
Abstract
The authors introduce a new kind of surface artificial biomimetic receptor, referred to as aptameric imprinted polymer (AIP), for separation of biological macromolecules. Highly dispersed magnetic nanoparticles (MNPs) were coated with silica and then functionalized with methacrylate groups via silane chemistry. The aptamer was covalently immobilized on the surface of nanoparticles via a “thiol-ene” click reaction. Once the target analyte (bovine serum albumin; BSA) has bound to the aptamer, a polymer is created by 2-dimensional copolymerization of short-length poly(ethylene glycol) and (3-aminopropyl)triethoxysilane. Following removal of BSA from the polymer, the AIP-MNPs presented here can selectively capture BSA with a specific absorbance (κ) as high as 65. When using this AIP, the recovery of BSA from spiked real biological samples is >97%, and the adsorption capacity is as high as 146 mg g−1. In our perception, this method has a wide scope in that it may be applied to the specific extraction of numerous other biomolecules.

Schematic presentation of the AIP (aptamer-imprinted polymer) introduced here. The surface of silica coated magnetic nanoparticles is modified with a polymer that is covalently modified with an aptamer against bovine serum albumin (BSA).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Artificial receptors, such as aptamers and molecular-imprinted polymers, demonstrate high antibody-like properties due to their molecular recognition ability. This binding ability has made them suitable for selective recognition. Artificial receptors mainly recognize guest molecules (i.e. proteins) through non-covalent interactions and also similarity of their three-dimensional structures.
Aptamers have been widely used in the fields of biosensing, diagnostics and therapeutics. Aptamers have higher binding affinity for small molecules, proteins, cells and tissues. Hydrogen bonding, hydrophobic and electrostatic interactions are the major chemical interactions, which lead to high affinity of aptamers to their respective targets. Such specificity allows them to even distinguish structurally related molecules such as proteins containing slightly different sequences. Aptamers are less expensive compared to antibodies and have been described as diagnostic tools for protein detection assays. Moreover, aptamers due to high affinity and specificity, might be ideal reagents for a wide range of analytical applications based on the capture, separation and biosensing of some ligands that cannot be recognized by antibodies, such as small molecules or macromolecules.
DNA and RNA aptamers and molecularly imprinted polymers (MIPs) have been recognized as promising chemical structures with natural-like molecular recognition properties. Although they are very different from each other, but their binding behavior is based on the lock-and-key principle and they are able to memorize the typical features of a guest molecule due to selective binding and shape of cavity [1]. MIPs are polymeric assembly that shows great selectivity towards a well-defined molecular structure or a group of related compounds. Functional monomers are organized around a template molecule by non-covalent forces such as hydrogen bonding, electrostatic or hydrophobic interactions. Molecular imprinting has been applied mostly for low-molecular weight molecules and their application for bio-macromolecules such as proteins has some limitations. One of the major challenges faced by bio-macromolecules for imprinting applications is diffusion limitation, which makes difficult the removal and uptake of template [2,3,4]. To resolve this problem, surface [5,6,7,8,9], epitope [10,11,12,13,14] and solid-phase imprinting [15, 16] have been proposed as feasible strategies for protein imprinting. It is expected that binding capacity and site availability of imprinted bio-macromolecules would be improved by these modifications.
Functionalized magnetic nanoparticles modified with protein surface-imprinted polymer were used for purification of peptides and proteins [17, 18]. By coat of a thin imprinted polymer layer on the surface of a nanoparticle with a small size, large surface-to-volume ratio, and well-defined material shape, the selectivity of such magnetic nanoparticles would be increased to selectively capture a protein in a complex matrix. Moreover, to improve the selectivity of magnetic nanostructures, they can be modified by silane or other functional groups (such as –COOH, –CHO, and –NH2) [19]. However, synthesis of MIPs with high selectivity toward bio-macromolecules remains great challenge. Poma and co-workers generated the novel hybrid aptamer–molecularly imprinted polymer based on the solid-phase imprinting technique [20]. They used this aptameric-imprinted polymer against the cocaine as a small molecule.
In addition, double recognition of small molecules with aptamer grafted at MIP cavities was used for biosensing of erofloxacin and oligonucleotide, respectively [21, 22]. In this study, a new generation of modern molecular imprinted polymers is introduced by applying specific aptamers in their structures for recognition of protein. This new modified nanoparticle is much more selective toward proteins in comparison with traditional MIPs. So, the resulting AIP-MNPs were able to selectively capture the bovine serum albumin (BSA) within a complex matrix. This AIP-MNP provides highly discriminative and adjustable tool for high-precision identification of a large amount of protein targets for diagnostic and proteomic applications.
