
Abstract
This paper presents a simple electrochemical approach for the detection of thrombin, using aptamer-modified electrodes. The use of gold nanoparticles results in significant signal enhancement for subsequent detection. 1,6-Hexanedithiol was used as the medium to link Au nanoparticles to a bare gold electrode. Anti-thrombin aptamers were immobilized on the gold nanoparticles’ surfaces by self-assembly. The packing density of aptamers was determined by cyclic voltammetric (CV) studies of redox cations (e.g., [Ru(NH3)6]3+) which were electrostatically bound to the DNA phosphate backbones. The results indicate that the total amount of aptamer probes immobilized on the gold nanoparticle surface is sixfold higher than that on the bare electrode, leading to increased sensitivity of the aptasensor and a detection limit of 1 pmol L−1. Based on the Langmuir model, the sensor signal displayed an almost perfect linear relationship over the range of 1 pmol L−1 to 30 nmol L−1. Moreover, the proposed aptasensor is highly selective and stable. In summary, this biosensor is simple, highly sensitive, and selective, which is beneficial to the ever-growing interest in fabricating portable bio-analytical devices with simple electrical readout procedures.

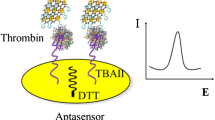
Schematic diagram showing biosensor fabrication
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Thrombin is a ubiquitous protein in mammals. Human thrombin is a dimer composed of an A chain (31 kDa) and a B chain (5.7 kDa), which are linked together by disulfide bonds. The enzyme is a glycoprotein containing 1.7% glucosamine, 1.8% sialic acid, 0.61% galactose, and 0.95% mannose. It has a molecular weight of 36.7 kDa, and an isoelectric point of 7.05. Thrombin is the last enzyme protease involved in the coagulation cascade, and it converts fibrinogen to insoluble fibrin that forms the fibrin gel both under physiological conditions and in a pathological thrombus [1]. Thrombin also has hormone-like properties, and is involved in thrombosis and platelet activation. Therefore, thrombin plays a central role in a number of cardiovascular diseases [2], and it is thought to regulate many processes in inflammation and tissue repair at the blood vessel wall. Because the high picomolar range of thrombin in blood is known to be associated with disease, it is important to be able to assess this protein at trace levels with high sensitivity [3].
Aptamers have advantages of high specificity, small molecular weight, simple structure, easy synthesis, and long-term stability. They have very promising prospects for applications in medical diagnosis, environmental monitoring [4], and biological analysis. Aptasensors for thrombin have been developed on the basis of different technologies, for example fluorescence [5], surface-enhanced Raman spectroscopy (SERS) [6, 7], and electrochemistry [8–23]. Among these, electrochemical methods have attracted most attention in the development of aptasensors because of their high sensitivity, simple instrumentation, low production cost, fast response, and portability.
Among the various electrochemical thrombin-biosensing techniques, each approach has its inherent advantages, for example, versatility, sensitivity, or selectivity, but some suffer from disadvantages including relatively high detection limits, or expensive labeling substances [14–19]. Recently, researchers described a new class of electronic aptamer-based (E-AB) thrombin sensor, based on the conformational change induced in aptamers on binding the target molecule; different signal changes were observed on adding differing concentrations of target molecules [14–23, 25, 26]. Cheng et al. developed an amplification strategy for an ultrasensitive electrochemical aptasensor for thrombin, with network-like thiocyanuric acid–gold nanoparticles [20]. Li et al. developed ultrasensitive electrochemical detection of thrombin by amplification of aptamer−nanoparticle bio barcodes [21]. In their approaches, very low detection limits were obtained, however, relatively complex multi-steps were involved in the fabrication process. Wang et al. described a quantum dot–aptamer-based ultrasensitive sensor for thrombin detection, but toxic heavy metal sulfides, such as CdS and PbS have to be introduced [22]. Electrochemiluminescence has also been used for thrombin sensing, however use of the ECL biosensor is restricted by the fact that significant loss of biomolecular activity results from mixing biomolecules with ruthenium(II) tris(bipyridine) (Ru(bpy) 2+3 ), the most frequently used as ECL substrate [23].
