
Abstract
Gold nanoparticles (AuNP) were deposited on the surface of multiwalled carbon nanotubes (MWCNT) by in-situ thermal decomposition of gold acetate under solvent and reducing agent free conditions to obtain a nanohybrid of AuNP-MWCNT. A glassy carbon electrode (GCE) was then modified with the nanohybrid and used for amperometric determination of hydrazine. The modified GCE showed an improved electrocatalytic effect towards hydrazine oxidation at a working potential of about 0.30 V (vs Ag|AgCl,KClsat). The calibration plot for hydrazine is linear in two concentration ranges, viz. from 0.1–100 μM and 0.2–1 mM. The respective sensitivities are 4.98 and 4.00 μA μM−1cm−2, and the limit of detection (at S/N = 3) is about 30 nM.

Schematic presentation of the electrocatalytic oxidation of hydrazine using a glassy carbon electrode modified with gold nanoparticle-decorated multiwalled carbon nanotubes at the potential of 0.303 V vs. Ag|AgCl, KClsat. This modified electrode shows high sensitivity and low detection limit by hydrodynamic amperometry.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrazine, N2H4, is a highly reactive base and reducing agent which has a wide range of industrial, medical, agricultural and military applications [1]. Hydrazine has been also recognized as a neurotoxic and carcinogenic substance. Thus, development of new methods for the sensitive determination of hydrazine has attracted the interest of many researchers. Electrochemical methods including amperometry and voltammetry are promising methods for the determination of hydrazine due to its simplicity, high reliability, high sensitivity and selectivity, low detection limit, low cost, compatibility for miniaturization, and ease of use. However, the oxidation of hydrazine at ordinary solid electrodes is kinetically slow and needs large overpotential. Several types of materials, including organic [2] and inorganic mediators [3, 4], metal nanoparticles [5,6,7], and carbonaceous nanomaterials [5, 8] have been used to modify the electrode surface in order to accelerate the rate of hydrazine oxidation and to decrease the extent of overpotential.
Nanohybrid materials as the result of the combination of two or more families of nanomaterials can greatly facilitate the rate of electron transfer, decline the extent of overpotential and increase the analytical current signal. These materials provide an efficient electrically conductive network with a much higher and more effective surface area. Among nanohybrid materials, hybrid of noble metal nanoparticles (NPs) with carbon nanotubes has received a great deal of attention for their use in the area of chemically modified electrodes. The intrinsic and unique properties of carbon nanotubes such as high electrical conductivity, good chemical stability, hollow geometry and a large surface area make them very fascinating materials which can be used as a support and/or a skeleton for the formation of metal nanoparticles assemblies [9]. On the other hand, the existence of the noble metal NPs causes a further increase on the effective surface area and the improvement of electron transfer rate due to their unusual electronic properties arisen by activated electron hopping and high catalytic activity. Noble metal nanoparticles such as Au, Ag, Pt and Pd together with CNTs have been successfully applied for the fabrication of new modified electrodes for the determination of hydrazine at the reduced redox potentials and low concentration levels [10,11,12,13,14].
Our previous report [14] revealed that PdNPs decorated MWCNTs nanohybrid had a significant enhancing effect on the electrochemical oxidation of hydrazine. Hence, it was meaningful to investigate and compare the synergetic effect of AuNP and MWCNT on the electro-oxidation of hydrazine. In the present work, AuNP were decorated on the surface of MWCNT by a low cost and solvent-free method which was similar to that used for the preparation of PdNPs-MWCNTs hybrid. After that, the surface of a glassy carbon electrode (GCE) was modified with AuNP-MWCNT using a simple casting method. The electrocatalytic behavior of AuNP-MWCNT/GCE was investigated towards hydrazine oxidation by cyclic voltammetry. The modified electrode was then used for detection of hydrazine by hydrodynamic amperometry and differential pulse voltammetry. The fabricated sensor showed improved electroanalytical performance in terms of linear dynamic range, sensitivity, response time and stability.