Experimental
Materials
Tetraethoxysilane (TEOS), 3-aminopropyltriethoxylsilane (APTES), γ-Methylmethacrylate tri-methoxy silane (γ-MAPS, 98%), PEG-diacrylate, poly(ethylene glycol) (PEG, MW = 400), BSA (pI = 4.9, MW = 66.0 kDa), HSA (pI = 4.7, MW = 66.5 kDa), RNase A (pI = 9.4, MW = 13.7 kDa) and Lyz (pI = 11.2, MW = 13.4 kDa) and all other chemicals were analytical grade and were purchased from Sigma Company (www.sigmaaldrich.com). Double distilled water was used in all experiments. The aptamer against BSA (5’-GCCCGCCGTGGCTGGGTCTTCCTTGGT CGGTCTACAAAAAAAAAA-SH-3′), was ordered by SBS Genetech Co. Ltd. (www.sbsbio.com).
Preparation of Fe3O4@SiO2@acrylate@aptamer
Hydrophilic Fe3O4 nanoparticles with about mean diameter of 20 nm were synthesized and covered with TEOS and mixture of γ-MAPS and PEG-diacrylate, respectively (explained in electronic supplementary material). The Fe3O4@SiO2@acrylate were decorated with aptamer by self-assembly method [23]. Summarily, 15 ml L of Fe3O4@SiO2@acrylate solution (5% w/v) mixed with thermal initiator V50 (1.0 mg) and then were reacted with the 7.5 μL of 10 μM aptamer (pH =7.4, 10 mM Tris.HCl) incubating at 55 °C for 5 h. Aptamer solution was previously activated with 100 μL 1,4-dithiothreitol (DTT) solution (10 mg mL−1) by being incubated at 30 °C for 30 min. The ss-aptamer concentration in supernatant solution before and after “thiol-ene” click reaction was monitored by UV–Vis spectrophotometer. Finally, the Fe3O4@SiO2@acrylate@aptamer were washed with buffer and stored at 4 °C before any use.
Preparation of NIPs, MIPs and AIP-MNPs
BSA was chosen as a template to immobilize on the core–shell structures of MIPs and AIP-MNPs. Fe3O4@SiO2@acrylate and Fe3O4@SiO2@acrylate@aptamer used as principal core-shell of MIPs and AIP-MNPs, respectively. In Brief, a volume of 2.5 mL of 1000 mg mL−1 BSA solution was added to the 10 mg of Fe3O4@SiO2@acrylate and Fe3O4@SiO2@ acrylate@aptamer, respectively. The mixture was incubated for 2 h at 25 °C and then washed with buffer and consequently dispersed in 7.5 mL of that. In this step, 15 μL APTES and 1.5 μL PEG400 were added dropwisely and it was orbitally rotated for 36 h. Then MIPs and AIP-MNPs were rinsed several times with water and buffer respectively till the supernatant was clear. To remove template BSA, a volume of 5.0 mL of NaOH 0.1 M was added and shake at 25 °C for 12 h. The products were then repeatedly washed using water and buffer and stored at 4 °C before any use. NIPs were produced without template BSA in the same way as MIPs and AIP-MNPs.
Binding capacity, reusability and reproducibility of MIPs and AIP-MNPs were measured and their performance and selectivity in extraction from complex matrix at presence of interfering protein was compared with NIP (explained in electronic supplementary material).
Results and discussion
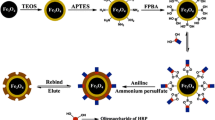
The schematic of AIP-MNPs synthesis is shown in Scheme 1. In this procedure, highly dispersed MNPs were synthesized by a modified protocol [24]. Then, to obtain Fe3O4@SiO2, the surface of the MNPs was coated with a tetraethoxysilane (TEOS) among sol-gel method [25]. A silica shell on the surface of MNPs prevented agglomeration of MNPs due to negatively charges of hydroxyl groups and also provided accessibility for easy modification with various groups and bio-conjugation. The surface hydroxyl groups of Fe3O4@SiO2 were further masked with acrylate-functionalized substances, γ-MAPS as precursor and PEGDA as a crosslinking agent by using sol–gel method. γ-MAPS provides accessible acrylate-functionalized groups on the surface of the MNPs to bind to thiol groups corresponding to aptamer and stabilizes it on the surface of MNPs. Moreover, coating the Fe3O4@SiO2 particles with PEGDA provides an excellent biocompatibility and hydrophobicity for particles, which is of great interest in medical applications. In addition, adding cross linker among sol-gel process inhibits MNPs from extra aggregation without reducing surface density of functional groups (Table S-1) [26]. Subsequently, aptamer was immobilized on the surface of Fe3O4@SiO2@acrylate via thiol attachment to acrylate groups. Therefore, the tertiary structure of target protein, BSA, was recognized by aptamer decorated on the surface of the Fe3O4@SiO2@acrylate. Then, a thin layer of silica by using APTES and PEG was polymerized around the BSA protein and stabilized track of BSA on the surface of Fe3O4@SiO2@acrylate@aptamer. Finally, the template protein was removed and the AIP-MNPs have imprinted cavities complementary to the target protein in shape, size, and functional group orientation.

