
Abstract
This work reports on the synthesis of nanocomposites from NiO and α-Fe2O3 by a hydrothermal route. The material was characterized in terms of structural and morphological features by X-ray diffraction and scanning electron microscopy. The nanocomposites were synthesized by growing α-Fe2O3 nanoparticles on the surface of flower-like NiO nanosheets, and then characterized by cyclic voltammetry and amperometric techniques. A glassy carbon electrode (GCE) modified with the nanocomposite displayed distinctly improved response to H2O2 compared to a GCE modified with bare NiO. The H2O2 sensor, best operated at a voltage of 0.4 V (vs. Ag/AgCl) has a sensitivity of 146.98 μA·μM−1·cm−2, a 0.05 mM lower detection limit, and a linear working range that extends from 0.5 to 3 mM of H2O2. The sensor is reproducible and long-term stable even in the presence of various interfering molecules such as ascorbic acid and uric acid.

Schematic of a non-enzymatic electrochemical platform for H2O2 detection using NiO/α-Fe2O3 nanocomposites. The sensor has a 50 µM detection limit and good selectivity over potentially interfering agents.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanoscale materials play a promising role in sensing applications due to their bio-compatibility, portability, rapid response time, low cost and specificity, which has made them to project their role in both clinical and non-clinical applications. Nanostructured metal oxides provide an efficient surface for biomolecule immobilization with their unique electrical, optical and magnetic properties. Metal and metal oxide nanomaterials have been reported to construct non enzymatic hydrogen peroxide sensors, such as gold [1, 2], platinum [3], palladium [4], iron oxide [5, 6], manganese oxide [7], titanium oxide [8], cobalt oxide [9, 10] and nickel oxide [11]. Due to the high cost of coinage metals, metal oxides have become a challenge for commercial applications, which dragged most of the researchers attention [12]. Metal oxide nanocomposite electrodes has been abundantly used in various sensing and bio-sensing applications for electro-oxidation of sugars [13], hydrogen peroxide [14,15,16,17], non-aromatic alcohols [18], carbohydrates [19] and other compounds due to its high surface-to-volume ratio and good chemical stability [20].
Hydrogen peroxide is an important biochemical molecule, which find applications in many areas like food [21], textiles [22], medical, industrial, environmental protection, biological system [23,24,25] and hence the detection of H2O2 is crucial for various such applications [26]. There are various methods to detect hydrogen peroxide viz. titration [27], spectrometry [28], chromatography [29], electrochemistry [30], chemiluminescence [31] and electrochemical techniques. Electrochemical techniques is used widely due to its accuracy, fast response, stability, ease operation, reproducibility and sensitivity [32, 33]. Basically electrochemical sensors are classified into two types as enzymatic and non-enzymatic sensors [34, 35]. The motive of our work is to present a non-enzymatic electrochemical sensor for H2O2 in view of its low cost, stability and reproducibility. Here, the redox reaction takes place between the electrocatalyst and hydrogen peroxide on the surface of the electrode. These exposed electrocatalytic active sites are responsible to improve the performance of hydrogen peroxide sensing. Though many materials have been developed for H2O2 sensing, new nanocomposite based materials are needed for improving the performance as nanocomposites have synergistic effect compared with nanoparticles like high surface area, increased electron transport etc., Iron oxide materials are biocompatible, non-toxic, stable, low-cost, possess superior magnetic properties and an abundant material. Thus the application of NiO/α-Fe2O3 nanocomposites will have the following advantages viz. rapid and non-chemical damaging regeneration of the sensor by applying an external magnetic field, high surface area, biocompatibility and electrode renewability using an external magnetic field.
In this work, NiO/α-Fe2O3 nanocomposites were synthesized by a hydrothermal route. The crystalline size and morphology was confirmed by XRD and FESEM analysis. Further, a detailed electrochemical analysis has been performed to study the catalytic activity of the composites towards sensing of H2O2. Results showed that the electrode modified with NiO/α-Fe2O3 possess appreciable electrocatalytic activity of hydrogen peroxide than bare NiO. Thus, the metal oxide nanocomposites sensing element can be used as a non-enzymatic sensor.
