
Abstract
Alzheimer’s disease (AD) is connected to aggregation of amyloid-β (Aβ) peptide and formation of insoluble plaques in the brain. Aβ level can be monitored as an AD early diagnosis route. In this study, an irregular shaped microporous gold nanostructure with a typical size of 150 × 250 nm was electrodeposited on a polycrystalline gold surface at 0 mV (vs. AgCl) using sodium alendronate. The nanostructure was then characterized by field-emission scanning electron microscopy. An electrochemical peptide-based biosensor was fabricated by immobilizing an Aβ(1–42)-binding peptide on the gold nanostructure. Binding of Aβ(1–42) by the peptide was followed electrochemically using ferro/ferricyanide as a redox probe. Differential pulse voltammograms in a potential range of 0–500 mV (vs. AgCl) with typical peak potentials at 224 mV are linear in the 3–7000 pg mL−1 Aβ(1–42) concentration range, with a 0.2 pg mL−1 detection limit. The biosensor is free of interferences and was applied to the quantitation of Aβ(1–42) in artificial cerebrospinal fluid and spiked serum samples.

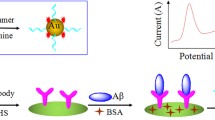
Schematic presentation of an immobilized amyloid-β(1-42)-specific peptide on the surface of a microporous gold nanostructure to fabricate an electrochemical biosensor for early diagnosis of Alzheimer’s disease. Aβ(1-42) capturing by the peptide led to repulsion of ferrocyanide/ferricyanide redox couple.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is related to aggregation of amyloid-β (Aβ) as β-sheet conformation and formation of insoluble plaques in the brain [1]. Aβ(1–40) and Aβ(1–42) are well-known forms of this oligopeptide. Aβ(1–42) has a higher tendency toward aggregate and neuronal cell death. Aβ level can be monitored in biofluids such as cerebrospinal fluid (CSF), whole blood, plasma and serum as AD early diagnosis before incidence symptoms of dementia. In patients suffering from AD, the concentration of Aβ in the CSF decreases due to accumulation in the brain (<500 pg mL−1) [2], and accumulated Aβ fibrils can be released in the bloodstream. So far, various methods were reported for early diagnosis of AD including enzyme-linked immunosorbent assay (ELISA) [3], electrochemiluminescence [4], surface-enhanced Raman spectroscopy [5], electroanalysis [6], fluorometry [7], localized surface plasmon resonance [8], and ultraviolet-visible spectrophotometry [9]. False-negative diagnoses, high cost, time consuming operation, feeble sensitivity, and low stability are some of disadvantages of these assays. Finding highly sensitive, selective and stable diagnosis methods to Aβ determination is essential and required.
Antibodies, aptamers, peptides and artificial receptors have been used extensively as biorecognition elements in biosensors fabrication [10,11,12]. The goal of diversity in the biorecognition element is usually to improve the sensitivity and specificity of the biosensors. However, antibodies suffer from some important disadvantages such as expensive production, inability to cause chemical modification, and irreversible thermal denaturation [13]. Although aptamers have important benefits including high stability, quick chemical production, none-immunogenicity, flexible conformation and easy chemical modification [14], there are several limitations such as nuclease degradability, tertiary structure dependence on the solution characters, renal filtration in some in-vivo applications, and interaction with some intracellular and similar-structure targets [15]. Synthetic peptides have some major advantages including easy C-terminus modification, small size, being chemically defined and none-immunogenicity [16].
The use of nanostructures in the design of biosensors has led to an increase in the surface concentration of the biorecognition element, creation of a specific location for the optimal immobilization of the biorecognition element, increment in binding the target into the immobilized biorecognition element and improvement in the electron transfer process [10, 11]. For preparation of a suitable nanostructured transducer surface via electrodeposition, the components of synthesis solution and electrodeposition conditions are important and effective, and should be controlled [17]. Gold nanostructures are multifunctional, conjugatable and morphologically tunable, and have specific localized surface plasmon resonance properties, controllable surface chemistry, a high surface-to-volume ratio, and high biocompatibility leading to a variety of applications in different areas including diagnostics, sensing, imaging, and therapy [18,19,20].
