
Abstract
We have prepared a surface imprinted polymer (SIP) film for label-free recognition of immunoglobulin G (IgG). The IgG-SIPs were obtained by covalent immobilization of IgG via a cleavable covalent bond and a suitable spacer unit to a gold electrode, followed by electrodepostion of a nm-thin film of polydopamine (PDA). The IgG was then removed by destruction of the cleavable bond so that complementary binding sites were created on the surface of the film. IgG-SIPs with various thicknesses of the PDA films were compared with respect to their affinity to IgG using a quartz crystal microbalance combined with flow injection analysis. The films were also characterized by cyclic voltammetry and scanning electron microscopy. The IgG-SIPs with a film thickness of around 17 nm showed the most pronounced imprinting effect (IF 1.66) and a binding constant of 296 nM.

A strategy for preparation of the IgG-Surface Imprinted Polymeric (IgG-SIP) thin films was developed. IgG was covalently immobilized via a cleavable cross-linker to a gold electrode surface followed by electrochemical deposition of a nanometer thin PDA film. After cleaving S-S bond in the linker the IgG was removed leaving behind the complementary binding sites confined in the surface of the polymer film. The prepared IgG-SIPs were applied for IgG recognition.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Today, the concept of molecular imprinting has been widely recognized as a promising strategy for developing robust molecular recognition materials with high specificity toward the analyte [1]. Molecular imprinting consists in polymerization of a mixture of functional monomers in the presence of a target molecule that acts as a template. During polymerization, the template induces binding sites in the reticulated polymer that are capable to selectively recognize the target molecules or similar structures after removal of the templates from the polymer. The main benefits of these so-called Molecularly Imprinted Polymers (MIPs), are related to their synthetic nature, i.e., excellent chemical and thermal stability associated with reproducible, cost-effective fabrication. Therefore, MIPs have a substantial potential for application in various fields such as chemical analysis and detection [2], separation and purification [3], drug delivery [4], and catalysis [5]. In addition, MIPs have been shown to be a promising alternative to natural biological receptors (e.g. enzymes, DNA, antibodies) in biosensors providing more stable and low-cost recognition elements [1].
However, there remain many unsolved issues in the development of MIP-based biosensors, especially concerning the imprinting of high molecular weight templates, proteins or whole cells. The main drawbacks in protein imprinting including permanent entrapment, poor mass transfer, denaturation, and heterogeneity in binding sites, are commonly attributed to the inherent properties of proteins such as conformational instability, large size, and complexity. Moreover the need of synthesis in aqueous media can present serious difficulties as aqueous solutions significantly reduce the binding strength of the non-covalent template—monomer interactions. The surface imprinting approach resulting in polymer with the imprinted sites situated at or close to the surface—Surface Imprinted Polymers (SIPs)—as well as introducing new biocompatible monomers are promising ways to overcome these difficulties. SIPs have many advantages such as monoclonal binding sites; faster mass transfer and hence stronger binding capacity as compared to traditional MIPs [6]. SIPs for selective protein recognition were realized by implementing various protocols based on immobilization of a target protein on different support materials acting either as a sacrificial [7], or a transfer material [6]. Li et al. has prepared the SIP nanowires exhibiting highly selective recognition for a variety of template proteins, including albumin, hemoglobin, and cytochrome c [8]. Qin et al. demonstrated a possibility to combine surface initiated living-radical polymerization with a protein SIP and prepared the lysozyme-SIP beads capable of separating the template from competitive proteins in a chromatographic column [9]. Sellergren’s group introduced hierarchical imprinting to produce MIP particles with surface-confined binding sites for a small molecule of amino acid [10], as well as for peptide [11] and protein [12]. Recently, we have achieved success in the development of surface imprinted poly(3,4-ethylenedioxithiophene) microstructures for avidin specific recognition [13, 14].