Experimental section
Reagents
Nickel chloride hexahydrate (NiCl2.6H2O,99%), Hexamethylenetetramine (HMT,99%), Ethanolamine (99%), Ferric chloride hexahydrate (FeCl3.6H2O,99%), Sodium sulfate anhydrous (Na2SO4,99%), Ethanol (99%), Nafion, Hydrogen Peroxide, Glucose (C6H12O6,99%), Ascorbic acid (C6H8O6,99.7%), potassium hydroxide (KOH,99%), Sucrose and Fructose were all purchased from Sigma Aldrich (http://www.sigmaaldrich.com). The concentration of phosphate buffer was optimized to 0.5 M, which was used an electrolyte for the whole electrochemical experiments. All the above mentioned chemicals were prepared by deionized water from Milli-Q system (Millipore) (http://www.merckmillipore.com).
Preparation of flowerlike NiO microsphere
NiO microflowers were prepared by a hydrothermal route. In brief, 2.375 g nickel chloride hexahydrate (NiCl2.6H2O) and 1.406 g of hexamethylenetetramine (HMT) were dissolved in 150 mL of solvent containing (75 mL of ethanol and 75 mL of DI water) and kept under stirring to form homogenous solution. Then 20 mL of ethanolamine (EA) was added dropwise to the above solution and stirred vigorously until the pale green turned clarified blue. The homogeneous solution were transferred into 200 mL Teflon-lined stainless steel autoclave and heated at 160οC for 12 h. Then the autoclave was allowed to cool at room temperature. The resultant green precipitate was collected and centrifuged with DI water and ethanol for several times and dried at 80οC in air for 10 h. Finally, the green precipitate was calcinated at 350οC for 1 h in air and gray-black hollow NiO microflowers were obtained.
Preparation of NiO/α-Fe2O3 nanocomposites
NiO/α-Fe2O3 nanocomposites were synthesized by hydrothermal route. In brief, 100 mg of NiO was taken from the above prepared powder and dispersed in 160 mL of DI water and stirred vigorously for 30 mins. Then, 86 mg of ferric chloride hexahydrate (FeCl3.6H2O) and 103 mg of sodium sulfate anhydrous (Na2SO4) were added to the above suspension under constant stirring. The above mixture was transferred in to Teflon-lined stainless steel autoclave of 200 mL and heated at 120οC for 2.5 h. The autoclave was made to cool at room temperature. The resultant product was collected and centrifuged with DI water and ethanol for several times and dried at 80οC in air for 10 h. NiO/α-Fe2O3 nanocomposites were calcinated at 450οC for 2 h.
Material characterization
The crystalline structure and phase purity of NiO and NiO/α-Fe2O3 nanocomposites were examined using X-ray diffractometer (D8 Advanced, Burker Diffractometer, Germany) using Cu Kα-1.54 Å X-ray source. The surface morphologies and EDS mappings of the composites were obtained with FESEM by using a Hitachi S-4800 and EDS respectively.
Electrochemical testing
All the electrochemical measurements were carried out with an electrochemical analyzer (CHI 600C work station) using a three electrode system at room temperature under ambient conditions. The potentials and current were measured with respect to Ag/AgCl (sat. KCl) as the reference.
Analytical procedures
The electrode was modified by a casting method as follows, the working electrode was chosen as glassy carbon electrode with a surface area of 3 mm diameter, polished with 0.3 μm alumina powder to provide mirror like surface, which was then rinsed gently with DI water. The polished surface was sonicated for 10 mins to remove impurities on the surface of the electrode with DI water, ethanol, acetone and the electrode was dried for 15 mins. 5 mg of NiO microflowers was dispersed in 250 μl of ethanol and from the above suspension, 5 μl was dropped on the cleaned surface of GCE and dried. 5 μl of nafion was dropped on the above suspension and dried for 30 mins and further electrochemical studies were carried (NiO/Nafion/GCE). The same was carried out for the modification of electrode using NiO/α-Fe2O3 nanocomposites (NiO/α-Fe2O3/Nafion/GCE). The standard three electrodes (GCE, platinum wire and Ag/AgCl) were immersed in an electrolyte containing 20 mL of 0.5 M phosphate buffer and a known volume of hydrogen peroxide was added to the electrochemical cell setup. Cyclic voltammetry was carried out to investigate the electrochemical behavior of the modified electrode in the potential range of −0.8 to 0.6 V at a scan rate of 0.1 V/s. H2O2 measurement was carried out in 0.5 M phosphate buffer (pH 7) at room temperature. For amperometric detection, all measurements were performed by applying an appropriate potential (vs. Ag/AgCl) to the working electrode and allowing the transient background current to decay to a steady-state value, prior to the addition of H2O2. The current response due to the addition of H2O2 was recorded. A stirred solution was employed to provide convective transport. In addition to this, amperometric studies were also carried out by adding a known amount of the analyte at different time intervals to the electrolyte under continuous stirring.