In this study, a specific peptide sequence creates a strong and successful bond with the surface of a microporous gold nanostructure through the thiol group of its cysteine residue to fabricate an Aβ(1–42) biosensor. In designing and selecting this peptide sequence, a high specificity to capture Aβ(1–42) was considered. For electrodeposition of the microporous gold nanostructure, alendronate was employed as an amine-bearing molecule and shape directing agent to attain a special morphology and uniform surface. Ferrocyanide/ferricyanide redox couple was employed as a redox marker to quantify Aβ(1–42) in clinical samples.
Materials and methods
Reagents and chemicals
Aβ(1–42), heparin, hemoglobin, human serum albumin (HSA), bilirubin, tetrachloroauric acid, alendronate sodium, potassium chloride, potassium ferricyanide and potassium ferrocyanide were purchased from Sigma (USA, sigmaaldrich.com). 6-Mercapto-1-hexanol (MCH), ethyl acetate, dithiothreitol (DTT), sulfuric acid, hydrochloric acid and hydrogen peroxide were purchased from Scharlau (Spain, scharlab.com). Deionized water (DW) was employed to prepare the solutions. A specific peptide sequence of CPPPPTHSQWNKPSKPKTNMK was purchased from PepTron (Korea, peptron.com).
Apparatus
The microporous gold nanostructure was evaluated by field-emission scanning electron microscopy (FESEM) with energy-dispersive X-ray spectroscopy (EDS) capability, using a TESCAN Mira 3-XMU microscope (Czech Republic, tescan.com).
A digital voltmeter of Mastech MS8340B (China, mastech-group.com) was used to measure the open circuit potential (OCP) of the working electrode during immobilization of the peptide. Voltammetric studies were performed using a potentiostat/galvanostat of μ-Autolab (the Netherlands, metrohm-autolab.com) equipped with the GPES software. A conventional three-electrode system comprising a gold disk (Au) electrode of 2 mm in diameter as the working, a platinum wire as the counter, and a silver/silver chloride, 3 mol L−1 potassium chloride as the reference electrode were employed. Differential pulse voltammograms (DPVs) at a pulse time of 50 ms, a potential sweep rate of 10 mV s−1 and a pulse width of 25 mV were recorded. All measurements were performed at room temperature otherwise stated.
Synthesis of microporous gold nanostructure
Firstly, the Au electrode (of 2 mm in diameter) was polished on a polishing pad and then on 0.05-μm alumina powder until a mirror-like surface was observed. Then, the electrode was rinsed with DW and then immersed in a 3:1 ethanol/water mixture and sonicated in an ultrasound bath for 8 min. The Au electrode was dipped in a solution containing 20 mmol L−1 tetrachloroauric acid, 500 mmol L−1 sulfuric acid and 150 mmol L−1 sodium alendronate. Electrodeposition was performed at 0 mV during 300 s and the resultant electrode was denoted as the MGN/Au electrode.
Fabrication and evaluation of the biosensor
The preformed S-S bond between the peptide molecules was broken by DTT. 50 μL of 10 μmol L−1 peptide solution was mixed with 10 μL 500 mmol L−1 DTT for 20 min. Then, 100 μL ethyl acetate was added and mixed by a vortex; then, the upper layer was discarded for three repeated times. 10 μL of the resultant solution was immediately dropped on the surface of the MGN/Au electrode and leaved at 4 °C for a desired time. After that, the electrode was rinsed with DW, and 10 μL 1 mmol L−1 MCH was dropped on the electrode surface and leaved at room temperature for 30 min. After final rinsing with DW, the biosensor was attained.
To optimize the immobilization time of the peptide on the microporous gold nanostructure surface, we electrodeposited the working electrode of the screen printed electrodes from DropSens (Spain, dropsens.com) followed by dropping the peptide, while OCP changes were measured over time at 4 °C.
To find the optimized time of binding of Aβ(1–42) into the immobilized peptide (the biosensor surface), we bound 100 pg mL−1 Aβ(1–42) at each 5-min interval at 37 °C, transferred the biosensor into a 20 mmol L−1 tris(hydroxymethyl)aminomethane hydrochloride solution, pH = 7.4 (Tris buffer) containing 0.5 mol L−1 potassium chloride and 0.5 mmol L−1 ferrocyanide/ferricyanide (1:1), and DPVs were measured at each binding time. This experiment (prolonging the binding time) was continued till the peak current in DPVs reached a constant value.
To quantify Aβ(1–42) using the biosensor, we captured different concentrations of Aβ(1–42) (0.003, 0.01, 0.03, 0.1, 0.3, 0.5, 1, 3, 5 and 7 ng mL−1) dissolved in Tris buffer by the biosensor and measured DPVs for each concentration.