Schematic process for AIP-MNPs synthesis
Choice of materials
In order to increase the adsorption capacity and decrease the none-specific adsorption of AIP, BSA should be imbedded in a thin and homogeneous matrix with 1) a surface polymerization instead volumetric polymerization which can increase protein penetration; 2) a positive surface charge which can increase protein adsorption and 3) a hydrophilic surface which decrease the none-specific protein binding and facilitate the correct aptamer folding. Therefore, a mixture of APTES and PEG were chosen. Due to the SH-aptamer and thiol-ene click reaction, the gender on substrate was limited and a combination of PEGDA and γ-MAPS was selected that be in harmony with surface matrix. In addition, this new combination of sol-gel component and short length PEG dramatically enhanced the sustainability of NPs. Therefore, homogeneous shell was covered on each stage and surface to volume ratio and protein binding capacity were significantly increased.
Characterization
Transmission electron microscopy (TEM) and scanning electron microscopy (FE-SEM) were performed to estimate size and shape of Fe3O4, Fe3O4@SiO2, and AIP-MNPs. The TEM images shown in Fig. 1a, d, g confirmed the homogeneity, uniformity, spherical shape and dispersity of prepared MNPs. Moreover, the size distribution diagram (Fig. 1c and f) was plotted and demonstrated the average diameter of 20 ± 4 and 40 ± 8 nm for Fe3O4 and Fe3O4@SiO2, respectively. It has been confirmed that the core–shell structures of Fe3O4@SiO2 were successfully prepared with thickness of about 20 nm. The diameter of the AIP-MNPs increased to approximately 50 nm after the imprinting process. The size comparison before and after coating thin layer of APTES and PEG on the Fe3O4@SiO2 confirmed that the thickness of the imprinted polymer layer was approximately 10 nm, which was optimized by the amount of APTES and PEG shown in electronic supporting material (Table S-2). The thickness of APTES layer has critical role in mass transport between the solution and the surface of AIP-MNPs for BSA. As expected, by increasing the thickness of silica coating, the protein cavities on the surface of the AIP-MNPs were imbedded in silicate lattice and the adsorption capacity of BSA decreased.

TEM, FE-SEM images of Fe3O4 (a) and (b), Fe3O4@SiO2 (d) and (e), and AIP-MNPs (g) and (h) and normal distribution diagrams of Fe3O4 (C) and Fe3O4@SiO2 (F)
The FE-SEM images (Fig. 1b, e, h) also indicates highly homogenous and uniform MNPs without agglomeration through polymeric matrix during the multi-coating steps. In addition, FE-SEM images show the similar size of nanoparticles as observed with TEM study. The benefit of these uniform highly distributed AIP-MNPs, is to provide extensive surface to adsorb high amount of target protein and improves overall permeability of final product.
The specific structures of the synthesized Fe3O4, Fe3O4@SiO2 and AIP-MNPs were investigated by XRD patterns and EDAX analysis shown in electronic supporting material (Fig. S-3). Moreover, elemental mapping analysis of these two nanoparticles confirmed the presence of nitrogen and sulfur in nucleotide structure when aptamer conjugation took place (Fig. 2a and b).

a Elemental mapping analysis of Fe3O4@SiO2@acrylate b Fe3O4@SiO2@acrylate@aptamer, each element is shown with different colors c Magnetization curve at 298 K with Fe3O4 (a), Fe3O4@SiO2 (b), and AIP-MNPs (c); insert: Separation of AIP-MNPs with a magnet and redispersion of AIP-MNPs after removal of the magnet. d FT-IR of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2@acrylate (c), Fe3O4@SiO2@acrylate@aptamer (d) and AIP-MNPs(e)
The magnetic properties of the synthesized magnetic nanoparticles were investigated using VSM. Results are presented in Fig. 2c (a, b and c). The saturation magnetization values at room temperature were 46.85, 38.82 and 36.52 emu g−1 for Fe3O4, Fe3O4@SiO2 and AIP-MNPs, respectively. The results confirmed that the AIP-MNPs were strongly magnetic and allowed for effective magnetic separation of trace protein. Figure 2c–insert shows the AIP-MNPs magnetically separated and redispersed due to presence and absence of external magnetic field. The superparamagnetism and essential surface properties of AIP-MNPs prevented them from aggregating and enabled rapid redispersion after the magnetic field was removed.