Results and discussion
Structural and morphological characteristics
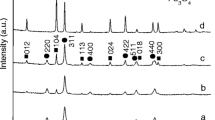
The crystalline structure and phase composition of the prepared samples were confirmed by X-ray diffraction (XRD). Figure 1 shows the XRD pattern of pure NiO microflowers, which exhibits diffraction peaks at 2θ = 37.095, 43.098, 62.590, 75.648 and 79.015 corresponding to (111), (200), (220), (311) and (222) planes. The sharp peaks show that the as-prepared NiO are high crystalline and pure without the presence of any impurities. The peaks show that the products are well matched with the face-centered cubic phase of NiO (JCPDS NO: 89–7130). The crystalline size of the prepared NiO has been calculated using Debbye Scherer formula with an average size of ~14 nm. Figure 1 shows the residual diffraction peaks of metal oxide nanocomposites of NiO/α-Fe2O3, which is consistent with the rhombohedral structure of α-Fe2O3 (JCPDS No: 33–0664). In addition to the NiO peaks, peaks pertaining to α-Fe2O3 were also observed indicating the composite formation. The diffraction peaks shows that no other impurities were observed, which strongly reveals that the synthesized product was a mixture of NiO/α-Fe2O3 with high purity.

X-Ray diffraction spectrum of a NiO. b NiO/α-Fe2O3
The morphology of the prepared metal oxide nanocomposites were observed by FE-SEM analysis as given in Fig.S1 and Fig.S2 (ESM). As observed from the images, α-Fe2O3 particles were uniformly dispersed on the NiO flowers.
Electrochemical analysis
Cyclic voltammetry studies
To assess the electrocatalytic performance of the prepared material to H2O2, a non-enzymatic sensor was constructed. In order to understand the nature of the modified electrode, cyclic voltammetry was carried out in 0.5 M phosphate buffer as shown in Fig. 2. The observed redox peaks are due to the electrochemical reduction of NiO (Fig. 2a). There are couple of well-defined reduction and oxidation peaks observed on the NiO/GCE electrode at a potential of 0.33 and 0.48 V respectively. Thus, the electrocatalytic process occurs on the surface of NiO/GCE. The obtained peaks are matching to the literatures reported before [36, 37]. Here the oxidation peaks occur due to the electrooxidation of NiO to NiOOH, and the peaks exhibited the valence transition of NiOOH to form NiO. There is an oxygen interference found to be more in the range of 0.35–0.45 V. This is because of the consumption of added H2O2, which resulted in the acceleration of the oxidation of NiO to NiOOH. The entire electrochemical process for the oxidation of H2O2 is shown below.

Cyclic voltammetry of a NiO. b NiO/α-Fe2O3 modified electrode in the (i) absence (ii) presence of 5 mM of H2O2 in 0.5 M phosphate buffer at a scan rate of 5 mV/s vs. Ag/AgCl
The electrocatalytic activity of NiO/α-Fe2O3 metal oxide nanocomposites sensor towards the reduction of H2O2 was investigated by voltammetry responses (Fig.2b). Here, the electrocatalytic reaction occurred on the surface of NiO/α-Fe2O3/NF/GCE electrode in the potential range (0.0 to 0.6 V) respectively. Here, (curve i&ii) shows the electrooxidation process towards 0.5 mM H2O2 and an increase in current response towards the addition of H2O2 (curve ii) was observed. The cathodic peak of NiO/α-Fe2O3 towards the hydrogen peroxide reduction was around 0.35 V. It could be clearly observed from the figure that the composite materials exhibits notable catalytic activity for H2O2 reduction than NiO.