In our work, the fundamental electrostatic interaction between redox-active cations (e.g., [Ru(NH3)6]3+) and the negatively charged phosphate backbone of DNA is the basis of our quantitative analysis for thrombin. Thrombin is positively charged and interacts with the negatively charged phosphate backbone, displacing the [Ru(NH3)6]3+ and resulting in a substantial decrease in the signal observed by CV measurements. Of note, in order to enhance the sensitivity of the aptasensor, gold nanoparticles 15 nm in diameter were used to immobilize more anti-thrombin DNA aptamers on the surface via self-assembly chemistry. The results indicate that the total amount of aptamer probes immobilized on the gold nanoparticles’ surfaces is sixfold higher than on the bare electrode, thus offering the very low detection limit of 1 pmol L−1. In the future this approach may be a highly sensitive, easy, and accurate method for detection of any protein.
Materials and methods
Apparatus and reagents
Cyclic voltammetric experiments were performed with CHI660C electrochemical workstation (Shang Hai Chen Hua Instruments, China). The three-electrode system consisted of a gold disk working electrode (2 mm in diameter; Shang Hai Chen Hua Instruments), a platinum wire auxiliary electrode and an Ag/AgCl (Sat. KCl) reference electrode.
The synthetic anti-thrombin oligonucleotides, ssDNA (5′-(SH)-(CH2)6-CCA TCT CCA CTT GGT TGG TGT GGT TGG-3′) [14, 24] were obtained from TaKaRa Biotechnology (Dalian, China). Thrombin, lysozyme, bovine serum albumin and bovine hemoglobin from lyophilized erythrocytes were purchased from CNS Bioservices. 6-Mercapto-1-hexanol (MCH) was obtained from J&K Chemical. Hexaammineruthenium(III) chloride ([Ru(NH3)6]Cl3), 1,6-hexanedithiol (HDT, 97%), and tris-(2-carboxyethyl) phosphine hydrochloride (TCEP) were obtained from Alfa Aesar (Tianjing, China). Hydrogen tetrachloroaurate(III) hydrate (HAuCl4), sodium citrate, and tris-base were purchased from Sigma–Aldrich.
All other reagents were of analytical reagent grade. All solutions were prepared with doubly distilled water. Phosphate buffer pH 7.0 containing 1 mmol L−1 K3Fe(CN)6 was chosen as the supporting electrolyte for electrode characterization, and I-B solution (20 mmol L−1 Tris-HCl–0.1 mol L−1 NaCl–0.2 mol L−1 KCl–5 mmol L−1 MgCl2, pH 7.41) was used as supporting electrolyte for thrombin assays.
Phosphate buffer pH 7.0 containing 1 mmol L−1 K3Fe(CN)6 was chosen as the supporting electrolyte.
Colloidal gold preparation
Colloidal Au particles were prepared by citrate reduction of HAuCl4 in aqueous solution [27]. Particle sizes were characterized by transmission electron microscopy (TEM) photographs, X-ray Diffraction (XRD), and UV–visible spectrophotometry (UVs). As shown in the Electronic Supplementary Material, Fig. S1a, average particle diameters were consistently in the range of ∼15 nm with standard deviations of 1–2 nm. Electronic Supplementary Material Fig. S1b shows the characteristic absorption peak at approx. 520 nm, consistent with the absorption of the Au-NPs of this size [28, 29]. X-ray diffraction (XRD) experiments were also carried out to investigate the purity and size distribution of the sample (data not shown). By the Debye–Scherrer equation, the calculated diameter of AuNPs is in good agreement with the result from the TEM.
DNA preparation
TCEP immobilization buffer (I-B) solution (99 μL) and 1 μL 100 nmol L−1 anti-thrombin aptamer were mixed together. Herein, TCEP was a reducing agent, which was intended to disrupt any disulfide bonds and ensure that free –SH groups are ready to react with the gold surface. Afterwards, the mixture heated to 90 °C and left to gradually cool to room temperature in I-B. This heating and cooling step helps to maintain the structural flexibility of the aptamers (for binding thrombin) [30]. Afterwards, a 20 μL droplet of anti-thrombin aptamer (1 μmol L−1 in I-B) was placed on the AuNP–HDT–Au electrode surface for later electrochemical analysis.