Experimental
Reagents and chemicals
High quality grade chemicals were used without further treatment. Gold(III) acetate, hydrazinium dichloride, sulfuric acid, sodium hydroxide and dimethylformamide (DMF) were obtained from Merck (Darmstadt, Germany, www.merck.com). Multi-wall carbon nanotubes (MWCNTs, 95% purity, OD = 10–30 nm, ID = 5–10 nm and length = 0.5–500 μm) were obtained from Aldrich (Steinheim, Germany, www.sigmaaldrich.com).
Apparatus
Voltammetric and amperometric measurements were performed using an Autolab potentiostat-galvanostat model PGSTAT30 (Utrecht, The Netherlands) with a conventional three-electrode set-up in which AuNP-MWCNT/GCE, an Ag|AgCl,KClsat electrode and a platinum rod served as the working, reference and auxiliary electrodes, respectively. The working potential was applied to the AuNP-MWCNT/GCE in the standard way using the potentiostat and the output signal was acquired by Autolab GPES software. For chronoamperometric studies, 10 mL of phosphate buffer (0.1 M, pH = 7) containing different concentrations of hydrazine (0.1–1 mM) were transferred into an electrochemical cell. Then, the anodic current response was measured at the potential of +0.350 V by the use of AuNP-MWCNT/GCE in the stationary condition for 30 s. For hydrodynamic amperometric measurements, the accurate volume of hydrazine standard solution was successively added into a cell containing 10 mL of phosphate buffer (0.1 M, pH = 7). The solution was then stirred by a rod stirrer of a Metrohm (663 VA stand) with a speed of 2000 rpm and the anodic current response was measured at the potential of +0.350 V. Differential pulse voltammetry was performed by the sweep of potential in the range between 0.0 and 0.6 V with the scan rate of 0.01 V s−1, pulse height of 25 mV and pulse width of 0.05 s. Electrochemical impedance spectroscopy (EIS) experiments were carried out using a Zahner Zennium workstation in a solution containing 5 mM Fe(CN)6 3−/4- couple and 0.1 M KCl. The excitation signal with the amplitude of 5 mV and frequency between 100 kHz and 0.1 Hz was applied and the output signal was acquired with the Thales Z (Zennium release) software.
Transmission electron microscopy (TEM) was performed using a Philips CM 120 Cryo-TEM instrument (Eindhoven, The Netherlands) at 120 kV. A Metrohm 691 pH meter was used for pH adjustments. All measurements were performed at room temperature.
Preparation of the AuNP-MWCNT
MWCNTs decorated with gold nanoparticle were prepared according to the method reported previously [15, 16]. In brief, the treatment of MWCNTs (500 mg) was conducted by refluxing with 70% HNO3 (25 mL) for 16 h, followed by filtering and thorough washing of the material with deionized water until pH of 7. The acid-treated MWCNTs were dried in a vacuum oven for 1 h. 100 mg of the dried MWCNTs (ca. 8 mmol carbon equivalent) was dry mixed with the powdered Au(CH3COO)3 (3 mg, 0.08 mmol) using a mortar and pestle under ambient conditions for 15 min. The solid Au(CH3COO)3/MWCNTs mixture was then transferred to a glass vial and heated in a nitrogen oven to 300 °C for 1 h and held isothermally for 3 h. The product was then collected as AuNP-MWCNT.
Fabrication of the AuNP-MWCNT/GCE
The surface of a glassy carbon electrode (GCE) was polished successively with 0.3 and 0.1 μm alumina paste (Struers, Copenhagen, Denmark) to obtain a mirror finish and then cleaned in water under ultrasonication. One milligram of AuNP-MWCNT was dispersed in 2 mL DMF with ultrasonic agitation for an hour to achieve a well dispersed suspension. 5 μL of the AuNP-MWCNT suspension was placed on the surface of GCE and dried in an oven at 50 °C. For comparison, MWCNT/GCE was prepared through a similar procedure.