Figure 2d curve a-e shows the FT-IR spectra of the Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@acrylate, Fe3O4@SiO2@acrylate@aptamer and AIP-MNPs, respectively. The presence of an absorption peak at 592 cm−1, which corresponds to the Fe–O of magnetite phase, and two peaks at 1383 and 1588 cm−1 corresponding to symmetric and asymmetric stretching carboxyl groups of sodium citrate verify the presence of citrate ions at surface of MNPs (Fig. 2d, curve a). The appearance of broad and sharp band at 1094 cm−1 confirmed the immobilization of SiO2 layer onto the surface of Fe3O4 (Fig. 2d, curve b). Replacement of surface functional groups of Fe3O4@SiO2 with acrylate group were confirmed by appearance the sharp bands at 1739 and 1630 cm−1, attributing to C=O and C=C, respectively (Fig. 2d curve c). Weakening of specific absorption peaks of acrylate at 945.2 and 1739 cm−1 corresponding to the C=C–H and C=O after “thiol-ene” click chemistry confirm assembly of single stranded aptamer on Fe3O4@SiO2@acrylate (Fig. 2d curve d). Finally, hydroxyl group of PEG, presented at the surface of AIP-MNPs can be approved by the absorption peak at 1426 cm−1 in Fig. 2d, curve e.
FT-IR results were more confirmed by study of zeta potential analysis for Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@acrylate, Fe3O4@SiO2@acrylate@aptamer and AIP-MNPs. The plain Fe3O4 demonstrates negative zeta potential value, −23.5 mV, due to hydroxyl and citrate groups surrounded around the MNPs. The high absolute zeta potential value for Fe3O4 is critical factor that provided adequate dispersity of MNPs through the next coating steps and was necessary to achieve the core-shell structure. After modification of MNPs surface by TEOS and γ-MAPS, the zeta potential value of the surface shifted to more positive values, −17.6 and −10.5 mV, respectively [27]. When aptamer immobilized on the surface of Fe3O4@SiO2@acrylate, the zeta potential value got more negative, −20.7, probably due to the phosphate groups of aptamer. Finally, the zeta potential value tended to more positive value, −15.6 mV, due to the presence of amino groups of APTES layer immobilized on the surface of AIP-MNPs. This negative surface charge can provide repulsive force which decrease the none-selective adsorption of negative protein at surface of AIP-MNPs. In addition, the settlement of hydrophilic functional groups, layer by layer on the surface of the MNPs provides higher adsorption tendency to aqueous solution. The hydrophilic radius value of Fe3O4@SiO2@acrylate and Fe3O4@SiO2@acrylate@aptamer which measured by DLS analysis clearly showed the increase in hydrophilic radius of Fe3O4@SiO2@acrylate from 295 up to 824 nm after binding to aptamer.
Aptamer binding characterizations
Lack of selectivity is the most important factor to limit MIPs application. In this study, to overcome this restriction, aptamer was used as extra constituent in designing of MIPs. The aptamer against the BSA protein was chosen by literature review [28]. Then, the aptamer sequence was modified at end by adding 10 A nucleotide to that which can act as a linker and preserved the unique folding of aptamer when attached to the surface of the Fe3O4@SiO2@acrylate. The location of 10A at 3` or 5` end of strain were evaluated by mfold Web Server on the basis of the calculation ΔG value for possible structures of these new sequences. The computational value of ΔG for modified aptamer with 10A at 3` end confirmed that this modification preserved the original folding without affecting the structural stability of the aptamer.
In addition, molecular docking studies (explained in electronic supplementary material) revealed that the aptamer can efficiently bind to an interface of BSA located between domain I and III, especially between sub-domains IA and IIIB (Fig. 3). Furthermore ten hydrogen bonds were formed between the aptamer and BSA indicating a strong binding affinity of aptamer to BSA.

Proposed model for interaction of the aptamer with BSA along with their corresponding binding sites. (a) and (b) Surface representation of BSA and examined aptamer structure along with their binding sites. (c) Cartoon representation of BSA-aptamer complex achieved from NPdock web tool. Sub-domains IA, IB, IIA, IIB, IIIA and IIIB of BSA are shown in dark red, bright red, dark blue, bright blue, dark green and bright green, respectively. The aptamer is indicated in orange color. (D) Ligplot result of BSA-aptamer complex. Ligand bonds, non-ligand bonds, H-bonds, external bonds and hydrophobic interactions are shown in gray, red, green, cyan and violet colors, respectively
According to the prediction results, using of aptamer would increase the selective affinity of MIPs related to BSA. As permeation of bio-macromolecules such as BSA into surface of MIPs can limit the application of final MIPs, the core-shell arrangement instead of the batch mixing, was used to imprint the surface of MIPs with the BSA. In this design, aptamer was immobilized on the surface of Fe3O4@SiO2@acrylate by “thiol-ene” click reaction and then trapped the BSA. 10A nucleotides at the beginning of the aptamer chain provided the good flexibility for BSA-aptamer complex at surface of nanoparticle during the imprinting the cavities. In addition, this 10A nucleotide can be hold the BSA in a few angstroms above the surface and prevent it from being hidden in deep. Therefore, BSA-cavities would be accessible for exchange with solution.
