capture microdissection (LCM) and complex integrated microfluidic platforms (e.g. Fluidigm C1) [4], which are constrained by cost, minimum sample input, sample loss, time and labour [1]. Recently, multiple products have been developed to allow massively parallel single-cell isolation and barcoding of individual cells genomic material which is then followed by cDNA synthesis. Currently, most commonly used single-cell isolation methods are microfluidic droplet generators that encapsulate single cells with a uniquely barcoded RNA capture beads in a tiny water-in-oil droplet. However, droplet-based techniques often face challenges when dealing with low sample input (i.e. due to initial stabilisation time of the system), while requiring specialised equipment and high setup and operational costs [9]. An alternative approach to droplets is the use of nanolitre wells that sample gets loaded and settles down by gravity which reduces the need for specialised equipment and operating dead volume (e.g. BD Rhapsody system). Wells are designed with specific dimensions to allow for single cell and bead occupancy at the right sample concentration. Although nanolitre wells overcome some of the issues associated with droplet generators, the lack of a mechanism to encapsulate/isolate the cells in the wells leads to lower capture efficiency and a significant cross-contamination [1].
Most of the current single-cell technologies are employed for processing a large population of cells such as tissue biopsies. While there has been a significant development in single-cell analysis platforms for such cases, less attention has been paid to the single-cell analysis of low input samples (≤104 cells) [1]. With advancements in cell enrichment methods, there has been a growing interest in analysing rare cells at a single-cell resolution which can unravel unique insights that are not revealed by bulk molecular analysis [10]. Typical examples of rare cell analysis include circulating tumour cells (CTCs), circulating foetal cells (CFCs), immune cells and stem cells. These cells are usually found in liquid biopsies (e.g. blood, urine) and are processed through an enrichment step to remove the majority of unwanted cells. In the past years, liquid biopsy has gained significant attention due to the critical information that can be obtained from the analysis of rare cells in a non-invasive manner [11,12,13]. In most of the enrichment cases, a low number of rare cells is found among thousands of undesired background cells (e.g. in CTC cases, usually 1–10 CTCs among thousands of white blood cells (WBCs) are found in 1mL of a cancer patient blood sample) [14]. Single-cell analysis of such rare cells is not easily possible through droplet or nanolitre well systems due to abovementioned limitations associated with these approaches, especially cells losses. The majority of the current single-cell studies on rare cells are done via conventional approaches including limiting dilution, micromanipulation and LCM, despite their disadvantages [15]. Besides, there has been multiple single-cell capturing and analysis methods reported in literature based on hydrodynamic or active trapping of cells [16,17,18,19,20]. However, these methods suffer from high complexity, reliant on specialised equipment, inability to process low number of cells (e.g. high cell loss) and lack of a simple retrieval method. Thus, there is a critical need for the development of a low-cost, rapid and simple single-cell analysis platform that is capable of processing a low number of input cells and does not require special equipment. In Table S2 (supplementary file), different single-cell isolation techniques are compared for isolation and analysis of rare cells.
To overcome these challenges, we have developed a simple and low-cost method for single-cell trapping, isolation and retrieval using a microfluidic static droplet array (SDA). The volume of SDA traps is 20 nL, and the whole process is designed to be operated using a handheld pipette and fluorescent microscope, making the method more accessible to general laboratories. Our novel retrieval method is based on temporary bonding of the chip on a substrate during the injection, followed by freezing and peeling off the chip. The freezing step allows the droplets to stick to the substrate after chip peel off, which can be picked and transferred to a PCR tube, without losing any molecular content. We demonstrated a proof-of-concept study for single-cell analysis of rare cells by injecting 300 cells and analysing them via quantitative reverse transcription PCR (RT-qPCR) to validate our protocol.
Material and method
Device design and fabrication
The proposed devices have pocket volumes of 20 nL designed with a hexagonal cross-section, optimised to trap single cells and sized accordingly to allow for pickup of the droplets with a 10 μL pipette tip after freezing the chip. Also, pocket sizes were designed to ensure that enough nutrient is provided for on-chip culturing of the cells. The main channel and trap entrance dimensions are 170 μm and 220 μm, respectively, which allows the fluid to fill the traps first. Height of the channel was designed at 100 μm to allow cell movements and avoid clogging of the channel.
In this work, we used two different strategies to fabricate master moulds of our devices. The first master mould was made using standard photolithography as follows. Initially, the SDA pattern was designed using AutoCAD software (AutoDesk, USA) and printed out on a glass mask. Then, photolithography was performed using nLOF2020 photoresist and Karl Suss MA6 mask aligner (SUSS Microtec, Germany) on a silicon on insulator (SOI) wafer with 100 mm diameter. Eventually, deep reactive ion etching (DRIE) was deployed to transfer the pattern into the wafer with an aspect ratio of 1:10.