Electrode pretreatment
Gold substrates were cleaned by immersion in a 3:1 mixture of concentrated H2SO4 and 30% H2O2 (piranha) for 5 min at 70 °C, followed by rinsing with copious amounts of water. CAUTION: piranha reacts violently with organic solvents, and should be handled with extreme caution. After this, the electrode was polished consecutively with 1.0, 0.3, and 0.05-μm alumina slurry on a polishing pad. The electrode was then ultrasonically cleaned in ethanol for 1 min and in water for 2 min. The electrode was subsequently voltammetrically cycled in 0.5 mol L−1 H2SO4 with the potential between −0.2 and +1.6 V at 0.1 V s−1 until a representative cyclic voltammogram of a clean gold electrode was obtained.
Experimental procedure
For preparation of the thrombin biosensor, the pre-cleaned Au electrode was initially dipped into ethanol solution containing 2 mmol L−1 HS(CH2)6SH(1,6-HDT) for more than 24 h at room temperature. After being thoroughly rinsed with ethanol and deionized water, the electrode was dried under an N2 stream and then dipped into the AuNP colloid for 10 h. The AuNP–1,6-HDT–gold electrode was rinsed with deionized water and dried under an N2 stream. Afterwards, the modified electrode was incubated with a 20-μL droplet of anti-thrombin aptamer (1 μmol L−1 in I-B) for 12 h at room temperature in 100% humidity. After being washed with the Tris-buffer (pH 7.41), the electrode was incubated for 1 h in 1 mmol L−1 MCH (in I-B) to fill any unoccupied gaps on the gold surface, in order to prevent subsequent non-specific adsorption. Finally, the gold surface was rinsed again with I-B and then with deionized water, followed by drying under an N2 stream. For the packing density analysis of aptamers, the anti-thrombin aptamer-modified electrode was incubated with 20-μL droplets of [Ru(NH3)6]Cl3, at concentrations ranging between 2 and 10 μmol L−1. until complete electrostatic saturation was achieved between the redox cations (e.g., [Ru(NH3)6]3+) and the negatively charged phosphate backbone of DNA. The charge densities of the aptamer-modified electrodes were monitored by the CV response of surface-bound [Ru(NH3)6]3+. For detection of thrombin, 20-μL droplets of thrombin in I-B of different concentrations were deposited on to the aptasensor and kept for 1 h at 37°C. Upon target molecule binding, a substantial CV current decrease was observed. The complete process used for fabrication of the aptasensors is shown in Scheme 1.

Schematic diagram showing biosensor fabrication
Results and discussion
Characterization of modified electrode
In order to clarify the electrochemical properties of the resulting aptasensor, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were used to monitor the process of fabrication of the aptasensor in each step. As expected, the behavior of K3[Fe(CN)6]/K4[Fe(CN)6] on a bare gold electrode was reversible with a peak-to-peak separation ∆E p of 70 mV (Fig. 1a, curve 1). After self-assembly of 1,6-hexanedithiol (HDT) was on the surface of the gold electrode, the shape of the CV changed dramatically (Fig. 1a, curve 2), indicating that HDT formed a well-packed monolayer on the surface of the gold electrode and was responsible for passivation of the electrode. After self-assembly of AuNPs on the surface of the gold electrode via HDT, the reversibility of the AuNP–HDT–gold electrode was markedly restored with a ∆E p of 63 mV, and the peak current even surpassed that of the bare gold electrode (Fig. 1a, curve 3). This suggests that the AuNPs provide the necessary conduction pathways and promote electron transfer between the redox marker and the electrode surface. After the DNA probe was immobilized on the surface of the AuNP–HDT–gold electrode, a significant increase in the ∆E p of the electrode was observed (Fig. 1a, curve 4), probably because of the established kinetics barrier between [Fe(CN)6]3−/4− and the negatively charged phosphate backbone of the DNA. After rinsing with MCH, the peak current increased, indicating that MCH removed nonspecific anti-thrombin DNA adsorption on the AuNPs surface.