Results and discussion
Characterization of AuNP-MWCNT
TEM image of AuNP-MWCNT (Fig. 1) revealed the formation of Au nanoparticles on the outer walls of carbon nanotubes with an average diameter of about 12 nm. When AuNP-MWCNT was immobilized on the surface of a bare GCE and its cyclic voltammogram was recorded in 0.5 M H2SO4 solution at the scan rate of 100 mV s−1, a typical anodic peak due to the oxidation of gold in the forward scan (starting at about 1.2 V) and cathodic peak in the reverse scan (at about 0.9 V) were observed similar to those reported previously [17, 18] (Fig. S1). In addition, a pair of oxidation-reduction peaks was appeared at about 0.4 V corresponding to the quasi-reversible electrochemical reaction of quinone-type carbon-oxygen functionalities on the surface of acid-treated MWCNTs [16,17,18] (Fig. S1). The results clearly confirmed that the thermal decomposition of Au(CH3COO)3 successfully created uniform AuNP on the outer surface of MWCNTs.

TEM image of the AuNP-MWCNT nanohybrid
Electrochemical impedance spectroscopy (EIS) was also done to investigate the interfacial properties of the bare and modified electrodes. Fig. S2 shows the Nyquist plots (Z im vs. Z re) of the EIS experiments sketched for the bare GCE, MWCNT/GCE and AuNP-MWCNT/GCE. Charge-transfer resistance (R ct) values for Fe(CN)6 3−/4- redox couple (5 mM in 0.1 M KCl) at bare GCE (190 Ω) was much higher than at MWCNT/GCE (34 Ω) and at AuNP-MWCNT/GCE (17 Ω). The results demonstrate that the modification of the GCE with MWCNTs and AuNP-MWCNT nanohybrid causes a significant decrease in R ct, indicating that the hybrid of AuNP and MWCNTs is an excellent electrically-conducting material and decoration of AuNP on the MWCNTs enhances the rate of charge transfer compared with that of MWCNTs.
Electrocatalytic activity of AuNP-MWCNT/GCE towards hydrazine
Figure 2 demonstrates cyclic voltammograms of a bare GCE (a, dotted line), MWCNT/GCE (b, dashed line) and AuNP-MWCNT/GCE (c, solid line) recorded in phosphate buffer (0.1 M, pH = 7) containing 1 mM hydrazine in the potential range between 0 and 1 V versus Ag|AgCl,KClsat at the scan rate of 50 mV s−1.

Cyclic voltammograms for a bare GCE (a), MWCNT/GCE (b) and AuNP-MWCNT/GCE (c) in the presence of 1 mM N2H4. Conditions: supporting electrolyte, phosphate buffer (0.1 M and pH = 7); scan rate, 50 mV s−1
As seen in Fig. 2, a well-defined and pronounced oxidation peak is observed for hydrazine with the peak potential (E p) at +0.303 V on AuNP-MWCNT/GCE. But, no obvious oxidation peak on bare GCE and a typical broad oxidation peak at E p = +0.640 V on MWCNT/GCE were recorded for hydrazine. It seems that the well distribution of AuNP on the surface of MWCNTs together with the formation of an efficient electrical network caused by direct binding of AuNP with MWCNTs create more active sites with the improved electrocatalytic activity for hydrazine oxidation [14]. Moreover, due to the increase of electroactive surface area, the charging current density was obviously enhanced by modification of GCE with MWCNTs and AuNP-MWCNT.
Figure S3 shows cyclic voltammograms recorded for hydrazine with the various concentrations on AuNP-MWCNT/GCE. As seen, the oxidation peak current intensity increases with increasing the concentration of hydrazine. At the same time, the increase of hydrazine concentration caused the oxidation peak potential shifted to the more positive potential which confirmed the catalytic effects of AuNP-MWCNT on hydrazine oxidation [19].
The influence of pH on electrocatalytic oxidation of hydrazine was investigated in the range between 5.5 and 8 by cyclic voltammetry (Fig. S4a). As seen in Fig. S4b, the peak current density increases with the increase of pH from 5.5 to 6.5 and then levels off. The decline of peak current density at pH less than 6.5 is attributed to the protonation of hydrazine as the protonated form is less electroactive [20]. Also, the oxidation peak potential of hydrazine shifted to more negative value with increasing pH, indicating that protons participated in the electrode reaction process (Fig. S4c). The observed potential shift was about 0.0736 V pH −1 in the pH range between 5.5 and 8, which was close to the expected value of 0.0737 V pH −1 for a 4e−/5H+ redox process [14].