Subsequently the BSA was immobilized on the surface of Fe3O4@SiO2@acrylate@aptamer. Then, the cavity imprinted around the BSA by copolymerization of APTES and PEG. The last layer components must be hydrolyzed around the BSA-aptamer complex in a controlled manner and prevented the agglomeration of AIP-MNPs. The selection of APTES instead of TEOS together with PEG400 was in this regard. The hydrolysis of APTES ideally provided thin 2D surface covering the Fe3O4@SiO2@acrylate@aptamer without masking the BSA cavities. In addition, PEG400 with low molecular weight lead to the minor aggregation of the product without increasing the thickness of final layer.
The immobilization of BSA on the surface of Fe3O4@SiO2@ acrylate@ aptamer was traced by UV-Vis analysis at 280 nm before and after reaction of target protein with Fe3O4@SiO2@ acrylate@aptamer. When the solution absorption was remained constant, the different amount of APTES and PEG was applied due to finally obtain optimized value of siloxane copolymerization materials. The results (Fig. S-1) clearly shows, by increasing the amount of APTES and PEG, the thickness of silica coating on the surface of Fe3O4@SiO2@acrylate @aptamer increased and decreased the adsorption capacity of AIP-MNPs. On the other hand, the lesser amount of APTES and PEG than the optimized ones causes the AIP-MNPs to have less adsorption capacity probably due to the thinner layer of silica coating on the surface of AIP-MNPs. After removing the template protein, AIP-MNPs complementary to the template protein in shape, size and functional group were obtained. Then, the binding capacity, selectivity and reusability of AIP-MNPs were investigated in comparison with the NIPs and MIPs in synthetic and complex matrix samples.
Binding properties of NIPs, MIPs and AIP-MNPs
The adsorption kinetics of 0.50 mg mL−1 BSA onto NIPs, MIPs and AIP-MNPs was shown in Fig. 4a. It has been shown that the adsorption capacity of AIP-MNPs increased with time by a fast adsorption rate. In the first 120 min, the adsorption increased rapidly and then almost reached equilibrium. Although, imprinted non-thin films, it takes generally 12–24 h to reach adsorption equilibrium [29]. The imprinted thin films needed only 30–120 min to reach adsorption equilibrium for bio-macromolecular templates [5]. Moreover, the binding isotherms of BSA to 20 mg of NIPs, MIPs and AIP-MNPs were determined in the concentration range of 0.05–0.80 mg mL−1 (initial concentration) and the results were shown in Fig. 4b. It can be seen that the amount of BSA bound to the AIP-MNPs increased quickly with increasing initial concentration below 0.50 mg mL−1. When the initial concentration was above 0.50 mg mL−1, the adsorption curve became relatively flat and reached saturation.

a Adsorption kinetics of 20 mg sample of NIPs, MIPs and AIP-MNPs which incubated in a solution of BSA at a concentration of 0.5 mg/mL at 25 °C (n = 3), b Adsorption isotherms of BSA onto NIPs, MIPs and AIPs. The 20 mg samples of NIPs, MIPs and AIP-MNPs were incubated at different concentration of BSA for 2 h at 25 °C (n = 3)
These results indicated that the adsorption capacity (Q, mg of protein/g of nanoparticle) of AIP-MNPs (146 ± 2.4 mg g−1) was higher than that of MIPs (96.31 ± 1.3) and NIPs (51.43 ± 0.47) at the same initial concentration. In all stages, the amount of BSA adsorbed onto the NIPs, MIPs and AIP-MNPs were confirmed by measuring residual concentration of BSA in supernatant with UV-Vis analysis at 280 nm.
The specific recognition property of AIP-MNPs is evaluated by imprinting factor (α), which is defined as:
Where QAIP-MNPs and QNIPs are the adsorption capacity of AIP-MNPs and NIPs, respectively. In addition, the specificity of the AIP-MNPs related to template was determined by the selectivity factor (β) determined according to the following formula:
Where αTem and αCompetitive Tem are the imprinting factor of template and competitive template proteins. Three proteins chosen as competitive templates for BSA (pI = 4.9), were HSA (pI =5.3), RNase A (pI = 9.4) and Lyz (pI =11.2). Additionally, specific absorbance (κ) was used to evaluate the selectivity properties of NIPs, MIPs and AIP-MNPs toward template protein and other proteins. The specific absorbance (κ) was calculated from the following equation:
The Q of AIP-MNPs with respect to Lyz, RNase A and HSA as competitive proteins were obtained 75.92, 53.07, and 41.58 mg g−1, respectively. Figure 5 demonstrates competitive adsorption of BSA and three other proteins by NIPs, MIPs and AIP-MNPs. The results represented in Table 1 show that the adsorption capacity (Q), imprinting factor (α) and specific absorbance (κ) of AIP-MNPs for target template protein, BSA, were all significantly higher than those one from three other competitive proteins (p < 0.001).