Amperometric detection of H2O2
Figure 3a displays the steady state i-t response of H2O2 oxidation at pure NiO/NF/GCE electrode after the successive addition of 0.5 mM of hydrogen peroxide under constant stirring with an applied potential at 0.4 V. As the addition of aliquots H2O2 into the electrolyte, the modified electrode responded rapidly and the oxidation current rose steeply till a stable value was reached. Figure 3b shows the current response of hydrogen peroxide obtained at the electrode surface, which increased linearly with the concentration of H2O2 over the range up to 4 mM with a sensitivity of 95.6 ± 3 μA.mM−1.cm−2 (n = 3), and a detection limit of 0.29 M at a S/N = 3. The corresponding calibration curve shown in Fig.3b can be fitted with the equation i (μA) = 9.595 × 10−5 μA.mM−1 + 2.935 × 10−5 with a correlation coefficient R2 = 0.9936.

a Current-time amperometric response of NiO upon subsequent injection of 0.5 mM H2O2 into 0.5 M phosphate buffer solution under stirring at 0.4 V and at a regular time interval of 50 s. b Linear response of hydrogen peroxide concentrations
Figure 4 shows the steady state i-t response of H2O2 oxidation using NiO/α-Fe2O3 nanocomposites after the successive addition of 0.5 mM of hydrogen peroxide under constant stirring at an applied voltage of 0.4 V. Here the current response increased rapidly and was stable than pure NiO. Figure 4b shows the current response of hydrogen peroxide at the electrode surface, which increased linearly with the concentration of H2O2 over the range up to 4 mM and a low detection limit of 0.05 mM. The nanocomposites exhibited high sensitivity and a low detection limit at S/N = 3. The corresponding calibration curve shown in Fig.4b can be fitted with the equation i(μA) = 1.469 × 10−4 μA.mM−1 + 1.878 × 10−4 with a correlation coefficient R2 = 0.97606. The value of sensitivity calculated from the slope is obtained as 146.982 ± 2 μA.mM−1.cm−2 (n = 3). In both the cases, a major limitation for these materials is the short linear range of detection, which needs to be improved.

a Current-time amperometric response of NiO/α-Fe2O3 upon subsequent injection of 0.5 mM H2O2 into 0.5 M phosphate buffer under stirring at 0.4 V and at a regular time interval of 50 s. b Linear response of hydrogen peroxide concentrations
A comparative table (Table 1) has been given to show our results with the earlier ones and as seen from the Table, compared with several H2O2 sensors reported previously, the present modified electrode displayed an appreciable and comprehensive performance, though its linear range needs to be improved further.
Selectivity, stability and reproducibility of H2O2 sensor
Reproducibility of NiO was analyzed from the amperometric current response to the addition of 0.5 M of H2O2 at 0.4 V, which is for eight successive measurements. Selectivity is yet another important factor for the H2O2 sensor. To investigate the selectivity of the material, the influences of some interferents have been studied. The typical amperometric response on successive additions of 0.5 M H2O2 and 0.5 mM interfering species under stirring is shown in Fig. 5. Fig. 5a shows the evident current step of pure NiO with the addition of 0.5 M H2O2 under continuous stirring. There was no apparent rise in current peaks when interfering species were added into the electrolyte. The same results could be seen in the Fig. 5b and hence no apparent rise in current peaks was observed when interfering species were added into the electrolyte. The below results confirmed that interference for the other species is negligible and hence shows that the fabricated electrode with NiO and NiO/α-Fe2O3 has an excellent selectivity for H2O2.

Interference response of a NiO. b NiO/α-Fe2O3 in 0.5 M phosphate buffer at 0.4 V
Conclusions
In conclusion, hydrothermal route was adopted to prepare metal oxide nanocomposites viz. NiO/α-Fe2O3 and further used for the direct electrocatalytic oxidation of H2O2. The fabricated NiO/NF/GCE and NiO/α-Fe2O3/NF/GCE sensor exhibited appreciable electrocatalytic activity towards H2O2 sensing, with the corresponding sensitivities of 95.6 μA.mM−1.cm−2and 146.98 μA.mM−1 cm−2 for NiO and NiO/α-Fe2O3. In summary, the prepared nanocomposites can be used for highly selective, sensitive and stable amperometric sensing of H2O2 among common coexisting electroactive interferents. Still, further studies needs to be done on improvement of the detection limit and the linear range of the sensor to extend its utility for fabrication of other sensors.