To explore the possibility of an alternative approach for master mould fabrication, we have utilised high-resolution SLA printing technology to make devices with bigger spacings between the traps. The 3D-printed mould was fabricated using MiiCraft Ultra 50 (MiiCraft, Taiwan) with a printing area of 57 × 32 × 120 mm and XY resolution of 30 μm as per previous works of our research group [21]. The 3D printer used in this work utilises a 385–405-nm UV wavelength to cure resin (BV-007) on a printing platform. Given the lower resolution of 3D printing compared to soft lithography, we adjusted the design parameters to accommodate for that. The design was made using computer-aided design modelling software, SolidWorks 2018 (Dassault Systemes, France), and the file was exported as an STL file to Miicraft printing software (MiiCraft 125, version 4.01, MiiCraft Inc). Slicing of 50 μm in z-axis was used with a curing time of 1.2 s, 2 base layers and 4 buffer layers, to ensure that the part is adhered to the picker during printing. After the printing was over, the mould was washed thoroughly using isopropanol alcohol to remove any residual resin and post-cured for enhancing the surface proprieties of the mould. Then to eliminate any uncured monomers and oligomers on the surface, the mould was dipped in isopropanol alcohol for 2 h.
Once the fabrication and preparation of both soft lithography and 3D printed moulds were completed, to avoid any polydimethylsiloxane (PDMS) sticking to the surfaces of the moulds, salinisation was done using trichloro (1H, 1H, 2H, 2H-perfluoro-octyl) silane (Sigma-Aldrich, Australia) in a pressure desiccator under vacuum for 2 h to produce a hydrophobic coating layer for easier release of cured PDMS. PDMS (Sylgard 184, Dow Corning, USA) was prepared by mixing elastomer and curing agent in a standard ratio of 10:1, followed by degassing in a vacuum chamber until all air bubbles are removed. The mixture was then poured onto the moulds and cured at 65 °C for 1 h before peeling. The PDMS device was then cut from the mould, and inlet and outlet access holes were made using 1-mm biopsy punch. The device was then pressed on Glass slide (Corning, Australia), which was pre-coated with 1% BSA (Sigma Aldrich) for 1 h, to create a temporary binding.
Cell culture
Breast cancer cell line, MCF-7, and monocyte cell line, THP-1, were cultured in complete media made of RPMI media (Gibco, Australia) with 10% foetal bovine serum (Gibco, Australia) and 1% Penicillin-streptomycin antibiotics (Gibco, Australia) in T-25 flasks (Corning, Australia). The cells were passaged at 80% confluency, and the seeding density was about 0.7 × 106 cells.
Cell/bead loading and capture rate counting
Once cells reached 80% confluency in the flask, the cells were detached by TrypLE (Gibco, Australia), and cells were resuspended in 1-mL culture media after centrifugation. Cells were then counted using a haemocytometer (Sigma-Aldrich, Australia), and cell viability of >90% was ensured by staining with 0.4% Trypan blue solution (Gibco, Australia). A range of different cell concentration solutions (5–50cells/μL) was prepared by diluting the cells in DPBS (Gibco, Australia), and after injection of the solutions, a fluorescent microscope was used to compare single-cell capture rates. Different injection speeds, between 10 and 90 μL/min, were tested by using a syringe pump as well as a handheld micropipette. Before injecting 5 μL of the sample into the microfluidic chips, the cell suspension was remixed by pipetting for 5 s. After injection, the device was imaged under a microscope, and cell occupancy in each chamber was identified.
The workflow of our method includes 5 steps (Fig. 1 and Movie S1): (1) cells were harvested, diluted to the appropriate concentration and injected by a handheld pipette into the device; (2) the remaining liquid in the main channel (if any) of the device was aspirated to create individual droplets (Movie S2); (3) the device was frozen by placing it in −80 °C fridge for 5 min; (4) the device was taken out, and the PDMS chip was peeled off; and (5) the droplets of interest were picked up by a hand-pipette for further studies (Movie S3).

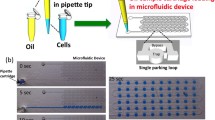
Single-cell capturing and retrieving in SDA. a Schematic illustration of the workflow developed in this study for single-cell retrieval using a static droplet array (SDA) microfluidics. The cells were then retrieved for single-cell RT-qPCR in this paper. b Picture of actual PDMS device which is filled with the red food dye. The close-up views show the architecture of the device, localisation indices and a single cancer cells which is stained with DAPI and anti-CK antibodies
Cell viability assay
The effect of different dimethyl sulfoxide (DMSO, Sigma-Aldrich, Australia) concentrations on the cellular viability post-freezing was examined. Firstly, MCF-7 cells were centrifuged and suspended in 1 mL fresh media in 1.5 mL microtubes. Two microlitres of live and dead stain (Abcam, Australia) was added into each tube. Then the cells were allocated into 5 different tubes to make 1, 3, 5, 7 and 10% DMSO content in the cell solutions, and the cells were injected into the device, sheathed with air, and the number of live and dead cells was counted under the microscope. After that, the chips were frozen in −80 °C freezer and taken out after 10 min. The device was thawed in 37 °C incubator for 5 min and imaged under the microscope to calculate the number of live cells post-freezing.