Analysis of real samples
50 mL artificial CSF was prepared by mixing desired amounts of the following materials to attained the reported concentrations. Artificial CSF contained potassium chloride of 2.5 mmol L−1, magnesium chloride of 1.2 mmol L−1, D-glucose of 10 mmol L−1, calcium chloride of 2.0 mmol L−1, sodium dihydrogen phosphate of 1.24 mmol L−1, sodium chloride of 126 mmol L−1 and sodium hydrogen carbonate of 26 mmol L−1, that adjusted at pH = 7.35 (by adding HCl or NaOH of 100 mmol L−1) and bubbled with 95% air and 5% CO2. Serum samples (from 5-mL blood samples) were obtained from healthy individuals. Five serum and five CSF samples were diluted (final dilution ratio of 1:1) with Tris buffer containing 0.5 mol L−1 potassium chloride and 0.5 mmol L−1 ferrocyanide/ferricyanide (1:1), spiked with stock Aβ(1–42) solutions of 20, 50, 200 or 1000 pg mL−1 prepared in Tris buffer to attain different concentrations of Aβ(1–42) (2, 5, 20, 50 and 200 pg mL−1). DPVs were measured for these spiked samples and analyzed similar to previous section.
Results and discussion
Choice of materials
Nanostructures of noble metals (especially gold) having diversity in shape, size and morphology are highly attracted because of unique physicochemical properties. Among the noble metals and nanostructures, anisotropic gold nanostructures represent high (re)activity and surface area making them suitable for fabrication of biosensors transducer. For synthesis of gold nanostructures, electrodeposition is a potentially interested route due to advantages of easy controllability, preparation of the nanostructures as pure and uniform deposits, and tenability for fabrication of a variety of zero to two dimension nanostructures [10, 11, 17]. Therefore, electrodeposition of a microporous gold nanostructure with a high surface area was followed in this study to immobilize a thiolated Aβ(1–42)-specific peptide with a high surface concentration. This led to amplification of the electrochemical signals arriving at increment in the detection sensitivity and specificity. As for the peptide, the last 16-amino acids row of the peptide sequence is specific to capture Aβ(1–42) [21]; the cysteine-terminal of the peptide acts as a linker to the surface of microporous gold nanostructure through the thiol (functional group of cysteine)-gold binding, and the PPPP row acts as a spacer to place a distance between the capturing part and the transducer surface.
Characterization of the microporous gold nanostructure
In order to investigate surface morphology and structure of the microporous gold nanostructure, FESEM images at different magnifications were recorded, and are shown in Fig. 1a, b. The images show that the gold nanostructure comprising of irregular shaped objects covers the entire surface with typical size of 150 × 250 nm. These nanostructures provided a high porosity surface full of “micro”scale pinholes at the surface. The EDS spectrum shown in Fig. 1c also confirmed the purity of the microporous gold nanostructure without deposition/adsorption/occupation of sodium alendronate. As to the role of sodium alendronate in the electrodeposition of the microporous gold nanostructure, it should be noted that different additives with different functional groups affect the morphology, structure and size of electrodeposited gold structures [10, 11, 17, 18]. The applied potential led to a rapid formation of gold nuclei that was also accelerated by the underlying gold substrate [22]. Alendronate at once coordinated and formed partial electrostatic complex with these gold nuclei [23] through its primary amine group [24] on the (1 1 1) gold plane surface. Further growth was then followed along this (1 1 1) direction.

a, b FESEM images of the microporous gold nanostructure at different magnifications. c an EDS spectrum of the nanostructure
To check the privilege of the microporous gold nanostructure to use as the biosensor transducer, we electrochemically determined the true surface area of the MGN/Au electrode (and the Au electrode as well) using the redox transition of ferrocyanide/ferricyanide (1:1, 0.5 mmol L−1) in 0.5 mol L−1 KCl and 20 mmol L−1 tris(hydroxymethyl)aminomethane hydrochloride, pH = 7.4 by recording cyclic voltammograms at different potential sweep rates. Supplementary material S1 demonstrates these voltammograms and the corresponding analysis. According to these results, the true surface area of the MGN/Au and Au electrodes was obtained as 0.185 and 0.032 cm−2, respectively, indicating a roughness factor of 5.7 (compared to the true surface area of the Au electrode) or 5.9 (compared to the geometric surface area of the Au electrode) for the MGN/Au electrode. As indicated above, and as expected, the microporous gold nanostructure can provide a high peptide surface concentration.