Immunoglobulin G (IgG) is the most plentiful class of antibodies present in human serum, and protects organism against bacterial and viral infections. Analyzing the presence of specific IgG molecules in the body fluids can be useful in diagnosing infections or certain illnesses. Despite the numerous successful reports on molecular imprinting of different kinds of proteins, only a few attempts have been made to imprint IgG [12, 15]. The difficulties in producing of the complementary cavities for IgG in a polymer matrix are mainly attributed to the complex Y-shape structure of an antibody molecule as well as the considerable amount of chemically identical groups in one molecule. A significant progress has been recently achieved by Dickert and co-workers who produced artificial antibody replicas by applying IgG-imprinted nanoparticles as templates in a surface imprinting process to generate SIP thin films directly on the sensor electrodes of a quartz crystal microbalance (QCM) [15].
QCM technique is a widely applied for the investigation of biomolecular interactions allowing the real-time analysis of reactions without labeling requirements [16]. Furthermore, a QCM sensor integrated in a FIA system (QCM-FIA) provides the advantage to monitor on-line the binding events of an analyte and could be applied to analysis of specific interactions such as antigen-antibody [17], protein-drug [18] as well as between MIP films and template molecules [19].
Although the significant achievements were reported for preparation of different protein MIPs formats, the use of thin MIP films seems to be preferred for sensing applications. MIP films can be easily integrated with different sensor transducers (e.g. optical, piezoelectric) allowing real-time label-free monitoring of protein binding events. Electrochemical polymerization is one of the prospective approaches for combining a MIP film with a sensor transducer surface. In this method, the conducting surface of a transducer is used as an electrode, where a polymeric film with well controlled thickness can be easily formed by varying amount of charge passed during the electrodeposition. There are a number of successful reports on the application of the electropolymerization method in molecular imprinting, where various types of polymer were used, such as overoxidized polypyrrole [19], poly(o-phenylenediamine) [20], and polydopamine (PDA) [21]. PDA appears to be especially attractive for protein-SIPs formation due to its high hydrophilicity and high biocompatibility as well as the existence of a wide variety of functional groups (amino-, hydroxy-, and π-π bonds), compatibility with aqueous solutions, the possibility to obtain an ultrathin compact polymer film by the controlled self-limiting growth. Until now there are only a few reports on PDA based MIP films and most of them however concern the PDA prepared by chemical or self-polymerization of dopamine at weak alkaline pH [22, 23]. The application of the electrosynthesized PDA based MIP film as a recognition element for the capacitive sensing of nicotine was firstly reported by Liu et al. [21].
The aim of this work was to elaborate a strategy that gives a possibility to create IgG surface imprinted polymer (IgG-SIP) thin films using PDA controlled electrodeposition on QCM sensor electrodes for real-time label-free IgG recognition. The specific rebinding of IgG to the prepared IgG-SIPs was analyzed by QCM-FIA technique. In order to optimize the strategy a series of the IgG-SIPs based on the various thicknesses of PDA films were compared in terms of their affinity to IgG molecule.
Materials and methods
Materials
IgG from human serum, IgA from human serum, 4-aminothiophenol (4-ATP), 2-mercaptoethanol, phosphate buffer saline tablets (PBS), sodium chloride and sodium dodecyl sulfate were obtained from Sigma-Aldrich (http://www.sigmaaldrich.com). 3,3′-dithiobis [sulfosuccinimidylpropionate] (DTSSP) was purchased from ThermoFisher Scientific Inc (http://www.thermoscientific.com). 4-(2-aminoethyl)benzene-1,2-diol (dopamine) was supplied from Fluka (http://www.sigmaaldrich.com). All chemicals were of analytical grade or higher and were used as received without any further purification. Ultrapure Milli-Q water (resistivity 18.2 MΩ · cm, Millipore, USA) was used for preparation of all aqueous solutions. PBS solution (0.01 M, pH 7.4) was prepared by dissolving one tablet in 200 mL of Milli-Q water.