Competitive adsorption of BSA with other proteins. Experimental conditions: 5.0 mL of 0.5 mg mL−1 proteins, incubated with 20.0 mg of the polymer particles for 2 h at 25 °C (n = 3). Significance: ns ≡ not significant (p ≥ 0.05), * ≡ significant (p < 0.05), ** ≡ very significant (p < 0.01), *** ≡ extremely significant (p < 0.001)
It is clear that the adsorption capacity of BSA is highly dependent on the three-dimensional distribution of functional groups existed on the surface of AIP-MNPs and overall complementary interactions, such as hydrogen bonds, Van der Waals forces, ionic bonds and hydrophobic interactions with protein target. The comparison between the imprinting factor (α) of BSA in NIPs and MIPs shows that the less than the 33% of BSA adsorption on AIP-MNPs surface is related to ionic interaction of positive charge of amino groups of APTES and negative surface charge of BSA which are nonspecific adsorption represented in NIPs. In addition, a similar comparison between the MIPs and AIPs appearances that the 33% of BSA adsorption on AIP-MNPs is due to presence of cavities generated on the surface of MIPs. Surprisingly, the immobilization of aptamer into cavities can synchronously increase the hydrophobic adsorption and selective detection of BSA. While according to theoretical calculation the protein adsorption capacity of aptamer is 132.96 mg g−1. So imprinting process results in the nano-masking effect which blocks approximately 50% of aptamer binding site.
Because of high protein binding capacity of aptamer (2nmolBSA/pmolAptamer) [28] the amount of BSA immobilized on AIP-MNPs (147 mg g−1) in this work was remarkably higher in comparison with previous works (70 mg g−1) [5, 30]. In addition, the AIP-MNPs (average diameter: 50 nm) had a small diameter in this study with an extremely high surface-to-volume ratio in comparison with previous works (average diameter: 220 nm) [6], so most of the template protein might have situated at the surface of the AIP-MNPs. Moreover, Table 2 shows that the imprinting factor (α) and specific absorbance (κ) increased in comparison with those one reported in literature. Therefore, AIP-MNPs were expected not only to improve the binding capacity and specific absorbance (κ) but also it provided great accessibility of the recognition sites to the targets.
Reusability of AIP-MNPs
Regeneration is the most important property for the application of AIP-MNPs. So, the adsorption–desorption cycle was repeated five times using NIPs, MIPs and AIP-MNPs. The results (Fig. S-2) show the amount of adsorbed BSA after five regeneration cycles, were lost only 10%, 12% and 11%, respectively. Therefore, the effect of adsorption–desorption cycles were not statistically significant while the adsorption capacity of NIPs, MIPs and AIP-MNPs were significantly different from each other (p < 0.05). As a result, NIPs, MIPs and AIP-MNPs were regenerated with maintaining the recovery efficiency after removal of BSA by eluting with 0.1 M sodium hydroxide. In addition, reusability of AIP-MNPs was reconfirmed with pre-concentration of BSA from fetal bovine serum (FBS) as real sample. Figure 6a shows that whole amount of BSA protein from FBS was extracted during the five adsorption-desorption cycles by using the same amount of AIP-MNPs. As the efficiency of NIPs and MIPs were lower than AIP-MNPs, since the adsorption capacity of NIPs and MIPs were dramatically lower than AIP-MNPs (p < 0.05).

a Reusability of the NIPs, MIPs and AIP-MNPs checked with real sample, FBS (n = 3). b Nature-PAGE analysis for 0.50 mg mL−1 BSA spiked in 100-fold dilution egg white sample and natural FBS, respectively. M: protein molecular weight marker; crude: real sample; supernatant: remained BSA solution after adsorption by AIP-MNPs; eluted sample: the BSA eluted by 0.1 M NaOH from AIP-MNPs. Significance: ns ≡ not significant (p ≥ 0.05), * ≡ significant (p < 0.05), ** ≡ very significant (p < 0.01), *** ≡ extremely significant (p < 0.001)
Furthermore, the reproducibility of NIPs, MIPs and AIP-MNPs was investigated using five batches of NIPs, MIPs and AIP-MNPs prepared on different days. Three independent replicates were used for each batch. The results (not shown) confirmed that the reproducibility of the five batches of NIPs, MIPs and AIP-MNPs were all satisfactory with a RSD less than 10%.