Fabrication of the biosensor and quantitation of Aβ(1–42)
To prepare the biosensor through immobilization of the peptide on the MGN/Au electrode surface, the time of peptide-microporous gold nanostructure incubation should be optimized. Changes in the OCP value of the MGN/Au electrode during immobilization of the peptide are shown in Supplementary material S2. For the peptide, values of the point of isoelectric of 9.24 [25], 10.78 [26] and 10.81 [27] can be calculated. In any case, the peptide had a net positive charge at physiological pH (working pH of the biosensor), and its immobilization progression led to decrement in the negative potential; it and reached the stable value after about 80 min. This time that was selected for the peptide immobilization was shorter than that reported for the immobilization of thiolated oligonucleotide on some different gold nanostructures [10] and a thiolated aptamer on an array of gold nanodumbbells [11].
Supplementary material S3 shows DPVs recorded using the biosensor after binding 0.1 ng mL−1 Aβ(1–42) at different times. Upon increment in the binding time, the peak current lowered and after 30 min reached a stable value and remained constant upon prolonging the binding time. Therefore, 30 min was selected as the optimized binding time. This time is shorter than the optimized binding time of thrombin-antibody and thrombin-aptamer [28], platelet-derived growth factor-aptamer [29], thrombin-aptamer [30] prostate-specific antigen-aptamer [31], and troponin I-aptamer [11]. Before capturing Aβ(1–42), the biosensor surface had a specific positive surface charge related to protonation of the peptide amino acids, and the redox marker had a desired access to the biosensor surface for electron exchange. The redox marker signal is, therefore, high due to electrostatic attraction of the marker and the biosensor surface; usually, the biorecognition elements bear negative charge at physiological pH. Upon Aβ(1–42) binding, the negative surface charge of the biosensor was increased because Aβ(1–42) has an isoelectric point of 5.31 [32], and in the working pH of the biosensor, it has a net negative charge. The biosensor fabrication procedure and Aβ(1–42) determination are schematically presented in Scheme 1.

The biosensor fabrication procedure and Aβ(1–42) determination
Figure 2a shows DPVs recorded using the biosensor before and after capturing Aβ(1–42) of different concentrations of 0.003 to 7 ng mL−1, and dependency of the peak current on the Aβ(1–42) concentration as a calibration plot is displayed in Fig. 2b. Aβ(1–42) concentration increment led to a regular decrement in the peak current due to repelling the redox marker from the biosensor surface after Aβ(1–42) binding. According the calibration plot, the regression equation is Ip (μA) = (−0.6017 ± 0.0104) log (CAβ(1–42)/ng mL−1) + (7.1451 ± 0.0129), R2 = 0.9976. Considering a signal decrement value equal to 3 × SD (where SD is the standard deviation of the blank signal) as the signal of the limit of detection (LOD) and the use of the slope of the calibration plot, LOD was obtained as 0.2 pg mL−1.

a DPVs recorded using the biosensor before and after capturing Aβ(1–42) of different concentrations of 0, 0.003, 0.01, 0.03, 0.1, 0.3, 0.5, 1, 3, 5 and 7 ng mL−1. b Dependency of the peak currents on the Aβ(1–42) concentration derived from panel (a) as a calibration plot. The peak currents were measured at 224 mV
Determination of Aβ(1–42) in plasma and CSF
In Alzheimer’s disease, Aβ(1–42) plasma and CSF levels are lowered [33,34,35] because of aggregation in the brain [35]. Results of a study indicated Aβ(1–42) plasma levels as 18.05 ± 5.1 and 27.11 ± 2.45 pg mL−1 for Alzheimer’s disease and elderly normal controls, respectively [34]. A cohort study was performed on normal subjects in three age ranges of ≤34, 35 ≤ age ≤ 64, and > 64 years [33]. The results showed that the normal plasma level of Aβ(1–42) had an average value of 17.65 ± 5.71 pg mL−1. In addition, Aβ(1–42) levels in CSF of healthy persons and Alzheimer’s patients were obtained as 1678 ± 436 and 709 ± 304 pg mL−1, respectively [35]. Therefore, all of these Aβ(1–42) levels can be placed within the linear range of the biosensor, and the biosensor can determine the Aβ(1–42) levels as low as those reported for the biological fluids of the Alzheimer’s patients. To compare the figure of the merit of the biosensor with those reported for other methods of Aβ(1–42) determination, a literature survey was performed and the result is presented in Supplementary material S4. This comparison indicated that the biosensor can detect a very low concentration of Aβ. Additionally, the reported methods were mainly immunosensing; the peptide-based biosensing had advantages such as high affinity of the peptide, simplicity over the antibody, lower cost, and resistance to immunogenicity.