IgG immobilization on the gold electrode surface
The gold electrode of a 5 MHz quartz crystal microbalance (QCM) sensor (Maxtek, Inc.) served as a substrate for IgG immobilization. The electrode was cleaned by immersion in fresh piranha solution (97 % H2SO4:30 % H2O2, 3:1 volume ratio) for 10 min and then rinsed abundantly with Milli-Q water. Then the electrode was electrochemically treated in 0.1 M H2SO4 by cycling the potential between −0.2 and +1.5 V (vs Ag/AgCl ) until the gold oxide formation region of the voltammograms displayed three distinct peaks and successive scans showed minimal to no change. Finally, the electrode was rinsed with distilled water and dried in a nitrogen stream. The modification of the QCM electrode surface with amino-groups was carried out by immersing the cleaned electrode in ethanolic solution of 0.1 M 4-ATP for 1 h to form the self-assembled monolayer, after which the electrode was thoroughly rinsed with ethanol to remove the unreacted thiols. The attachment of DTSSP crosslinker to the 4-ATP- modified electrode was achieved by subsequent incubation in PBS buffer (pH 7.4) containing 10 mM of DTSSP for 30 min followed by rinsing with PBS buffer. Then, 100 μL of PBS containing 1 mg mL−1 of IgG was dropped onto the modified electrode surface and allowed to react for 30 min, following which the unbound IgG was washed out from the electrode surface by additional rinsing with PBS buffer.
Electrochemical measurements
Gold electrode modification steps were characterized electrochemically by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). Electrochemical measurements were made in a custom-made 8 mL Teflon electrochemical cell connected with Reference 600 Potentiostat (Gamry Instruments, USA). A conventional three-electrode system was used with QCM sensor electrode as working electrode, a rectangular shaped platinum plate (4 × 1.5 cm2) as counter electrode, and a Ag/AgCl/KClsat reference electrode. Also, gold disk electrodes with diameter either 2 mm or 7 mm were used as working electrodes for CV and EIS studies (see experimental details in ESM).
Electrochemical deposition of PDA film
Dopamine electrochemical polymerization on the IgG-modified electrode was monitored by the Electrochemical Quartz Crystal Microbalance (EQCM). EQCM measurements were performed using the QCM100 system (Stanford Research Systems, Inc., Sunnyvale, CA, USA) connected to the Reference 600TM potentiostat (Gamry Instruments, Inc.) and PM 6680B counter (Fluke Corporation) as described [19]. Electropolymerization was carried out on the IgG-modified QCM sensors by cycling the potential between −0.45 and +0.55 V at a scan rate of 50 mV · s−1 in PBS buffer solution containing 5 mM of dopamine until the resonant frequency dropped a designated value. The thickness of the deposited PDA film T f was believed to be uniform and estimated from the resonant frequency shift ∆f according to the equation Eq. 1:
where C f is the sensitivity factor for a 5 MHz quartz crystal resonator, C f = 56.6 Hz cm2 μg−1; the PDA film density ρ is assumed to be 1.2 g cm−3 as polyaniline density [24].
After polymer film electrochemical deposition, the electrode was rinsed with distilled water and dried in a nitrogen stream. When not in use, the IgG-PDA modified QCM sensors were stored under nitrogen atmosphere in the refrigerator.
Template protein removal
Removal of the template protein, IgG, from polymeric matrix of the electrodeposited PDA film was carried out in order to create IgG-SIP films. For this purpose the IgG-PDA modified electrode was immersed in 0.1 M solution of 2-mercaptoethanol in ethanol, and heated in water bath up to 100 °C. The reaction was maintained under stirring for 15 min. After rinsing with ethanol and distilled water, the electrode was immersed into 3 M NaCl aqueous solution containing 0.1 % sodium dodecyl sulfate and the same parameters of heating were applied. The protein washing out procedure was maintained under stirring for 15 min and repeated twice. Finally, the resulting IgG-SIP sensor was washed thoroughly with distilled water and subjected to protein rebinding studies. To compare the IgG-SIPs in terms of their affinity to IgG molecule, a non-imprinted polymer (NIP) structure was also created. The NIP was formed under very same conditions as the IgG-SIP, excluding protein subsequent removal. In this case the PDA film still contains the target protein, but has no cavities on its surface.
IgG rebinding studies by QCM-FIA technique
Template protein rebinding on the prepared IgG-SIP films modified QCM sensors was studied by a QCM-FIA technique. The analysis was carried out in a QCM-FIA system comprising programmable precision pump (M6, VICI® Valco Instruments Company Inc., USA), a motorized six-way port injection valve controlled by a microelectric actuator (C22-3186EH, VICI® Valco Instruments Company Inc., USA) and a small volume (150 μL) axial flow cell attached to the QCM sensor holder (Stanford Research Systems, Inc.) [19]. All elements of the system were connected to a PC and controlled by software written in Labview. A constant flow of degassed PBS buffer solution (pH = 7.4) flowed over the sensor at a flow rate of 12 μL · min−1 until a constant baseline of the QCM sensor resonance frequency was reached. Subsequently, various concentrations of analyte samples (IgG solution in PBS buffer) was injected into the flow stream via an injection loop (250 μL) and allowed to interact with the IgG-SIP or NIP modified QCM sensor for 2,500 s.