References
Luo JH, Jiao XX, Li NB, Luo HQ (2013) Sensitive determination of cd(II) by square wave anodic stripping voltammetry with in situ bismuth-modified multiwalled carbon nanotubes doped carbon paste electrodes. J Electroanal Chem 689:130–134
He J, Zhao J, Run Z, Zheng S, Pang H (2014) Sub-3nmPd-graphene Nanosheets with outstanding Electrocatalytic oxidation of formic acid. Int J Electrochem Sci 9:7351–7358
Wang X, Li Z, Li L, Wang J, Fu H, Chen Z (2014) Determination of ascorbic acid in individual liver cancer cells by capillary electrophoresis with a platinum nanoparticles modified electrode. J Electroanal Chem 712:139–145
Thevenot DR, Toth K, Durst RA, Wilson GS (2001) Electrochemical biosensors: recommended definitions and classification. Biosens Bioelectron 16:121–131
Siimenson C, Kruusma J, Anderson E, Merisalu M, Sammelselg V, Lust E, Banks CE (2012) Prussian blue modified solid carbon Nanorod whisker paste composite electrodes: evaluation towards the Electroanalytical sensing of H2O2. Int J Electrochem. doi:10.1155/2012/238419
Ju H, Zhang X, Wang J (2011) Nanostructured mimic enzymes for Biocatalysis and Biosensing, NanoBiosensing, Springer New York, 85–109
Zhang R, Chen W (2017) Recent advances in graphene-based nanomaterials for fabricating electrochemical hydrogen peroxide sensors. Biosens Bioelectron 89:249–268
Jiang LC, Zhang W (2009) Electrodeposition of TiO2 nanoparticles on Multiwalled carbon nanotube arrays for hydrogen peroxide sensing. Electroanalysis 21:988–993
Karuppiaha C, Palanisamya S, Chena S, Veeramani V, Periakaruppan P (2014) A novel enzymatic glucose biosensor and sensitive non-enzymatic hydrogen peroxide sensor based on graphene and cobalt oxide nanoparticles composite modified glassy carbon electrode. Sensors Actuators B Chem 196:450–456
Pang H, Gao F, Chen Q, Liu R, Lu Q (2012) Dendrite-like Co3O4 nanostructure and its applications in sensors, supercapacitors and catalysis. Dalton Trans 41:5862–5868
Shamsipur M, Pashabadia A, Molaabasib F (2015) A novel electrochemical hydrogen peroxide biosensor based on hemoglobin capped gold nanoclusters–chitosan composite. RSC Adv 5:61725–61734
Niu X, Lan M, Zhao H, Chen C (2013) Highly sensitive and selective Nonenzymatic detection of glucose using three-dimensional porous nickel nanostructures. Anal Chem 85:3561–3569
Berchmans S, Gomathi H, Prabhakara G (1995) Electrooxidation of alcohols and sugars catalysed on a nickel oxide modified glassy carbon electrode. J Electroanal Chem 394:267–270
Baghayeri M, Nazarzadeh Zare E, Lakouraj M (2014) A simple hydrogen peroxide biosensor based on a novel electro-magnetic poly(p-phenylenediamine) @Fe3O4 nanocomposite. Biosens Bioelectron 55:259–265
Baghayeri M, Nazarzadeh Zare E, Lakouraj M (2015) Monitoring of hydrogen peroxide using a glassy carbon electrode modified with hemoglobin and a polypyrrole-based nanocomposite. Microchim Acta 182:771–779
Baghayeri M, Veisi H (2015) Fabrication of a facile electrochemical biosensor for hydrogen peroxide using efficient catalysis of hemoglobin on the porous Pd@Fe3O4-MWCNT nanocomposite. Biosens Bioelectron 74:190–198
Chen X, Wu G, Cai Z, Oyama M, Chen X (2014) Advances in enzyme-free electrochemical sensors for hydrogen peroxide, glucose, and uric acid. Microchim Acta 181:689–705
Lin Q, Wei Y, Liu W, Yu Y, Hu J (2017) Electrocatalytic oxidation of ethylene glycol and glycerol on nickel ion implanted-modified indium tin oxide electrode. Int J Hydrog Energy 42:1403–1411
Casella IG, Cataldi TRI, Salvi AM, Desimoni E (1993) Electrocatalytic oxidation and liquid chromatographic detection of aliphatic alcohols at a nickel-based glassy carbon modified electrode. Anal Chem 65:3143–3150