(a) CVs obtained from 0.10 mol L−1 phosphate buffer (pH 7.0) containing 1.0 mmol L−1 Fe(CN) 3−6 at bare Au (1), HDT-modified Au electrode (2), AuNP–HDT-modified Au electrode (3), ssDNA–AuNPs–HDT-modified Au electrode (5), and cyclic voltammogram after incubation in MCH (4), (b) Nyquist plots obtained from 0.10 mol L−1 phosphate buffer (pH 7.0) containing 1.0 mmol L−1 Fe(CN) 3−6 at bare Au (1), Au-NP–HDT-modified Au electrode (2), ssDNA–AuNP–HDT-modified Au electrode (3), and the impedance diagram after incubation in MCH (4). The inset shows the Nyquist plot of the HDT-modified Au electrode. The scan rate was 100 mV s−1
Figure 1b shows the Nyquist plots of impedance spectra obtained at each preparation step. The change in semicircle diameter is a result of the change in the interfacial resistance, R et, because of electron transfer from the modified electrode to ferricyanide in solution. Curve 1 represents the electrochemical impedance of the bare gold electrode, which is almost a straight line. After the self-assembly of HDT (Fig. 1b, inset), the diameter of the semicircle increased dramatically and the R et was 500 kΩ. Figure 1b (curve 2) shows that the electrochemical response was almost a straight line, which is very similar to curve 1, when AuNPs were self-assembled on the surface of the HDT modified gold electrode. This can be attributed to the fact that AuNPs can further enhance the electron transfer rate and improve the adsorption capacity of the electrode, and again indicates that the AuNPs can increase the effective electrode surface area and thus have the potential to anchor more DNA probes to amplify the signal. After anti-thrombin aptamers were self-assembled on to the surface of the AuNP–HDT–gold electrode, the Ret markedly increased to 4 kΩ (Fig. 1b, curve 3). This can be attributed to the fact that the redox couple of K3[Fe(CN)6]/K4[Fe(CN)6] was affected by electrostatic repulsive forces from the self-assembled anti-thrombin aptamer. After being passivated by MCH, the impedance decreased apparently (Fig. 1b, curve 4), indicating that MCH removed nonspecific anti-thrombin DNA adsorption on the AuNPs surface and dramatically reduced the capacitance of the electrode [31, 32].
A bare Au electrode and an AuNP-modified Au electrode with the same geometric surface area were characterized by SEM; the results are shown in Electronic Supplementary Material Fig. S2. One can observe that the AuNPs have been incorporated on to the surface of the Au electrode. Herein, the active surface areas of a bare Au electrode and an AuNP-modified Au electrode were measured by CV in 0.5 mol L−1 H2SO4 (Electronic Supplementary Material Fig. S3). Assuming that a specific charge of 386 μC cm−2 is required for gold oxide reduction [33], the AuNP-modified Au electrode had a total active surface of 10.95 mm2, whereas that of the corresponding bare Au electrode was 3.88 mm2, indicating that the active electrode surface area had been enlarged approximately three times by AuNP modification.
Packing density of aptamers
When the aptamer-modified surface was exposed to a solution containing [Ru(NH3)6]3+, the redox cations bound electrostatically to the negatively charged phosphate backbone by replacing the native charge compensation ions (Na+), and reached an ion-exchange equilibrium. Thus, the packing density of anti-thrombin aptamer probe can be calculated by the measured charge acquired from the reduction of the [Ru(NH3)6]3+ that is electrostatically associated with the negatively charged backbone of the aptamer. Figure 2 shows the cyclic voltammograms obtained from anti-thrombin aptamer–AuNPs–HDT–gold electrodes incubated in different concentrations of [Ru(NH3)] 3+6 from 2 μmol L−1 to 8 μmol L−1. As shown in Fig. 2, the peak current increases with the increasing concentration, and at 8 μmol L−1 [Ru(NH3)] 3+6 the redox current reaches its maximum value which ensures saturation of the DNA-modified surface with the redox-active complex. Under the saturation conditions, the surface concentration of [Ru(NH3)6]3+, Г Ru, can be calculated by use of Eq. 1:
where Q is the charge obtained by integrating the reduction peak area of surface-bound [Ru(NH3)6]3+, n is the number of electrons involved in the redox reaction, F is Faraday’s constant, and A is the active electrode area. The calculated value of Г Ru can be converted to the surface density of DNA, Г DNA, by use of the equation:
where m is the number of nucleotides in the anti-thrombin DNA, z is the valence of the redox cations, and NA is Avogadro’s number [34].

Cyclic voltammograms obtained from anti-thrombin aptamer–AuNPs–HDT–gold electrodes in 10 mmol L−1 Tris buffer (pH 7.41) in the presence of [Ru(NH3)] 3+6 at different concentrations: (1) 2 μmol L−1, (2) 4 μmol L−1, (3) 6 μmol L−1, (4) 8 μmol L−1. The scan rate was 500 mV s−1
By use of Eqs. 1 and 2 the packing density of the DNA probes is estimated to be 2.317 ± 0.002 × 1012 molecules cm−2 for the AuNP-modified electrode surface, thus the total amount of DNA probes immobilized on the gold surface is 2.537 ± 0.002 × 1011 molecules, which is sixfold higher than that obtained from the bare gold electrode. This further confirms that AuNPs can increase the amount of immobilized aptamer, and thus aptasensors with AuNPs modification can capture many more target molecules, which greatly enhances the signal response.