Immunofluorescence staining and imaging
MCF-7 cells and THP-1 cells were harvested, centrifuged at 400 g for 5 min and resuspended in 100 μL methanol (Sigma-Aldrich, Australia) in two 1.5 mL Eppendorf tubes. The tubes were then incubated at −20 °C for 5 min for permeabilization and fixation. After two times washing by DPBS, the cells were resuspended in a cocktail of 96 μL DPBS, 2 μL DAPI solution and 2 μL anti-cytokeratin antibody or anti-CD45 (Miltenyi Biotech, Australia), respectively. The tubes were then incubated in 4°C fridge for 1 h. To wash off unbounded antibodies, the cells were centrifuged and washed with 1 mL DPBS for two times. Then, the mixed cell solution was diluted to 60 cells/μL and injected into the microfluidic device. The device was imaged using an Olympus IX 73 invented fluorescent microscope (Olympus, Japan). Panorama bright field and fluorescence images were taken by defining the imaging starting point and endpoint.
Single-cell RT-qPCR
To verify the preservation of genetic material in our proposed method, known number of MCF-7 cells were injected to the chip using a handheld pipette and then frozen quickly as mentioned above. The chip was peeled off from the substrate, and the droplets were retrieved and transferred into a 200 μL PCR tube under two conditions: (1) frozen droplet (by placing the chip on an ice block) and (2) melted droplet. As a control, individual MCF-7 cells were isolated with serial dilution and assessed using the same PCR protocol. The cells were lysed, extracted, reverse-transcribed and pre-amplified by NEBNext® Single Cell/Low Input complementary DNA (cDNA) Synthesis & Amplification Module (New England Biolab, Australia). Briefly, the cells were mixed with 0.5 μL 10× lysis buffer, 0.25 μL RNase inhibitor and 4.25 μL nuclease-free water and incubated at room temperature for 5 min. Then, 1 μL of Single Cell RT Primer Mix and 3 μL of nuclease-free water were added into the lysed cells and were incubated at 70 °C for 5 min to synthesise first-strand cDNA. To make cDNA strands for PCR, reverse transcription was carried out by adding 5 μL Single Cell RT Buffer, 1 μL Template Switching Oligo, 2 μL Single Cell RT Enzyme Mix and 3 μL nuclease-free water into the lysed cells. The solution was then incubated at 42°C for 90 min and 70 °C for 10 min. Lastly, in order to increase the amount of cDNA for detection with RT-qPCR, cDNA pre-amplification was performed with adding 50 μL Single-Cell cDNA PCR Master Mix, 2 μL of Single Cell cDNA PCR Primer and 28 μL of nuclease-free water to reach a final volume of 100 μL. The sample was then incubated in thermal cycler, as shown in Table S1.
Then, RT-qPCR was performed with HER2 and GAPDH (Life Technologies, Australia) primers to confirm the loss of genetic material of cells. Briefly, PCR was performed with SuperScript® III Platinum ® SYBR® Green One-Step qRT-PCR Kit (Invitrogen, Australia). Twenty-five μL of Master mix, 1 μL of Taq Mix, 1 μL of forward and reverse primers, 2 μL of samples and 20 μL of nuclease-free water were added into the PCR well plate (Biorad clear well PCR plate; HSL9601) to make a final volume of 50 μL. The CFX96 Touch (Biorad, Australia) thermocycler was set following the manufacturer’s guideline. Enzymes were activated at 95 °C for 5 min, and the following cycle was repeated for 40 times: 15 s at 95 °C, 30 s at 60 °C and 40 °C for 1 min (Table S1). The results were then analysed with CFX Maestro software (Biorad, Australia).
Numerical simulation
To better understand the flow behaviour of the fluid in our device as well as the heat transfer during the freezing and thawing process, numerical simulations were performed using ANSYS Fluent 2020R2 (ANSYS, USA) [22, 23]. The fluid flow was performed using a 2D and 3D laminar single-phase solver to understand the flow rate through the trap opening compared to the main channel. Boundary conditions, including inlet velocity, were assigned similar to the real-case scenario, as mentioned in the “Results and discussions” section. Furthermore, for the heat transfer, a 3D transient analysis was performed on a single droplet with the size and shapes of our chip with a 1.5-mm glass base and PDMS on the 3 other sides. The bottom face of the glass was set at a constant temperature of −80°C, and the droplet was initially set at 23°C. Phase transitions were not taken into account as the main focus of this study was to understand the rate of heat transfer during the freezing process [24]. Fluid properties of the water were set as water density = 998 kg/m3, dynamic viscosity = 1.002×10−3, thermal conductivity = 0.5 W/m·K and heat capacity at constant pressure = 4218 J/kg·K.