Figure S5a shows cyclic voltammograms recorded for 1 mM hydrazine in phosphate buffer (0.1 M, pH = 7) using AuNP-MWCNT/GCE at different scan rates (v). The peak current density (j p) for hydrazine oxidation increased linearly with v 1/2 , which revealed the presence of a diffusion-controlled process (Fig. S5b). Tafel slope for a totally irreversible diffusion controlled process [21] was determined using eq. 1:
The plot of E p versus log ν (Fig. S5c) showed a linear relationship with a Tafel slope, b = 2.3RT/αn α F, of about 145 mV decade−1, which indicated that the rate-determining step of electro-oxidation reaction was a one-electron transfer process, i.e. n α = 1, assuming a transfer coefficient of α = 0.59.
The electrocatalytic oxidation of hydrazine on AuNP-MWCNT/GCE was also studied by chronoamperometry. Fig. S6a represents the current density (j)-time profiles recorded for the various concentrations of hydrazine on AuNP-MWCNT/GCE at a working electrode potential of +0.350 V vs. Ag|AgCl,KClsat. The plot of j versus t-1/2 (Fig. S6b) for different concentrations of hydrazine showed a series of straight line with different slopes. The plot of slopes versus hydrazine concentration provided a line (Fig. S3c) which from its slope, using Cottrell eq. [19] and assuming n = 4, the value of diffusion coefficient (D) for hydrazine was calculated. The calculated value (2.05 × 10−5 cm2 s−1) was in good agreement with the values reported in the literature [22].
Electroanalytical characteristics of the fabricated hydrazine sensor
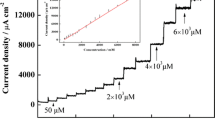
Figure 3 shows the current density-time plot obtained at the applied potential of +0.350 V for the successive addition of hydrazine solution using AuNP-MWCNT/GCE. The relationship between current density and hydrazine concentration was linear in two concentration ranges of 0.1–100 μM (Fig. 7Sa) and 0.2–1 mM (Fig. 7Sb) with the correlation coefficients better than 0.9995. The slope of calibration curves for the first and second concentration range was 4.98 and 4.00 μA μM−1 cm−2, respectively. The detection limit for hydrazine measurement (S/N = 3) was estimated to be 30 nM. The relative standard deviation (RSD) for ten times determination of 1 μM hydrazine was better than 4%. The response time of the fabricated hydrazine sensor was less than 1 s. The current signal of the sensor for 50 μM hydrazine was extremely stable and no decrease in signal was observed after 30 min.

Typical current density-time responses of AuNP-MWCNT/GCE to the successive addition of N2H4 (a;0.1, b;1, c;10, d;100 μM). Conditions: supporting electrolyte, phosphate buffer (0.1 M and pH = 7); operating potential, +0.350 V versus Ag|AgCl|KClsat; rotating speed, 2000 rpm
The analytical characteristics of the proposed hydrazine sensor were also evaluated using differential pulse voltammetry (DPV). For this purpose, DPV responses were recorded in phosphate buffer (0.1 M, pH = 7) containing hydrazine at different concentration (Fig. 4). The relationship between peak current density and hydrazine concentration was linear in the concentration range between 0.06 and 1 mM (Fig. 4, inset) with a correlation coefficient better than 0.9995. The slope of calibration curve and the limit of detection (S/N 3) for hydrazine were 0.21 mA mM−1 cm−2 and 23 μM, respectively. The analytical features for the determination of hydrazine by hydrodynamic amperometry were better those obtained by DPV. The advanced analytical features of the hydrodynamic amperometry were attributed to the hydrodynamic condition of measuring method. However, DPV is superior to the hydrodynamic amperometry when electroactive interfering compounds with the redox potentials close to hydrazine oxidation potential are present in the measuring medium. Table 1 summarizes the analytical characteristics reported for some electrodes modified with different nanomaterials and those achieved in this study for the measurement of hydrazine.