Casella IG, Desimoni E, Cataldi TRI (1991) Study of a nickel-catalysed glassy carbon electrode for detection of carbohydrates in liquid chromatography and flow injection analysis. Anal Chim Acta 248:117–125
Zhao H, Ju H (2006) Multilayer membranes for glucose biosensing via layer-by-layer assembly of multiwall carbon nanotubes and glucose oxidase. Anal Biochem 350:138–144
Tan XC, Zhang JL, Tan SW, Zhao DD, Huang ZW, Mi Y, Huang ZY (2009) Amperometric hydrogen peroxide biosensor based on immobilization of hemoglobin on a glassy carbon electrode modified with Fe3O4/chitosan Core-Shell microspheres. Sensors 9:6185–6199
Abbas ME, Luo W, Zhu L, Zou J, Tang H (2010) Fluorometric determination of hydrogen peroxide in milk by using a Fenton reaction system. Food Chem 120:327–331
Landoulsi J, El Kirat K, Richard C, Féron D, Pulvin S (2008) Enzymatic approach in microbial-influenced corrosion: a review based on stainless steels in natural waters. Environ Sci Technol 42:2233–2242
Piwkowska A, Rogacka D, Jankowski M, Kocbuch K, Angielski S (2012) Hydrogen peroxide induces dimerization of protein kinase G type Iα subunits and increases albumin permeability in cultured rat podocytes. J Cell Physiol 227:1004–1016
Palanisamya S, Chen SM, Sarawathi R (2012) A novel nonenzymatic hydrogen peroxide sensor based on reduced graphene oxide/ZnO composite modified electrode. Sensors Actuators B 166-167:372–377
Liu W, Zhang H, Yang B, Li Z, Lei L, Zhang X (2015) A non-enzymatic hydrogen peroxide sensor based on vertical NiO nanosheets supported on the graphite sheet. J Electroanal Chem 749:62–67
Klassen NV, Marchington D, McGowan HCE (1994) H2O2 determination by the I3− method and by KMnO4 titration. Anal Chem 66:2921–2925
Pinkernell U, Effkemann S, Karst U (1997) Simultaneous HPLC determination of Peroxyacetic acid and hydrogen peroxide. Anal Chem 69:3623–3627
Peng Y, Wei C, Liu YN, Li J (2011) Nafion coating the ferrocenyl alkanethiol and encapsulated glucose oxidase electrode for amperometric glucose detection. Analyst 136:4003–4007
Anees Y, Bandyopadhyaya R (2014) Silver nanoparticle impregnated mesoporous silica as a non-enzymatic amperometric sensor for an aqueous solution of hydrogen peroxide. J Electroanal Chem 727:184–190
Zhan K, Liu H, Zhang H, Chen Y, Ni H, Wu M, Sun D, Chen Y (2014) A facile method for the immobilization of myoglobin on multi-walled carbon nanotubes: poly (methacrylic acid-co-acrylamide) nanocomposite and its application for direct bio-detection of H2O2. J Electroanal Chem 724:80–86
Xu CY, Zhang LP, Liu L, Shi YF, Wang HJ, Wang XB, Wang FF, Yuan BQ, Zhang DJ (2014) A novel enzyme-free hydrogen peroxide sensor based on polyethylenimine-grafted graphene oxide-Pd particles modified electrode. J Electroanal Chem 731:67–71
Wu YH, Qiao PW, Chong TC, Shen ZX (2002) Carbon Nanowalls grown by microwave plasma enhanced chemical vapor deposition. Adv Mater 14:64–67
Li GH, Wang XW, Liu L, Liu R, Shen FP, Cui Z, Chen W, Zhang T (2015) Controllable synthesis of 3D Ni(OH)2 and NiO Nanowalls on various substrates for high-performance nanosensors. Small 11:731–739
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The author(s) declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 187 kb)
Rights and permissions
About this article

Cite this article
achari, D.S., Santhosh, C., Deivasegamani, R. et al. A non-enzymatic sensor for hydrogen peroxide based on the use of α-Fe2O3 nanoparticles deposited on the surface of NiO nanosheets. Microchim Acta 184, 3223–3229 (2017). https://doi.org/10.1007/s00604-017-2335-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-017-2335-8