Reproducibility and repeatability of the biosensor
In order to evaluate the reproducibility and repeatability of the biosensor, Aβ(1–42) solutions of three different concentrations were quantified through independent measurements over a same day (for intra-day assay) as well as three different days (for inter-day assay); the results are given in Table 1. In addition, an Aβ(1–42) solution of 0.1 ng mL−1 was quantified three times using one biosensor. A relative standard deviation (RSD) of 2.95% was attained.
Fabrication reproducibility of the biosensor was evaluated by 6 times fabrication of similar biosensors and after each fabrication time, a DPV was recorded. The data are presented in Supplementary material S5. The peak current change in the voltammograms had a RSD value of 1.33%, indicating the reproducibility of the fabrication procedure.
To inspect the ability of the biosensor for re-analysis of a sample or multiple uses for analysis of several samples, we recorded a DPV using the biosensor. Then, it was bound with an Aβ(1–42) solution of 0.1 ng mL−1 and a DPV was measured. The biosensor was then dipped into hot DW at 90 °C for 5 min to release the bound Aβ(1–42). The biosensor was then rinsed with Tris buffer, and a DPV was recorded, rinsed with Tris buffer, and re-bound with the same Aβ(1–42) solution. This cycle was repeated five times, and the two five-set of DPVs are shown in Supplementary material S6. Change in the peak current difference before and after binding with Aβ(1–42) had a RSD value of 3.76%, indicating the good regeneration ability of the biosensor.
Stability, selectivity and applicability of the biosensor
The biosensor stability was determined by recording DPVs after binding with an Aβ(1–42) solution of 0.1 ng mL−1 in consecutive days. During this analysis, the biosensor was stored in Tris buffer at 4 °C. Changes in the peak current over time are displayed in Supplementary material S7. It was observed that the peak current was decreased to 86% of its initial value after 14 days, and 70% after 17 days.
Selectivity of the biosensor was appraised by recording DPVs for binding different concentrations of bilirubin, hemoglobin, heparin and HAS, and are shown in Fig. 3. Values of the peak currents in these voltammograms are reported in Supplementary material S8. It should be noted that due to the high level of HSA in the serum, a higher concentration of 50 ng mL−1 was also checked. The results indicated that these interfering species did not significantly affect the biosensor signal with a very good selectivity.

DPVs recorded using the biosensor in the presence of a bilirubin, b hemoglobin, c heparin and d HSA at different concentrations
To investigate the applicability of the biosensor in determination of Aβ(1–42) in real matrices, spiked serum and artificial CSF with Aβ(1–42) were quantified. The values added and obtained for Aβ(1–42) are displayed in Table 2. The values found by the biosensor and those added were in good accord confirming the applicability of the biosensor.
Conclusion
We used a simple electrodeposition procedure for preparation of a microporous gold nanostructure. The microporous nanostructure was checked for immobilization of an Aβ(1–42) specific peptide and detection of its binding with a high sensitivity. The high sensitivity would be due to favorable conformation of the immobilized peptide, deflection ability of the peptide for an efficient binding, higher peptide surface concentration, and/or enhanced diffusion regime of the marker. Simplicity, low-cost and lack of interference were other advantages of the biosensor. The biosensor would be applicable for point-of-care diagnosis of Alzheimer’s disease.