Scanning electron microscopy
Scanning Electron Microscope Zeiss Ultra 55 (Carl Zeiss AG, Germany, Oberkochen) with ultra high resolution imaging was used for the surface investigation of the IgG-SIP and NIP modified as well as bare Au electrodes. A series of images at 100,000× magnification were obtained and analyzed.
Results and discussion
Strategy for preparation of IgG-SIP thin films
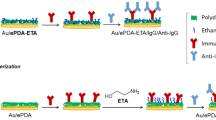
Our approach for an IgG-SIP thin film formation is based on covalent immobilization of the target protein using a cleavable cross-linker to a gold electrode surface followed by electrochemical deposition of a nanometer thin PDA film. Figure 1 sketches the concept of the strategy and gives theoretically estimated values of attached layers thicknesses. Thus, a gold electrode surface was modified with amino-groups by formation a self-assembled monolayer of 4-ATP (a). Then, a homobifunctional crosslinker with a cleavable disulfide bond and a suitable spacer unit, DTSSP, was attached to the amino-modified surface (b). The target protein was immobilized by the formation of covalent amide bond between succinimide group of DTSSP and amino-group of lysine residues of IgG (c). A polymer matrix was formed by electrochemical polymerization of dopamine around the immobilized IgG (d). It was expected that the multiple non-covalent interactions (hydrogen bonding, van der Waals interactions, electrostatic and hydrophobic) between PDA and IgG molecule ensure the formation of complementary cavities in the growing polymer films. After subsequent removal of the protein by destruction of the DTSSP cleavable bond using 2-mercaptoethanol, and non-covalent bonds by the surfactant containing NaCl solution, the complementary binding sites of the target protein confined in the surface of the polymer film were created (e).

The strategy for preparation of the IgG-SIP thin films: (a) 4-ATP self-assembled monolayer creation Au electrode (b) attachment of DTSSP cleavable linker (c) IgG covalent immobilization (d) PDA electrodepostion (e) washing out the IgG molecules (f) rebinding of IgG
In order to choose the appropriate thickness of the electrodeposited PDA layer the length of immobilized structure—4-ATP/DTSSP/IgG—was theoretically estimated. The previously reported AFM and ellipsometry studies indicated a perpendicular orientation of 4-ATP molecules to the metal surface [25], suggesting thus that thickness of a 4-ATP monolayer would be close to the length of the 4-ATP molecule (0.7–0.8 nm). The DTSSP linker has a spacer arm length of 1.2 nm. IgG covalent attachment to a succinimidyl group of the DTSSP proceeds through lysine residues that are abundant in antibody, resulting in random immobilization with multiple orientations of IgG on the surface. Therefore, two limiting cases of antibody possible orientations were considered to estimate the size of the immobilized IgG by RasMol software: “end-on” (Fc fragment closer to the surface) or “head-on” (Fab fragment closer to the surface) orientations with the size approx. 18 nm and “side-on” orientation (Fc and one of the Fabs closer to the surface while another Fab is away from the surface) with size approx. 8.5 nm. Thus, the theoretically estimated height of the whole immobilized structure can vary from approx. 10.5 to 20 nm. The choice for the range of thicknesses of the electrodeposited PDA film needed to achieve surface imprinting of the target protein is determined, from one side, by the linker system length, and, from the other side, by the height of the whole immobilized structure. In this work the polymer films with thicknesses ranging from 4.4 to 22.1 nm (Fig. 1d) were applied in order to evaluate the different extent of polymer coverage on the IgG-modified surface.