Theory of static droplet array
The channel dimensions (170–800 μm) and operating flow rates (10–90 μL/min) result in a laminar flow regime across the device, which is a key operating parameter for effective cell trapping. A Reynold’s number of 2.9 was calculated for this chip, which refers to a laminar flow by Re = ρuDh/μ, where ρ is density of the fluid, u is the mean velocity of the fluid (calculated by flow rate/cross section area), Dh is the hydraulic diameter of the channel and μ is the dynamic viscosity of the fluid. In this case, the fluid density and viscosity were estimated with the values of PBS. The larger inlet dimension of side chambers forces majority of the fluid to fill the chambers first due to the lower resistance of this pathway. Injection flow rate plays an essential role in the behaviour of fluid and performance of this device. The subsequent pressure drop (due to resistance) across a pathway is caused by friction and fitting losses. Friction losses are dependent on the flow regime and length of the channel, and fitting losses can be described by \( {P}_{drop}=\frac{1}{2}\rho {V}^2C \), where ρ is the density, V is the fluid mean velocity and C is the pressure drop coefficient obtained experimentally and is dependent on geometrical features of the channel (not the flow velocity) [25]. In parallel fluidic networks, the flow rate across each pathway is adjusted based on their resistances, in a way that total energy loss over each of the pathways is equal. Therefore, lower resistance of the side chambers compared to the main channel allows the liquid to fill the pockets first, and once the pockets are full, the liquid flows through the main channel to the next pair of pockets. However, at higher velocities (flow rates), the bending resistance (pathway to side chambers) increases that might result in all the fluid travelling through the main channel (i.e. side chambers remain empty). The 10-μm air gap designed at the end of each chamber allows air to escape but stops the liquid from proceeding by utilising surface tension properties of the media-air interface on a hydrophobic channel [26]. This phenomenon is often referred to as bursting pressure which provides a large enough pressure barrier for the chamber pathway that the rest of fluid would favour moving along the main channel, to the next empty pockets, and hence, no other cells would enter the filled chambers. The bursting pressure is highly dependent on the fluid viscosity, fluid surface tension, channel material and dimensions. Bursting pressure in a channel can simply be found using \( {P}_{Capillary}=\frac{Surface\ Tension\ Force}{Area}=\frac{\sigma cos\theta \times perimeter\ }{Area} \), where σ is the surface tension constant of the liquid-air and θ is the contact angle which is 107° for PDMS [25, 26]. In this case, the air gap provides a minimum of 4600 Pa pressure barrier. Once the chambers are filled, other cells in the fluid do not enter the chambers. Considering that cells only enter while the chambers are being filled, the trapping rate across device remains unchanged as the fluid flows through the device. The particle-particle interaction was ignored in this experiment for two reasons: firstly, the particle-to-particle interaction created by Brownian effect is negligible when the diameter of the cells and beads is bigger than 1–2 μm. Secondly, the concentration of cells and beads are too low to create an influential interaction [27].
Results and discussions
Device performance—trapping
In order to characterise the device performance, we first used 15-μm fluorescent polystyrene beads during the design process to mimic the trapping behaviour of cancer cells. Once the design was finalised, the device performance was optimised using MCF-7 cells. The effect of different flow rates and cell concentration on the trapping efficiency of the device was investigated. As shown in Fig. 2a, the experimental results have shown that the single-cell occupation follows Poisson distribution trend line, for instance, at the sample concentration of 50 cells/μL (1cell/20 nL), the single-cell encapsulation rate was recorded at 32% (average of 80 single cells trapped out of 250 injected cells). Furthermore, the single-cell encapsulation can be increased up to 88% by lowering the sample cell concentration to 5 cells/μL (average of 22 single cells trapped out of 25 cells injected). The single-cell encapsulation results were repeated a minimum of 5 times to minimise the potential experimental errors, and the associated variability of the results is shown in the figures based on the standard deviation. Generally, lowering the injection concentration would significantly increase the single-cell encapsulation rate; however, it would result in greater number of empty chambers. Also, it is noteworthy that greater number of cells could be processed by increasing the number of traps and/or running multiple devices in parallel, especially for potential integration of the proposed workflow for clinical pipelines.