Detection of thrombin using [Ru(NH3)6]3+ as a redox marker on the surface
Upon incubation in a solution containing thrombin, there are significant changes in the exposed charged surface. Most notably, because of the positive nature of thrombin, there is a neutralization of the negatively charged phosphate backbone, thus thrombin displaces [Ru(NH3)6]3+ and allows it to bind only where thrombin does not. This results in a substantial decrease in the signal observed by CV measurements, which is indicative of the redox behavior of surface-bound [Ru(NH3)6]3+. This decrease in CV response can be quantified by use of Eq. 1 and different concentrations of thrombin (assuming the surface packing densities of aptamer are similar) and thus, the system can be used to quantify the amount of thrombin in an unknown sample.
In Fig. 3a, we fixed the concentration of [Ru(NH3)6]3+ at 8 μmol L−1 (under saturation conditions), and kept increasing the concentration of thrombin from 1 pmol L−1 to 30 nmol L−1. It can be clearly seen that the CV peak decreases gradually as the concentration of thrombin is increased. In order to quantitatively characterize the detection limit and response range of the thrombin biosensor, we took the relative decrease in the integrated charge of the reduction peak (Fig. 3a). In particular, we showed the dependence of the relative decrease in the charges obtained by integration of the reduction peak of [Ru(NH3)6]3+ in the cyclic voltammograms before and after incubation with thrombin, ∆Q/Q=|(Q − Q i)|/Q, (where Q and Q i denote, respectively, the charges obtained before and after incubation with the different concentration of thrombin). As shown in Fig. 3a, the redox current in CV decreases gradually as the concentration of thrombin is increased from 1 pmol L−1 to 30 nmol L−1. Figure 3b shows that as the thrombin concentration rises, the relative decrease in the reduction signal is greater and more rapid, and the signal seems to saturate at 10 nmol L−1 thrombin. This would make sense if the following assumptions were made: the ratio between the number of molecules of protein and the number of molecules of aptamer is 1:1, indicating a very small dissociation constant of the aptamer–ligand complex. A surface density of aptamer of 2.317 ± 0.02 × 1012 molecules cm−2 immobilized on the AuNP–HDT–gold electrode gives a value of 2.537 ± 0.02 × 1011 anti-thrombin aptamers on the electrode surface. Based on the stoichiometric ratio of 1:1 and an incubation volume of 20 μL, the maximum detection limit of thrombin is estimated to be 2.1 × 10−8 mol L−1, which is of similar magnitude to the experimental value of 10−8 mol L−1.

(a) Cyclic voltammograms obtained from sensing system for different concentrations of thrombin: (1) 0 nmol L−1, (2) 0.001 nmol L−1, (3) 0.01 nmol L−1, (4) 0.02 nmol L−1, (5) 0.06 nmol L−1, (6) 0.1 nmol L−1, (7) 1 nmol L−1, (8) 3 nmol L−1, (9) 10 nmol L−1, (10) 20 nmol L−1, and (11) 30 nmol L−1. (b) Decrease in the integrated charge (reduction peak) as a function of concentration of thrombin (experimental conditions as for Fig. 2). Aptamer-modified electrodes with surface density 2.317 ± 0.02 × 1012 molecules cm−2 were used. The error bars indicate the standard deviation of four successive measurements for each incubation. Inset is linearized adsorption isotherm of thrombin binding to anti-thrombin aptamers on AuNPs surfaces based on the Langmuir model. The line is the best linear fit to the experimental data
Of note, the adsorption isotherm of thrombin binding to anti-thrombin aptamers can be regarded as arising from protein–aptamer interaction on AuNP surfaces, if the following assumptions are made: every binding site is equivalent and the ability of the molecule to bind is independent of whether adjacent sites are occupied, because DNA aptamer monolayers rarely form islands of very dense aggregates because of electrostatic repulsion of the phosphate backbones. Based on the classical Langmuir model, a linearized form of the adsorption isotherm is shown:
in which c is the concentration of thrombin in solution, ∆Q/Q is the sensor signal, (∆Q/Q)sat is the saturated sensor signal, and K D is the dissociation constant. As shown in Fig. 3b inset, the experimental results for c/(∆Q/Q) are in an almost perfect linear relationship over the range 1 pmol L−1 to 30 nmol L−1 thrombin with R = 0.9996. Based on the linear fit of the above equation, the K D of thrombin and the anti-thrombin aptamer can be calculated as 0.1738. This small dissociation constant indicates that the protein is strongly bound to the anti-thrombin aptamer. Additionally, this AuNPs-modified aptasensor has a very low detection limit of 1 pmol L−1. Compared with the above AuNP-modified electrode, the bare electrode has much lower ∆Q/Q values when both were incubated in the same concentration of thrombin (as shown in Table 1). The results indicate that the sensitivity of the AuNP-modified electrode is much higher than that if the bare electrode.