IgG immobilization on the gold electrode surface. Cyclic voltammetry and electrochemical impedance spectroscopy studies
IgG immobilization on the QCM gold electrode was accomplished by a multistep procedure including electrode modification with 4-ATP self-assembled monolayer, DTSSP crosslinker attachment, and finally antibody covalent immobilization. CV and EIS techniques were employed to evaluate the changes of gold electrode electrochemical behavior after each modification step. It was already shown that CV and EIS techniques are effective methods for testing the blocking ability towards the electron transfer reactions across a surface modified electrode/electrolyte interface using the ferro/ferri cyanide redox couple as a probe molecule [26].
Figure 2 shows cyclic voltammograms of the Au electrode after the different modification stages. It can be seen that the bare Au electrode (Fig. 2a) shows a reversible peak for the redox couple indicating that the electron transfer reaction is under diffusion-controlled. Reversibility of the reaction at the Au/4-ATP is almost the same as that at the bare Au electrode, indicating that the 4-ATP monolayer exhibits rather poor blocking ability. The poor barrier properties of a 4-ATP monolayer towards the redox reaction were reported earlier [26]. The CV and EIS studies of aromatic thiols SAMs on a gold surface performed by the authors demonstrated that the 4-ATP monolayer is formed with low surface coverage implying the existence of a large number of pinholes and defects within the monolayer. After subsequent attachment of the DTSSP linker to the 4-ATP modified electrode no well-defined current peaks were observed on the voltammogram indicating a significant barrier to a charge-transfer reaction involving the redox couple due to partial surface blocking and thicker layer formation. It is supposed that some DTSSP molecules were disrupted through S-S bond cleavage by contact with uncovered gold surface thus filling the defects of the 4-ATP monolayer and producing so called mixed monolayer, while the intact DTTSP molecules are attached directly to the amino-group of 4-ATP forming the outerlayer intended for IgG covalent binding. When IgG was immobilized through covalent binding between the succinimidyl groups of DTSSP and the amino-groups of the protein, a denser insulating layer is formed on the electrode surface inhibiting more strongly the faradic process as indicates the featureless voltammogram (Fig. 2d).

Cyclic voltammograms of the bare Au (a), 4-ATP (b), 4-ATP/DTSSP (c), and 4-ATP/DTSSP/IgG (d) modified electrodes. The data were recorded in 1 M KCl containing 4 mM Fe(CN)6 3−/Fe(CN)6 4− at scan rate of 50 mV · s−1
The EIS spectra (Fig. S1 in the ESM) are in well agreement with the CV measurements, which further confirm the successful immobilization of the IgG via the DTSSP linker on the electrode surface. In spite of significant perturbation to electronic transport at the modified electrode surface, it is evidently not completely blocked, since an electronic current is still observed on the cyclic voltammogram (Fig. 2d). Thus, polymer matrix formation around the immobilized IgG by dopamine electropolymerization is expected to be feasible.
PDA films electrodeposition. Electrochemical QCM study
The PDA films on the 4-ATP/DTSSP/IgG-modified QCM electrodes were formed by electrochemical polymerization of dopamine in order to provide a more accurate control of polymer film growth. The electrochemical QCM technique was used to simultaneously monitor voltamperometric and gravimetric responses during the polymer growth. Electrochemical QCM has proved to be a valuable in-situ tool to study electropolymerization processes, redox behaviour of electroactive polymers etc., which relates the mass changes during the electrochemical transformations at a QCM electrode to the observed changes in resonant frequency [27]. Considering the theoretical estimations for the range of a polymer bulk needed (Strategy for preparation of IgG-SIP thin films section), the PDA films with thicknesses of 4.4, 8.8, 13.3, 17.7 and 22.1 nm that correspond to the QCM frequency changes of 30, 60, 90, 120 and 150 Hz respectively, were electrodeposited on the 4-ATP/DTSSP/IgG modified QCM sensor electrode. Figure 3 shows a typical resonance frequency response of the QCM during PDA electrodeposition on its electrode. As it can be seen from Fig. 3a the PDA film grows non-linearly demonstrating a faster growth in the beginning of the process, following by its gradual slowing-down until the constant resonant frequency value is reached indicating the self-limiting polymerization. For comparison, the PDA film that is formed on a bare Au electrode, grows significantly faster as shown by Fig. 3b, due to lack of any layer hindering electron transfer at the electrode. In addition, it should be noted that due its insulating nature the PDA film electrochemical growth is self-limited achieving the maximum value of 22 nm in our experimental conditions. The SEM micrographs in Fig. S4 (see ESM) provide additional evidence for the PDA film deposition on the 4-ATP/DTSSP/IgG modified electrode, where this polymer with morphology of uniformly sized agglomerates of fine particles can be seen.