The SDA device performance for trapping cells. a and b Single-cell capturing rate of the device with respect to cell concentration and flow rate to find the optimum capture rate of this device. It was found that at 5 k cells/mL, the single-cell capture rate of the device was the highest, and about 88% of cells were captured individually. The flow rate plays a smaller role in single-cell capture rate; at 5 k/mL concentration, the variation of capture rate was trivial, and there was no significant difference between handheld pipette injection and syringe pump injection. c Device performance tested with MCF-7 and THP-1 cell lines in a 1:100 ratio (92 single cell were trapped). The green and red circles on traps were drawn manually to indicate the position of cells. Five traps were shown with higher magnification for better illustration of the single and double occupied traps. No cell was found to stick to the main channel, inlet and outlet of the device
Besides, as shown in Fig. 2b, no major difference was found on the capturing rate by adjusting the flow rate, given the flow rate stays within the operational limit (up to 90 μL/min) of the device, and does not cause leakage in the system (considering the temporary binding). Also, the injection via a handheld pipette showed similar capturing rate which indicates the independency of this device on specialised equipment for sample injection, such as syringe pumps. Furthermore, to lower systematic losses during operation of our SDA, a minimum of 5 μL of the solution was injected, just to cover volume of the pockets (4.8 μL). The 5-μL injected sample would fill more than half of the chip, which then air was injected to push the remaining of the sample from the main channel to fill the remaining pockets. Once all chambers are filled, any excess solution (if more than 4.8 μL) could be collected from the outlet.
To further explore the potential of using this device for single-cell analysis of rare cells, we injected MCF-7 and THP-1 cell lines in a ratio of 1:100 to mimic the scenario of CTC isolation post enrichment from peripheral blood, based on previously published works of our research group [28,29,30,31]. First, the harvested cells were stained using DAPI, anti-cytokeratin and CD-45 and then a mixture of both cell types was made at the optimum concentration. Then 5 μL of the sample was injected to our device (consisting of ~297 THP-1 cells and 3 MCF-7 cells), and we observed that average of 92 cells were captured individually (38.3% ± 2.0%, N = 3). Generally, the washing steps involved in CTC enrichment is highly optimised to remove most debris and soluble proteins to a level which cells are well identifiable from background noise under fluorescent microscope. As shown in Fig. 2c, two traps were found to contain MCF-7 cells, which one was a singlet, and the other two were trapped in a double-occupancy chamber (with a THP-1 cells). This illustrated the potential of using our proposed workflow to analyse rare cells at single-cell resolution. It is also worth mentioning that increasing the number of pockets and consequently lowering the cell concentration would allow for a more efficient analysis (increasing the single-cell trapping rate) within a single device.
Retrieval through quick freezing
When analysing rare cells (including CTCs), current single-cell isolation methods (e.g. micromanipulation, FACS, LCM and droplet barcoding) are constrained by the low input cell number processing and hence suffer from low yield, high cost, damage to cells, poor repeatability and over-reliance on operator skills. Besides, these platforms do not allow for collection and monitoring of live single cells for on-chip studies. To overcome abovementioned challenges of single-cell devices, we developed a simple and stand-alone approach for retrieval of single cells based on a low-cost SDA microfluidic device that is capable of isolation and on-chip analysis of single cells.
The retrieval is done through freezing the temporary bonded SDA device against a glass or parafilm substrate, followed by a peel-off step. The freezing step allows all frozen droplets to stick to the surface with a greater hardness—i.e. using glass as substrate results in frozen droplets to stick to the glass, while parafilm substrate allows the droplets to stick to the PDMS chip. Once the chip is peeled off, we covered the surface with oil to avoid merging or evaporation of droplets during the retrieval process. Then droplet of interest would be picked up using a handheld pipette and transferred to a PCR tube for downstream analysis. The entire process can take less than 15 min from injection to retrieval of the cell of interest. For a better illustration, food dye and dextran were used in Fig. 3 a and b to show the freezing process and droplet retrieval using this method. −80 °C freezer could be used for rapid freezing (~5 min) or long-term storage of the chip. Furthermore, Fig. 3 c and d show the fluid flow and heat transfer of the proposed approach during the injection and freezing process through computational fluid dynamic simulation. An average cooling rate of 0.3 °C/s was obtained with most of the heat transfer occurring through conduction of the 1.5-mm glass substrate.

Illustration of the device throughout the freezing and retrieval process along with simulation of the device during injection and freezing. a Frozen droplets of food dye tend to all stick to the glass substrate (more rigid surface compared to PDMS chip) after peeling off the device. If the PDMS device was temporarily bonded to a softer substrate than PDMS (e.g. Parafilm), the droplets tend to stick to the PDMS device. b Pickup of dextran droplet before and after freezing, showing no liquid remaining in the trap. c CFD simulation of the flow across the device indicating that the flow fills up the traps before proceeding through the main channel. d Heat transfer simulation of the droplet placed in −80 °C environment with respect to time, elaborating on the rapidness of this method. Simulation indicates that the droplets reach −80 °C in 5 min and are frozen in about 1 min
In terms of user-friendliness during retrieval, glass substrate allows easier handling of the chip throughout the injection process due to its rigidity, while parafilm substrate allows an easier pickup since the droplets stay on the chip and geometrical features of the trap (arms) assists with keeping the droplet in its original space even if melting occurs. To further enhance this method, we designed an SDA device with the same trap shape and size but with 2-mm spacing between each trap to avoid droplet merging during the pickup process. The new device was fabricated using a 3D printer, as explained in the “Material and method” section, and consisted of 88 traps in total in a 15 mm × 35 mm moulding area (Fig. 4a). The droplet generation process was recorded in Movie S4, showing the same droplet formation theory in this spaced chip.