Real sample analysis
The practical applicability of the proposed aptamer sensor was investigated by spike and recovery experiments. The Au–AuNPs–aptamer-based sensor did not detect thrombin in the serum sample. This was because thrombin is not present in the blood and plasma of healthy subjects when coagulation is not occurring [35]. However, to examine the applicability of this aptamer sensor in serum samples, we performed spike and recovery experiments. As shown in Table 2, when serum samples were spiked with 0.02, 0.22, 3.22, and 8.22 nmol L−1 thrombin, recoveries from standard additions were 90.0%, 92.3%, 91.9%, and 96.9% respectively, which clearly indicates the potential of this aptamer sensor for thrombin detection in real biological samples.
The specificity and the stability of the aptasensor
The specificity of the aptasensor was determined by challenging it with three other similar proteins, each at 10 μmol L−1 concentration. As shown in Fig. 4a, the CV signals for the other proteins are only as small as the background (in the absence of thrombin). In Fig. 4b, the value on the y-axis can be normalized by the CV current decrease for thrombin versus background, the relative signals for lysozyme, bovine hemoglobin, and bovine serum at concentrations of 10 μmol L−1 are below ∼5%. This excellent selectivity arises from the high specificity of the anti-thrombin aptamer.

(a) Cyclic voltammograms obtained from the sensing system on exposure to three nucleosides in 10 mmol L−1 Tris-HCl solution: (1) 8 μmol L−1 [Ru(NH3)6]3+, (2) lysozyme, (3) bovine hemoglobin, (4) bovine serum albumin, (5) thrombin. The concentration of thrombin was 1 nmol L−1. All the other enzymes were 10 μmol L−1. Other conditions were the same as for Fig. 2. (b) Relative response of the system to the different enzymes
The stability of the aptasensor fabricated for the detection of thrombin is also important in practical applications. Therefore, the storage stability of the aptasensor was checked. The results show that the CV signal is only reduced by 6.07% of the original value after one-month of storage in buffer solution.
Nevertheless, our proposed sensor suffers from the limitations associated with being a “signal-off” architecture, that is, at most, only 100% of the signal can be suppressed. Moreover, for the signal-off sensor, contaminants that degrade the aptamer can give rise to “false positives” that are difficult to distinguish from signals arising from the binding of authentic analyte. Thus, recently, the signal-on architecture has been further investigated in our work.
Conclusions
In this paper we propose a novel signal-off electrochemical aptasensor for detection of thrombin by anchoring 15 nm AuNPs on to a gold electrode with self-assembled monolayers of HDT. Anti-thrombin DNA aptamer is used as a molecular recognition element, and surface-bound [Ru(NH3)] 3+6 as a redox marker. The positively charged thrombin attaches to the aptamer, repelling the [Ru(NH3)6]3+ which was electrostatically attracted to the negatively charged DNA phosphate backbone. [Ru(NH3)6]3+ ions enter the solution, leading to a substantial decrease in the CV current. The proposed approach, in which AuNPs with a large specific surface area provide a new platform with sixfold more aptamer immobilization for target molecule capture than the aptasensor without AuNP modification, exhibits remarkably high sensitivity, stability, and selectivity. This biosensor has been used to detect thrombin with the very low detection limit of 1 pmol L−1, and an almost perfect linear relationship can be speculated on the basis of the adsorption isotherm equation. In addition, stability is maintained for over one month when the aptasensor is stored in buffer solution. This procedure could potentially be applied to the aptamer-based detection of other proteins and small molecules.