Extraction from complex matrix
The MIPs were known as power solid phase extraction substrate which their application limited by their specification level in real sample analysis. Therefore, the ability of AIP-MNPs to distinguish the BSA as target protein from other proteins in complicated real matrix was investigated by spiking the determined amount of BSA in diluted egg white solution and FBS. The Nature-PAGE analysis (Fig. 6b) clearly shows that the intensity of the BSA band was significantly weaker (line supernatants) after treated by AIP-MNPs. Since, the BSA was almost completely captured by AIP-MNPs and removed from the both real samples (line crudes) and successfully released after eluting (line eluted samples). The results of real sample analysis together with reusability results surprisingly revealed that the addition of aptamer in MIPs-cavities correlated with the specific structural design dramatically increase the potential of AIP-MIPs in comparison with traditional MIPs for real sample analysis. Therefore, the AIP-MNPs can improve the ability of MIPs for selective recognition of macro-biomolecules such as proteins among the complex biological samples.
Conclusions
Artificial lock-and-key systems such as traditional MIPs cannot provide sufficient selectivity in bio-macromolecules recognition. Here, a thin molecularly imprinted polymers including the aptamer as recognizing agent was successfully coated on the surface of MNPs. Multi-self-assembly coating was chosen as main strategy to prepare a uniform core–shell structure for the recognition and enrichment of BSA. In this way, creating a maximum surface-to-volume ratio at each step and preventing the accumulation of particles led us to use a complicated precursor. In order to increase the availability of aptameric cavities at surface of AIP, the thickness of PEI-APTES was critical ratio. So BSA was imprinted at surface of AIPs by two-dimensional copolymerization of PEI-APTES instead of unlimited polymerization. In addition, due to presence of short length PEG at surface coating of AIP, none selective protein adsorption decreased. The results showed that the strategy to immobilize aptamer on magnetic molecular polymer is of great potential to achieve high sensitive recognition and detection of target proteins in therapeutics and clinical applications. However, achieving this goal depends on the existence of a well-known sequence for related aptamer.
References
Rezaei B, Mirahmadi-Zare S (2011) Nanoscale manipulation of prednisolone as electroactive configuration using molecularly imprinted-multiwalled carbon nanotube paste electrode. Electroanalysis 23(11):2724–2734
Whitcombe MJ, Chianella I, Larcombe L, Piletsky SA, Noble J, Porter R, Horgan A (2011) The rational development of molecularly imprinted polymer-based sensors for protein detection. Chem Soc Rev 40(3):1547–1571
Bossi A, Bonini F, Turner A, Piletsky S (2007) Molecularly imprinted polymers for the recognition of proteins: the state of the art. Biosens Bioelectron 22(6):1131–1137
Zhou X, Li W, He X, Chen L, Zhang Y (2007) Recent advances in the study of protein imprinting. Sep Purif Rev 36(4):257–283
Gao R, Kong X, Wang X, He X, Chen L, Zhang Y (2011) Preparation and characterization of uniformly sized molecularly imprinted polymers functionalized with core–shell magnetic nanoparticles for the recognition and enrichment of protein. J Mater Chem 21(44):17863–17871
Gai Q-Q, Qu F, Zhang T, Zhang Y-K (2011) The preparation of bovine serum albumin surface-imprinted superparamagnetic polymer with the assistance of basic functional monomer and its application for protein separation. J Chromatogr A 1218(22):3489–3495
Lin Z, Xia Z, Zheng J, Zheng D, Zhang L, Yang H, Chen G (2012) Synthesis of uniformly sized molecularly imprinted polymer-coated silica nanoparticles for selective recognition and enrichment of lysozyme. J Mater Chem 22(34):17914–17922
Li L, He X, Chen L, Zhang Y (2009) Preparation of core-shell magnetic molecularly imprinted polymer nanoparticles for recognition of bovine hemoglobin. Chem Asian J 4(2):286–293
Zhou W-H, Lu C-H, Guo X-C, Chen F-R, Yang H-H, Wang X-R (2010) Mussel-inspired molecularly imprinted polymer coating superparamagnetic nanoparticles for protein recognition. J Mater Chem 20(5):880–883
Lu C-H, Zhang Y, Tang S-F, Fang Z-B, Yang H-H, Chen X, Chen G-N (2012) Sensing HIV related protein using epitope imprinted hydrophilic polymer coated quartz crystal microbalance. Biosens Bioelectron 31(1):439–444
Nishino H, Huang CS, Shea KJ (2006) Selective protein capture by epitope imprinting. Angew Chem Int Ed 45(15):2392–2396