References
Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, Bakardjian H, Benali H, Bertram L, Blennow K (2016) Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement 12(3):292–323
Almdahl IS, Lauridsen C, Selnes P, Kalheim LF, Coello C, Gajdzik B, Moller I, Wettergreen M, Grambaite R, Bjornerud A, Brathen G, Sando SB, White LR, Fladby T (2017) Cerebrospinal fluid levels of amyloid beta 1-43 mirror 1-42 in relation to imaging biomarkers of Alzheimer’s disease. Front Aging Neurosci 9:9. https://doi.org/10.3389/fnagi.2017.00009
Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S, Lannfelt L, Pettersson FE (2007) Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J Neurochem 103(1):334–345
Song X, Li X, Wei D, Feng R, Yan T, Wang Y, Ren X, Du B, Ma H, Wei Q (2019) CuS as co-reaction accelerator in PTCA-K2S2O8 system for enhancing electrochemiluminescence behavior of PTCA and its application in detection of amyloid-β protein. Biosens Bioelectron 126:222–229. https://doi.org/10.1016/j.bios.2018.10.068
Yu X, Hayden EY, Xia M, Liang O, Cheah L, Teplow DB, Xie YH (2018) Surface enhanced Raman spectroscopy distinguishes amyloid Beta-protein isoforms and conformational states. Protein Sci 27(8):1427–1438. https://doi.org/10.1002/pro.3434
Zhou Y, Zhang H, Liu L, Li C, Chang Z, Zhu X, Ye B, Xu M (2016) Fabrication of an antibody-aptamer sandwich assay for electrochemical evaluation of levels of beta-amyloid oligomers. Sci Rep 6:35186. https://doi.org/10.1038/srep35186
Han X, Man Z, Xu S, Cong L, Wang Y, Wang X, Du Y, Zhang Q, Tang S, Liu Z, Li W (2019) A gold nanocluster chemical tongue sensor array for Alzheimer’s disease diagnosis. Colloids Surf B: Biointerfaces 173:478–485. https://doi.org/10.1016/j.colsurfb.2018.10.020
Haes AJ, Hall WP, Chang L, Klein WL, Van Duyne RP (2004) A localized surface plasmon resonance biosensor: first steps toward an assay for Alzheimer’s disease. Nano Lett 4(6):1029–1034. https://doi.org/10.1021/nl049670j
Hu T, Chen C, Huang G, Yang X (2016) Antibody modified-silver nanoparticles for colorimetric immuno sensing of Aβ(1–40/1–42) based on the interaction between β-amyloid and Cu2+. Sensors Actuators B Chem 234:63–69. https://doi.org/10.1016/j.snb.2016.04.159
Rahi A, Sattarahmady N, Heli H (2015) Zepto-molar electrochemical detection of Brucella genome based on gold nanoribbons covered by gold nanoblooms. Sci Rep 5:18060
Negahdary M, Behjati-Ardakani M, Sattarahmady N, Yadegari H, Heli H (2017) Electrochemical aptasensing of human cardiac troponin I based on an array of gold nanodumbbells-applied to early detection of myocardial infarction. Sensors Actuators B Chem 252:62–71. https://doi.org/10.1016/j.snb.2017.05.149
Tondro GH, Dehdari Vais R, Sattarahmady N (2018) An optical genosensor for enterococcus faecalis using conjugated gold nanoparticles-rRNA oligonucleotide. Sensors Actuators B Chem 263:36–42. https://doi.org/10.1016/j.snb.2018.02.097
Altintas Z (ed) (2017) Biosensors and nanotechnology: applications in health care diagnostics. Wiley, Hoboken, NJ
Hayat A, Marty JL (2014) Aptamer based electrochemical sensors for emerging environmental pollutants. Front Chem 2:41
Rozenblum GT, Lopez VG, Vitullo AD, Radrizzani M (2016) Aptamers: current challenges and future prospects. Expert Opin Drug Discovery 11(2):127–135
Zhang Q, Lu Y, Li S, Wu J, Liu Q (2018) Peptide-based biosensors. In: Koutsopoulos S. Peptide applications in biomedicine, biotechnology and bioengineering, 1st edn. Elsevier, pp 565-601
Dehdari Vais R, Sattarahmady N, Heli H (2016) Green electrodeposition of gold nanostructures by diverse size, shape, and electrochemical activity. Gold Bull 49(3–4):95–102
Sattarahmady N, Firoozabadi V, Nazari-Vanani R, Azarpira N (2018) Investigation of amyloid formation inhibition of chemically and biogenically from Citrus aurantium L. blossoms and Rose damascena oils of gold nanoparticles: toxicity evaluation in rat pheochromocytoma PC12 cells. Int J Biol Macromol 112:703–711