Illustration of the device with greater spacing along with the effect of freezing on cell viability and molecular analysis of the retrieved droplets. a Picture of the spaced SDA design with 88 droplets generated using red food dye. This PDMS device was cast in a 3D-printed resin mould. Since the space between traps is bigger, this device allows easier pickup of frozen droplets without a microscope. b Cell viability after freezing indicated a positive relationship between DMSO content and viability of cells after freezing. Small droplet size and fast cooling reduce the damage of the cells. c Comparison between the cycle of the threshold of GAPDH and HER2 genes across the fresh, frozen and melted droplet sample. Compared to a melted droplet, the frozen droplet has closer Ct value to the fresh cell control, indicating better preservation of the genetic materials after retrieval
The ice crystals formed during freezing and thawing of cells cause the cell membrane to burst [32]. For applications where cells need to be retrieved after freezing, e.g. on-chip preservation and/or culturing, viability of cells post-freezing becomes crucial. Therefore, DMSO could be employed as one of the most commonly used cell cryoprotectant, which prevents water crystallisation during the freezing. We investigated the viability of cells after freezing and thawing the chip with different DMSO content (1%, 3%, 5%, 7% and 10%) in −80°C freezer. The viability was investigated by imaging the chip before freezing and after thawing, using live and dead cells staining. As shown in Fig. 4b, all cells frozen without DMSO were dead after thawing, and no cells could maintain the shape. In the 1% DMSO groups, none of the cells exploded due to water crystal formation, but the viability of the cells was low compared to the other DMSO groups. It was shown that the post-thawing viability is directly proportional to the DMSO concentration and a maximum viability of 86% (N = 5) was recorded when 10% DMSO was used. It is worth mentioning that in our previous works, we have shown that the cell viability is not affected during the injection and trapping due to the low operational pressure of the device and minimal stress being applied through hand injection of cell solution via a handheld pipette. Also, the potentials of culturing cells in the device over 48 h period with less than 5% drop in cell viability were shown [26, 33].
RNA analysis through RT-qPCR
To show the applicability of our method for single-cell molecular analysis, we performed single-cell RT-qPCR of some major genes such as GAPDH (housekeeping gene) and HER2 (epithelial marker). Although through the freezing process the cells would naturally burst, the RNA content will be preserved in the frozen droplets which can be retrieved. Therefore, we investigated the amount of RNA retrieved from (1) frozen and peeled-off chip placed on ice block where the frozen droplets were picked before melting occurs and (2) frozen and peeled-off chip placed on bench and picked up after melting occurred. We compared the retrieved RNA content of both cases to a fresh cell in a tube. Figure 4c and S4 show that the RT-qPCR results and the cycle of threshold (Ct) value of GAPDH and HER2 in the fresh single-cell were 26.16 and 31.06 (N = 3) in comparison with the single cell of the frozen droplet sample that were 27.28 and 32.43 (N = 3). The single cell of the melted droplet sample showed a Ct value of 33.23 and 39.06 (N = 3), respectively, while there was no Ct value for the negative controls. By comparing the (Ct) values across both genes, we observed a similar amount of RNA detected in the frozen and fresh sample, while a significant loss occurred when the droplets were melted. Once the droplet melts, some of the RNA content would attach to the surface of the chip which would not be recovered. The ice block allows extra time for handling and pickup of the cell without causing the droplets to melt. While the Ct values do not indicate the actual concentration or copy number of the genetic material content (i.e. RNA), the minor difference between the Ct values of the fresh and frozen sample shows a high recovery of the RNA throughout our retrieval approach.
Cell loss during retrieval
To quantify the cell loss throughout the retrieval process, we tested the entire method for retrieval and transfer of 10 single cells (frozen and thawed with 10% DMSO content) to a 96-well plate by using a handheld pipette. The pickup was repeated for 5 times, and an average of 80% cell transfer (8 out of 10 cells) was recorded. The cell loss was mainly due to the manual handling of the 20 nL droplet using a conventional 2-μL pipette, which can be improved by using more accurate equipment. As mentioned above, spacing out the droplets can significantly improve the handling of the chip during thawing and picking up, since droplets have almost no chance of merging during the pickup, as they are separated apart.