References
Holland CA, Whinna HC, Church FC, Henry AT (2000) FEBS Lett 484:87–91
Stubbs MT, Bode W (1993) Thromb Res 69:1–58
Centi S, Tombelli S, Minunni M, Mascini M (2007) Anal Chem 79:1466–1473
Xiao Y, Rowe AA, Plaxco KW (2007) J Am Chem Soc 129:262–263
Stojanovic MN, Prada P de, Landry DW (2001) J Am Chem Soc 123:4928–4931
Bizzarri AR, Cannistraro S (2009) Anal Biochem 393:149–154
Cho HS, Baker BR, Pagba CV, Laurence TA, Lane SM, Lee LP (2008) Nano Letters 8:4386–4390
Li XX, Shen LH, Zhang DD (2008) Biosens Bioelectron 2:1624–1630
Pavlov V, Xiao Y, Willner I (2004) J Am Chem Soc 126:11768–11769
Floch FL, Ho HA, Leclerc M (2006) Anal Chem 78:4727–4731
Li XM, Xia JP, Li W (2010) Chemistry-AN Asian Journal 5:294–300
Lu Y, Zhu NN, Yu P (2008) Analyst 133:1256–1260
Huang YC, Ge BX, Sen D (2008) J Am Chem Soc 130:8023–8039
Xiao Y, Piorek BD, Plaxco KW, Heeger AJ (2005) J Am Chem Soc 127:17990–17991
Radi A, Sanchez JLA, Baldrich E, O’Sullivan CK (2006) J Am Chem Soc 128:117–124
Kang D, Zuo XL, Yang RQ, Xia F, Plaxco KW, White RJ (2009) Anal Chem 81:9109–9113
Xiao Y, Lubin AA, Heeger AJ, Plaxco KW (2005) Angew Chem Int Ed 44:5456–5459
Bang GS, Cho SY, Kim BG (2005) Biosens Bioelectron 21:863–870
Cai H, Lee T Ming-Hung, Hsing I-Ming (2006) Sens Actuators B 114:433–437
Zheng J, Feng WJ, Lin L, Zhang F, Cheng GF, He PG, Fang YZ (2007) Biosens Bioelectron 23:341–347
He PL, Shen L, Cao YH, Li DF (2007) Anal Chem 79:8024–8029
Hansen JA, Wang J, Kawde AN, Xiang Y, Gothelf KV, Collins G (2006) J Am Chem Soc 128:2228–2229
Wang XY, Dong P, Yun W, Xu Y, He PG, Fang YZ (2009) Biosens Bioelectron 24:3288–3292
Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ (1992) Nature 355:564–566
Polsky R, Gill R, Kaganovsky L, Willner I (2006) Anal Chem 78:2268–2271
Willner I, Zayats M (2007) Angew Chem Int Ed 46:6408–6418
Frens G (1973) Nature Phys Sci 241:20–22
Doron A, Katz E, Willner I (1995) Langmuir 11:1313–1317
Brown KR, Walter DG, Natan MJ (2000) Chem Mater 12:306–313
Bini A, Minunni M, Tombelli S, Centi S, Mascini M (2007) Anal Chem 79:3016–3019
Herne TM, Tarlov MJ (1997) J Am Chem Soc 119:8916–8920
Levicky R, Herne TM, Tarlov MJ, Sajita SK (1998) J Am Chem Soc 120:9787–9792
Szamocki R, Velichko A, Holzapfel C (2007) Anal Chem 79:533–539
Yu HZ, Luo CY, Sankar CG, Sen D (2003) Anal Chem 75:3902–3907
Shuman MA, Majerus PW (1976) J Clin Invest 58:1249–1258
Acknowledgments
This project was financially supported by National Natural Science Foundation of China (nos 20903008, 20973019, and 50725208) and the State Key Project of Fundamental Research for Nanoscience and Nanotechnology (2006CB932300). The authors also acknowledge M. Wyatt for comments on manuscript.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1804 kb)
Rights and permissions
About this article
Cite this article
Li, L., Zhao, H., Chen, Z. et al. Aptamer-based electrochemical approach to the detection of thrombin by modification of gold nanoparticles. Anal Bioanal Chem 398, 563–570 (2010). https://doi.org/10.1007/s00216-010-3922-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-3922-2