Differential pulse voltammograms of AuNP-MWCNT/GCE in 0.1 M phosphate buffer (pH = 7) containing different concentrations of hydrazine (from inner to outer 0.06, 0.08, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1 mM). Inset: The plot of the peak current density versus N2H4 concentration. Conditions: pulse height, 25 mV; scan rate, 0.01 V s−1; pulse width of 0.05 s; supporting electrolyte, phosphate buffer (0.1 M and pH = 7)
The effect of the presence of potent interfering cations and anions in water samples such as Na+, K+, NH4 +, Ca2+, Mg2+, Cl−, NO3 −, NO2 −, SO4 2− and C2O4 2 on the signal intensity of the fabricated sensor was investigated. A series of solutions containing 10 μM hydrazine plus potent interfering compounds (100 folds) were prepared. The response of test solutions were recorded and compared with that of standard hydrazine solution (10 μM). At 5% error criterion, no obvious interference was observed from the mentioned species.
The fabricated modified electrode was successfully applied for determination of hydrazine in tap and mineral water samples under optimized conditions. Tap and mineral samples were mixed with phosphate buffer (0.1 M and pH = 7.0) and spiked with certain amount of hydrazine and analyzed using the fabricated sensor. Recovery for the each spiked sample was calculated by comparing the results obtained in the absence and presence of certain amount of hydrazine. The recovery of the analysis was better than 95%.
Conclusions
A glassy carbon electrode was modified with the nanohybrid material of AuNP-MWCNT which had been prepared by a thermal method without using any extra chemicals. The synergistic effect between AuNP and MWCNTs caused to observe an improved electrocataltyic property towards hydrazine oxidation at the reduced potential. The modified electrode was successfully used as a sensor for the determination of hydrazine by hydrodynamic amperometry and differential pulse voltammetry methods with the improved electroanalytical features.
References
Davis IIWE, Li Y (2008) Analysis of hydrazine in drinking water by isotope dilution gas chromatography/tandem mass spectrometry with derivatization and liquid−liquid extraction. Anal Chem 80(14):5449–5453. https://doi.org/10.1021/ac702536d
Ojani R, Raoof J-B, Norouzi B (2008) Acetylferrocene modified carbon paste electrode; a sensor for electrocatalytic determination of hydrazine. Electroanalysis 20(12):1378–1382. https://doi.org/10.1002/elan.200704187
Zhang J, Gao W, Dou M, Wang F, Liu J, Li Z, Ji J (2015) Nanorod-constructed porous Co3O4 nanowires: highly sensitive sensors for the detection of hydrazine. Analyst 140(5):1686–1692. https://doi.org/10.1039/C4AN02111H
Yang Z, Sheng Q, Zhang S, Zheng X, Zheng J (2017) One-pot synthesis of Fe3O4/polypyrrole/graphene oxide nanocomposites for electrochemical sensing of hydrazine. Microchim Acta 184(7):2219–2226. https://doi.org/10.1007/s00604-017-2197-0
Wang C, Zhang L, Guo Z, Xu J, Wang H, Zhai K, Zhuo X (2010) A novel hydrazine electrochemical sensor based on the high specific surface area graphene. Microchim Acta 169(1):1–6. https://doi.org/10.1007/s00604-010-0304-6
Bharath G, Naldoni A, Ramsait KH, Abdel-Wahab A, Madhu R, Alsharaeh E, Ponpandian N (2016) Enhanced electrocatalytic activity of gold nanoparticles on hydroxyapatite nanorods for sensitive hydrazine sensors. J Mater Chem A 4(17):6385–6394. https://doi.org/10.1039/C6TA01528J
Rahman MM, Khan A, Marwani HM, Asiri AM (2016) Hydrazine sensor based on silver nanoparticle-decorated polyaniline tungstophosphate nanocomposite for use in environmental remediation. Microchim Acta 183(5):1787–1796. https://doi.org/10.1007/s00604-016-1809-4