Conclusion
Single-cell analysis provides novel insights on answer comprehensive biological questions that fail to be solved by bulk sample analysis and reveals the heterogeneity among a subpopulation of cells. The number of techniques and instruments for single-cell analysis has been soaring in recent years, and data generated from single-cell studies show significant potentials in revealing heterogeneity among the various biological systems. However, the high cost and technical expertise required have hugely limited the use of single-cell devices, especially in clinical settings. Besides, current single-cell technologies are incapable of analysing rare cells (e.g. CTCs) at single-cell resolution in an efficient and cost-effective manner. We have demonstrated a simple, highly efficient and cost-effective method for single-cell isolation, followed by fluorescence microscopy and molecular analysis using RT-qPCR with minimal cell loss throughout the process by only using common laboratory tools. Nonetheless, the application of the proposed workflow is not limited only to molecular studies, and it enables the users to undertake various studies including cell secretion, drug testing, cloning and culturing studies. Lastly, this methodology is applicable to all SDA devices and overcomes the challenge of cell retrieval after injection [26, 34, 35]. The method we reported here provides a simple and effective procedure for obtaining multiplexed visual and molecular analysis in the single-cell level by using common laboratory tools. Although our droplet retrieval method is simple and overcomes shortcomings of previously proposed methods, the total number of static droplets in the device should be maximised before it can be integrated in real-case applications, including clinical settings. Besides, automation of this process, e.g. use of automated micromanipulators for labour-free transfer of cells, could provide significant advantages for the use of this workflow in clinical or research settings by lowering the human error, minimising cell loss and/or increasing the throughput.
References
Gierahn TM, Wadsworth MH II, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, Shalek AK (2017) Seq-well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods 14(4):395–398
Tang X, Huang Y, Lei J, Luo H, Zhu X (2019) The single-cell sequencing: new developments and medical applications. Cell & Bioscience 9(1):53
Wiedmeier JE et al (2019) Single-cell sequencing in precision medicine. Cancer Treat Res 178:237–252
Wu X, Yang B, Udo-Inyang I, Ji S, Ozog D, Zhou L, Mi QS (2018) Research techniques made simple: single-cell RNA sequencing and its applications in dermatology. The Journal of investigative dermatology 138(5):1004–1009
Au-Peterson SM, Au-Freeman JL (2009) RNA isolation from embryonic zebrafish and cDNA synthesis for gene expression analysis. JoVE 30:e1470
Hwang B, Lee JH, Bang D (2018) Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 50(8):96
Hu, P., et al. (2016) Single Cell Isolation and Analysis. Frontiers in Cell and Developmental Biology. 4(116)
Valihrach L, Androvic P, Kubista M (2018) Platforms for single-cell collection and analysis. Int J Mol Sci 19(3):807
Nguyen A, Khoo WH, Moran I, Croucher PI, Phan TG (2018) Single cell RNA sequencing of rare immune cell populations. Front Immunol 9:1553
Sabnis AJ, Bivona TG (2019) Principles of resistance to targeted cancer therapy: lessons from basic and translational cancer biology. Trends Mol Med 25(3):185–197
Mishima Y, Paiva B, Shi J, Park J, Manier S, Takagi S, Massoud M, Perilla-Glen A, Aljawai Y, Huynh D, Roccaro AM, Sacco A, Capelletti M, Detappe A, Alignani D, Anderson KC, Munshi NC, Prosper F, Lohr JG, Ha G, Freeman SS, van Allen EM, Adalsteinsson VA, Michor F, San Miguel JF, Ghobrial IM (2017) The mutational landscape of circulating tumor cells in multiple myeloma. Cell Rep 19(1):218–224
Rezaei M, Winter M, Zander-Fox D, Whitehead C, Liebelt J, Warkiani ME, Hardy T, Thierry B (2019) A reappraisal of circulating fetal cell noninvasive prenatal testing. Trends Biotechnol 37(6):632–644
Winter M, Hardy T, Rezaei M, Nguyen V, Zander-Fox D, Ebrahimi Warkiani M, Thierry B (2018) Isolation of circulating fetal trophoblasts using inertial microfluidics for noninvasive prenatal testing. Advanced Materials Technologies 3(7):1800066
Nelep C, Eberhardt J (2018) Automated rare single cell picking with the ALS cellcelector™. Cytometry. Part A : the journal of the International Society for Analytical Cytology 93(12):1267–1270