Tai D-F, Lin C-Y, Wu T-Z, Chen L-K (2005) Recognition of dengue virus protein using epitope-mediated molecularly imprinted film. Anal Chem 77(16):5140–5143
Yang Y-Q, He X-W, Wang Y-Z, Li W-Y, Zhang Y-K (2014) Epitope imprinted polymer coating CdTe quantum dots for specific recognition and direct fluorescent quantification of the target protein bovine serum albumin. Biosens Bioelectron 54:266–272
Kempe M, Glad M, Mosbach K (1995) An approach towards surface imprinting using the enzyme ribonuclease a. J Mol Recognit 8(1–2):35–39
Poma A, Guerreiro A, Whitcombe MJ, Piletska EV, Turner AP, Piletsky SA (2013) Solid-phase synthesis of molecularly imprinted polymer nanoparticles with a reusable template–“plastic antibodies”. Adv Funct Mater 23(22):2821–2827
Poma A, Guerreiro A, Caygill S, Moczko E, Piletsky S (2014) Automatic reactor for solid-phase synthesis of molecularly imprinted polymeric nanoparticles (MIP NPs) in water. RSC Adv 4(8):4203–4206
Kan X, Zhao Q, Shao D, Geng Z, Wang Z, Zhu J-J (2010) Preparation and recognition properties of bovine hemoglobin magnetic molecularly imprinted polymers. J Phys Chem B 114(11):3999–4004
Gai Q-Q, Qu F, Liu Z-J, Dai R-J, Zhang Y-K (2010) Superparamagnetic lysozyme surface-imprinted polymer prepared by atom transfer radical polymerization and its application for protein separation. J Chromatogr A 1217(31):5035–5042
Mirahmadi-Zare SZ, Allafchian A, Aboutalebi F, Shojaei P, Khazaie Y, Dormiani K, Lachinani L, Nasr-Esfahani M-H (2016) Super magnetic nanoparticles NiFe2O4, coated with aluminum–nickel oxide sol-gel lattices to safe, sensitive and selective purification of his-tagged proteins. Protein Expr Purif 121:52–60
Poma A, Brahmbhatt H, Pendergraff HM, Watts JK, Turner NW (2015) Generation of novel hybrid aptamer–molecularly imprinted polymeric nanoparticles. Adv Mater 27(4):750–758
Liu X, Ren J, Su L, Gao X, Tang Y, Ma T, Zhu L, Li J (2017) Novel hybrid probe based on double recognition of aptamer-molecularly imprinted polymer grafted on upconversion nanoparticles for enrofloxacin sensing. Biosens Bioelectron 87:203–208
Brahmbhatt H, Poma A, Pendergraff H, Watts J, Turner N (2016) Improvement of DNA recognition through molecular imprinting: hybrid oligomer imprinted polymeric nanoparticles (oligoMIP NPs). Biomater Sci 4(2):281–287
Wang Z, J-c Z, H-z L, H-y C (2015) Aptamer-based organic-silica hybrid affinity monolith prepared via “thiol-ene” click reaction for extraction of thrombin. Talanta 138:52–58
Hui C, Shen C, Yang T, Bao L, Tian J, Ding H, Li C, Gao H-J (2008) Large-scale Fe 3O 4 nanoparticles soluble in water synthesized by a facile method. J Phys Chem C 112(30):11336–11339
Stöber W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26(1):62–69
Kryscio DR, Peppas NA (2012) Surface imprinted thin polymer film systems with selective recognition for bovine serum albumin. Anal Chim Acta 718:109–115
Kim K-M, Kim HM, Lee W-J, Lee C-W, T-i K, Lee J-K, Jeong J, Paek S-M, Oh J-M (2014) Surface treatment of silica nanoparticles for stable and charge-controlled colloidal silica. Int J Nanomedicine 9(Suppl 2):29
Ruff P, Pai RB, Storici F (2012) Real-time PCR-coupled CE-SELEX for DNA aptamer selection. ISRN Mol Biol 2012:1–9
Ye L, Cormack PA, Mosbach K (1999) Molecularly imprinted monodisperse microspheres for competitive radioassay. Anal Commun 36(2):35–38
Li X, Zhang B, Li W, Lei X, Fan X, Tian L, Zhang H, Zhang Q (2014) Preparation and characterization of bovine serum albumin surface-imprinted thermosensitive magnetic polymer microsphere and its application for protein recognition. Biosens Bioelectron 51:261–267
Xue Z, Xi-Wen H, Lang-Xing C, Wen-You L, ZHANG Y-K (2009) Optimum conditions of separation selectivity based on molecularly imprinted polymers of bovine serum albumin formed on surface of Aminosilica. Chin J Anal Chem 37(2):174–180
Ran D, Wang Y, Jia X, Nie C (2012) Bovine serum albumin recognition via thermosensitive molecular imprinted macroporous hydrogels prepared at two different temperatures. Anal Chim Acta 723:45–53
Acknowledgments
The authors gratefully acknowledge the financial support from the National Elite’s Foundation (BMN) and Iran National Science Foundation (INSF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author(s) declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 367 kb)
Rights and permissions
About this article

Cite this article
Shoghi, E., Mirahmadi-Zare, S.Z., Ghasemi, R. et al. Nanosized aptameric cavities imprinted on the surface of magnetic nanoparticles for high-throughput protein recognition. Microchim Acta 185, 241 (2018). https://doi.org/10.1007/s00604-018-2745-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-018-2745-2