Zhao Y-D, Zhang W-D, Chen H, Luo Q-M (2002) Anodic oxidation of hydrazine at carbon nanotube powder microelectrode and its detection. Talanta 58(3):529–534. https://doi.org/10.1016/S0039-9140(02)00318-1
Wildgoose GG, Banks CE, Compton RG (2006) Metal nanoparticles and related materials supported on carbon nanotubes: methods and applications. Small 2(2):182–193. https://doi.org/10.1002/smll.200500324
Kim SK, Jeong YN, Ahmed MS, You J-M, Choi HC, Jeon S (2011) Electrocatalytic determination of hydrazine by a glassy carbon electrode modified with PEDOP/MWCNTs–Pd nanoparticles. Sens Actuators B 153(1):246–251. https://doi.org/10.1016/j.snb.2010.10.039
Kim SP, Lee SG, Choi MY, Choi HC (2015) Highly sensitive hydrazine chemical sensor based on CNT-PdPt nanocomposites. J Nanomater 2015:1–10. https://doi.org/10.1155/2015/120485
Gao G, Guo D, Wang C, Li H (2007) Electrocrystallized ag nanoparticle on functional multi-walled carbon nanotube surfaces for hydrazine oxidation. Electrochem Commun 9(7):1582–1586. https://doi.org/10.1016/j.elecom.2007.02.026
Guo DJ, Li HL (2005) Highly dispersed ag nanoparticles on functional MWNT surfaces for methanol oxidation in alkaline solution. Carbon 43(6):1259–1264. https://doi.org/10.1016/j.carbon.2004.12.021
Haghighi B, Hamidi H, Bozorgzadeh S (2010) Sensitive and selective determination of hydrazine using glassy carbon electrode modified with Pd nanoparticles decorated multiwalled carbon nanotubes. Anal Bioanal Chem 398(3):1411–1416. https://doi.org/10.1007/s00216-010-4049-1
Lin Y, Watson KA, Fallbach MJ, Ghose S, Smith JG, Delozier DM, Cao W, Crooks RE, Connell JW (2009) Rapid, solventless, bulk preparation of metal nanoparticle-decorated carbon nanotubes. ACS Nano 3(4):871–884. https://doi.org/10.1021/nn8009097
Haghighi B, Bozorgzadeh S, Gorton L (2011) Fabrication of a novel electrochemiluminescence glucose biosensor using au nanoparticles decorated multiwalled carbon nanotubes. Sens Actuators B 155(2):577–583. https://doi.org/10.1016/j.snb.2011.01.010
Alexeyeva N, Tammeveski K (2008) Electroreduction of oxygen on gold nanoparticle/PDDA-MWCNT nanocomposites in acid solution. Anal Chim Acta 618(2):140–146. https://doi.org/10.1016/j.aca.2008.04.056
Alexeyeva N, Kozlova J, Sammelselg V, Ritslaid P, Mandar H, Tammeveski K (2010) Electrochemical and surface characterisation of gold nanoparticle decorated multi-walled carbon nanotubes. Appl Surf Sci 256(10):3040–3046. https://doi.org/10.1016/j.apsusc.2009.11.070
Bard AJ, Faulkner LR (eds) (2001) Electrochemical methods, fundamentals and applications, 6th edn. Wiley, New York
Li J, Xie H, Chen L (2011) A sensitive hydrazine electrochemical sensor based on electrodeposition of gold nanoparticles on choline film modified glassy carbon electrode. Sens Actuators B 153(1):239–245. https://doi.org/10.1016/j.snb.2010.10.040
Harrison JA, Khan ZA (1970) The oxidation of hydrazine on platinum in acid solution. J Electroanal Chem Interfacial Electrochem 28(1):131–138. https://doi.org/10.1016/S0022-0728(70)80288-1
Karp S, Meites L (1962) The voltammetric characteristics and mechanism of electroöxidation of hydrazine. J Am Chem Soc 84(6):906–912. https://doi.org/10.1021/ja00865a006
Wang Y, Yang X, Bai J, Jiang X, Fan G (2013) High sensitivity hydrogen peroxide and hydrazine sensor based on silver nanocubes with rich {100} facets as an enhanced electrochemical sensing platform. Biosens Bioelectron 43:180–185. https://doi.org/10.1016/j.bios.2012.10.099
Karuppiah C, Palanisamy S, Chen S-M, Ramaraj SK, Periakaruppan P (2014) A novel and sensitive amperometric hydrazine sensor based on gold nanoparticles decorated graphite nanosheets modified screen printed carbon electrode. Electrochim Acta 139:157–164. https://doi.org/10.1016/j.electacta.2014.06.158