Keller L, Pantel K (2019) Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat Rev Cancer 19(10):553–567
Kim HS, Devarenne TP, Han A (2015) A high-throughput microfluidic single-cell screening platform capable of selective cell extraction. Lab Chip 15(11):2467–2475
Li Y, Motschman JD, Kelly ST, Yellen BB (2020) Injection molded microfluidics for establishing high-density single cell arrays in an open hydrogel format. Anal Chem 92(3):2794–2801
Zhang P, Yao J, Wang B, Qin L (2020) Microfluidics-based single-cell protrusion analysis for screening drugs targeting subcellular mitochondrial trafficking in cancer progression. Anal Chem 92(4):3095–3102
Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M (2010) Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci 107(43):18392–18397
Lu X, Martin A, Soto F, Angsantikul P, Li J, Chen C, Liang Y, Hu J, Zhang L, Wang J (2019) Parallel label-free Isolation of cancer cells using arrays of acoustic microstreaming traps. Advanced Materials Technologies 4(2):1800374
Shrestha J, Ghadiri M, Shanmugavel M, Razavi Bazaz S, Vasilescu S, Ding L, Ebrahimi Warkiani M (2019) A rapidly prototyped lung-on-a-chip model using 3D-printed molds. Organs-on-a-Chip 1:100001
Mohammadpour J, Lee A (2020) Investigation of nanoparticle effects on jet impingement heat transfer: a review. J Mol Liq 316:113819
Mohammadpour J, Rajabi-Zargarabadi M, Mujumdar AS, Ahmadi H (2014) Effect of intermittent and sinusoidal pulsed flows on impingement heat transfer from a concave surface. Int J Therm Sci 76:118–127
Akiyama Y, Shinose M, Watanabe H, Yamada S, Kanda Y (2019) Cryoprotectant-free cryopreservation of mammalian cells by superflash freezing. Proc Natl Acad Sci 116(16):7738–7743
White, F.M. (2011) Fluid Mechanics.: McGraw Hill
Hassanzadeh-Barforoushi A, Law AMK, Hejri A, Asadnia M, Ormandy CJ, Gallego-Ortega D, Ebrahimi Warkiani M (2018) Static droplet array for culturing single live adherent cells in an isolated chemical microenvironment. Lab Chip 18(15):2156–2166
Karimi A, Yazdi S, Ardekani AM (2013) Hydrodynamic mechanisms of cell and particle trapping in microfluidics. Biomicrofluidics 7(2):21501
Warkiani ME, Khoo BL, Wu L, Tay AKP, Bhagat AAS, Han J, Lim CT (2016) Ultra-fast, label-free isolation of circulating tumor cells from blood using spiral microfluidics. Nat Protoc 11(1):134–148
Hou HW, Warkiani ME, Khoo BL, Li ZR, Soo RA, Tan DSW, Lim WT, Han J, Bhagat AAS, Lim CT (2013) Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci Rep 3(1):1259
Warkiani ME, Guan G, Luan KB, Lee WC, Bhagat AAS, Kant Chaudhuri P, Tan DSW, Lim WT, Lee SC, Chen PCY, Lim CT, Han J (2014) Slanted spiral microfluidics for the ultra-fast, label-free isolation of circulating tumor cells. Lab Chip 14(1):128–137
Winter M, Cai Z, Winkler K, Georgiou K, Inglis D, Lavranos T, Rezaei M, Warkiani M, Thierry B (2019) Circulating tumour cell RNA characterisation from colorectal cancer patient blood after inertial microfluidic enrichment. MethodsX 6:1512–1520
Jang TH, Park SC, Yang JH, Kim JY, Seok JH, Park US, Choi CW, Lee SR, Han J (2017) Cryopreservation and its clinical applications. Integrative Medicine Research 6(1):12–18
Hassanzadeh-Barforoushi A, Warkiani ME, Gallego-Ortega D, Liu G, Barber T (2020) Capillary-assisted microfluidic biosensing platform captures single cell secretion dynamics in nanoliter compartments. Biosens Bioelectron 155:112113
Shemesh J, Ben Arye T, Avesar J, Kang JH, Fine A, Super M, Meller A, Ingber DE, Levenberg S (2014) Stationary nanoliter droplet array with a substrate of choice for single adherent/nonadherent cell incubation and analysis. Proc Natl Acad Sci 111(31):11293–11298
Boukellal H, Selimović Š, Jia Y, Cristobal G, Fraden S (2009) Simple, robust storage of drops and fluids in a microfluidic device. Lab Chip 9(2):331–338
Acknowledgements
M. E. W. would like to acknowledge the support of the Australian Research Council through Discovery Project Grants (DP170103704 and DP180103003) and the National Health and Medical Research Council through the Career Development Fellowship (APP1143377). The authors would like to acknowledge the assistance of Mr. Hanjie Wu during the early days of the project inception.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ding, L., Radfar, P., Rezaei, M. et al. An easy-to-operate method for single-cell isolation and retrieval using a microfluidic static droplet array. Microchim Acta 188, 242 (2021). https://doi.org/10.1007/s00604-021-04897-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-021-04897-9