Rahimi-Moghaddam F, Azarpira N, Sattarahmady N (2018) Evaluation of a nanocomposite of PEG-curcumin-gold nanoparticles as a near-infrared photothermal agent: an in vitro and animal model investigation. Lasers Med Sci 33(8):1769–1779. https://doi.org/10.1007/s10103-018-2538-1
Sattarahmady N, Heidari M, Zare T, Lotfi M, Heli H (2016) Zinc-nickel ferrite nanoparticles as a contrast agent in magnetic resonance imaging. Appl Magn Reson 47(8):925–935. https://doi.org/10.1007/s00723-016-0801-9
Rushworth JV, Ahmed A, Griffiths HH, Pollock NM, Hooper NM, Millner PA (2014) A label-free electrical impedimetric biosensor for the specific detection of Alzheimer’s amyloid-beta oligomers. Biosens Bioelectron 56:83–90. https://doi.org/10.1016/j.bios.2013.12.036
Wang J, Duan G, Li Y, Liu G, Dai Z, Zhang H, Cai W (2013) An invisible template method toward gold regular arrays of nanoflowers by electrodeposition. Langmuir 29(11):3512–3517. https://doi.org/10.1021/la400433z
Kumar A, Mandal S, Selvakannan PR, Pasricha R, Mandale AB, Sastry M (2003) Investigation into the interaction between surface-bound alkylamines and gold nanoparticles. Langmuir 19(15):6277–6282. https://doi.org/10.1021/la034209c
Ross RD, Roeder RK (2011) Binding affinity of surface functionalized gold nanoparticles to hydroxyapatite. J Biomed Mater Res A 99A(1):58–66. https://doi.org/10.1002/jbm.a.33165
Kozlowski LP (2016) IPC - Isoelectric Point Calculator. Biol Direct 11:55. http://isoelectric.org/calculate.php. Accessed 02 Oct 2019
Innovagen AB (2015) PepCalc.com - Peptide property calculator. https://pepcalc.com. Accessed 02 Oct 2019
GenScript (2002) Peptide Molecular Weight Calculator. https://www.genscript.com/tools/peptide-property-calculator. Accessed 02 Oct 2019
Kang Y, Feng K-J, Chen J-W, Jiang J-H, Shen G-L, Yu R-Q (2008) Electrochemical detection of thrombin by sandwich approach using antibody and aptamer. Bioelectrochemistry 73(1):76–81. https://doi.org/10.1016/j.bioelechem.2008.04.024
Huang Y, Nie X-M, Gan S-L, Jiang J-H, Shen G-L, Yu R-Q (2008) Electrochemical immunosensor of platelet-derived growth factor with aptamer-primed polymerase amplification. Anal Biochem 382(1):16–22. https://doi.org/10.1016/j.ab.2008.07.008
Li L, Zhao H, Chen Z, Mu X, Guo L (2010) Aptamer-based electrochemical approach to the detection of thrombin by modification of gold nanoparticles. Anal Bioanal Chem 398(1):563–570. https://doi.org/10.1007/s00216-010-3922-2
Rahi A, Sattarahmady N, Heli H (2016) Label-free electrochemical aptasensing of the human prostate-specific antigen using gold nanospears. Talanta 156-157:218–224. https://doi.org/10.1016/j.talanta.2016.05.029
Hortschansky P, Schroeckh V, Christopeit T, Zandomeneghi G, Fändrich M (2005) The aggregation kinetics of Alzheimer’s β-amyloid peptide is controlled by stochastic nucleation. Protein Sci 14(7):1753–1759. https://doi.org/10.1110/ps.041266605
Zecca C, Tortelli R, Panza F, Arcuti S, Piccininni M, Capozzo R, Barulli MR, Barone R, Cardinali R, Abbrescia D (2018) Plasma β-amyloid1–42 reference values in cognitively normal subjects. J Neurol Sci 391:120–126
Pesaresi M, Lovati C, Bertora P, Mailland E, Galimberti D, Scarpini E, Quadri P, Forloni G, Mariani C (2006) Plasma levels of beta-amyloid (1–42) in Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27(6):904–905
Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, Vanderstichele H, Vanmechelen E, Blennow K (1999) Cerebrospinal fluid β-amyloid (1-42) in Alzheimer disease: differences between early-and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol 56(6):673–680
Acknowledgments
The financial support of the Iran National Science Foundation Grant NO. 96005985 is gratefully acknowledged. We would also like to thank the Research Councils of Shiraz University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 464 kb)
Rights and permissions
About this article

Cite this article
Negahdary, M., Heli, H. An electrochemical peptide-based biosensor for the Alzheimer biomarker amyloid-β(1–42) using a microporous gold nanostructure. Microchim Acta 186, 766 (2019). https://doi.org/10.1007/s00604-019-3903-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-019-3903-x