Gupta R, Rastogi PK, Ganesan V, Yadav DK, Sonkar PK (2017) Gold nanoparticles decorated mesoporous silica microspheres: a proficient electrochemical sensing scaffold for hydrazine and nitrobenzene. Sens Actuators B 239:970–978. https://doi.org/10.1016/j.snb.2016.08.117
Ismail AA, Harraz FA, Faisal M, El-Toni AM, Al-Hajry A, Al-Assiri MS (2016) A sensitive and selective amperometric hydrazine sensor based on mesoporous au/ZnO nanocomposites. Mater Des 109:530–538. https://doi.org/10.1016/j.matdes.2016.07.107
Zhao S, Wang L, Wang T, Han Q, Xu S (2016) A high-performance hydrazine electrochemical sensor based on gold nanoparticles/single-walled carbon nanohorns composite film. Appl Surf Sci 369:36–42. https://doi.org/10.1016/j.apsusc.2016.02.013
Zhao Z, Sun Y, Li P, Zhang W, Lian K, Hu J, Chen Y (2016) Preparation and characterization of AuNPs/CNTs-ErGO electrochemical sensors for highly sensitive detection of hydrazine. Talanta 158:283–291. https://doi.org/10.1016/j.talanta.2016.05.065
Zhang C, Wang G, Ji Y, Liu M, Feng Y, Zhang Z, Fang B (2010) Enhancement in analytical hydrazine based on gold nanoparticles deposited on ZnO-MWCNTs films. Sens Actuators B 150(1):247–253. https://doi.org/10.1016/j.snb.2010.07.007
Madhu R, Dinesh B, Chen S-M, Saraswathi R, Mani V (2015) An electrochemical synthesis strategy for composite based ZnO microspheres–au nanoparticles on reduced graphene oxide for the sensitive detection of hydrazine in water samples. RSC Adv 5(67):54379–54386. https://doi.org/10.1039/c5ra05612h
Madhu R, Veeramani V, Chen S-M (2014) Fabrication of a novel gold nanospheres/activated carbon nanocomposite for enhanced electrocatalytic activity toward the detection of toxic hydrazine in various water samples. Sens Actuators B 204:382–387. https://doi.org/10.1016/j.snb.2014.07.068
Yang J, Zhao F, Zeng B (2016) Well-defined gold nanoparticle@N-doped porous carbon prepared from metal nanoparticle@metal-organic frameworks for electrochemical sensing of hydrazine. RSC Adv 6(28):23403–23410. https://doi.org/10.1039/C6RA00096G
Qin X, Li Q, Asiri AM, Al-Youbi AO, Sun X (2014) One-pot synthesis of au nanoparticles/reduced graphene oxide nanocomposites and their application for electrochemical H2O2, glucose, and hydrazine sensing. Gold Bull 47(1):3–8. https://doi.org/10.1007/s13404-013-0094-9
Zhu J, Chauhan DS, Shan D, Wu X-Y, Zhang G-Y, Zhang X-J (2014) Ultrasensitive determination of hydrazine using a glassy carbon electrode modified with pyrocatechol violet electrodeposited on single walled carbon nanotubes. Microchim Acta 181(7):813–820. https://doi.org/10.1007/s00604-014-1168-y
Kr K, Devasenathipathy R, Wang S-F (2017) A glassy carbon electrode modified with graphene oxide decorated silver phosphate nanodentrites for amperometric determination of dissolved hydrazine. Microchim Acta 184(8):2569–2577. https://doi.org/10.1007/s00604-017-2237-9
Acknowledgements
The authors acknowledge the Islamic Azad University (Zanjan Branch), the National Elites Foundation of Iran (INEF, Dr. Ashtiyani’s Research grant, grant number 1393-15/66597) and the Institute for Advanced Studies in Basic Sciences (IASBS, grant number G2016IASBS119) for their financial supports.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
We declare that we have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 400 kb)
Rights and permissions
About this article

Cite this article
Hamidi, H., Bozorgzadeh, S. & Haghighi, B. Amperometric hydrazine sensor using a glassy carbon electrode modified with gold nanoparticle-decorated multiwalled carbon nanotubes. Microchim Acta 184, 4537–4543 (2017). https://doi.org/10.1007/s00604-017-2480-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-017-2480-0