The resonant frequency responses during dopamine electropolymerization on bare Au (b) and the Au/4-ATP/DTSSP/IgG modified (a) electrodes. The data were recorded with the QCM sensor upon potential cycling between −0.45 and +0.55 V at scan rate of 50 mV · s−1in a PBS buffer solution containing 5 mM of dopamine
Evaluation of IgG-SIPs films rebinding capability
IgG rebinding capability of the IgG-SIPs prepared with the different PDA film thicknesses was studied by the QCM-FIA technique. To ascertain that the interaction between IgG and the IgG-SIP surface was specific, a control experiment was performed with the NIP modified electrode for each tested PDA film thickness. The sensor-to-sensor reproducibility reported as the relative standard deviation of the resonance frequency changes at saturation (6.7 μM IgG) for the different sensors was 14.5 %. Figure 4 shows typical frequency responses of the IgG-SIP and NIP modified QCM sensors with the thickness of PDA film of 8.8 nm upon consecutive injections of the solutions with increasing concentration of IgG in PBS buffer. A frequency drop associated with IgG binding is observed for the IgG-SIP modified QCM sensor already after the injection of 6.4 · 10−5 mg · mL−1 IgG concentration, while the NIP modified sensor shows the non-significant frequency change. With the increasing concentration of IgG the frequency drop becomes more pronounced for both SIP and NIP films but at the same time the noticeably difference between the frequency responses of the IgG-SIP and NIP modified sensors is still observed. The higher rebinding capability of the IgG-SIP is more likely attributed to the presence of the imprint sites on the surface. However, there is also evidence of the significant non-specific adsorption appeared as the considerable frequency decrease recorded by the NIP modified QCM sensor.

The resonant frequency response of the IgG-SIP (a) and NIP(b) modified QCM Au electrode. The IgG-SIP and NIP are prepared with PDA film thickness of 8.8 nm. The data were upon successive injections of the different concentrations of IgG in 10 mM PBS buffer
Considering the experimental conditions of the QCM-FIA rebinding study, in particular, the low flow rate (12 μL · min−1) and the construction of the axial flow cell, where an injected solution flows about stagnation point, it can be assumed that the adsorption equilibrium is achieved after each successive injection of IgG. Therefore, the adsorption isotherm can be generated by plotting the appropriate accumulated frequency changes that are related to the amount of the bound protein on the Y axis, and the IgG solution concentrations on the X axis (Fig. 5). Adsorption isotherms are useful in understanding the adsorption interaction mechanism of a template with a MIP surface.

The IgG adsorption isotherms for the IgG-SIP (black circle) and NIP (hollow circle) of the PDA various thicknesses. The curves represent fits of the data to Freundlich-Langmuir (FL) isotherm model
A number of different models such as Langmuir [28], bi-Langmuir [29], Freundlich [30] and Freundlich-Langmuir (FL) [31] was used in order to obtain comparable and physically interpretable parameters, which describe the adsorption process. Since most of MIPs have heterogeneous binding sites, the models that account for heterogeneity (Freundlich and FL) should be considered primarily [32]. According to Shimidzu and co-workers [33] FL model (Eq. 2) is more universally applicable in characterizing MIPs because it can provide heterogeneity information and is able to model adsorption behavior over the entire concentration range up to saturation:
where C is the concentration of a protein in a solution, B and B max the fraction of bound protein and its saturation value, respectively, m is the heterogeneity index, which varies from 0 to 1 and with values <1, the material is heterogeneous. K FL is the association constant, which relates to the equilibrium dissociation constant (KD) as KFL = 1/KD. In this work K D was used to assess the affinity of the prepared IgG-SIP film to the template protein. The experimental data of the adsorption isotherms were fitted to the FL model using a nonlinear regression and plotted in both normal and logarithmic forms (Figs. 5 and S5 in ESM for log plot). The fitted parameters are presented in the Table 1. The imprinting factor (IF) was calculated according to the following equation [34]:
It can be seen from the graphical evaluation that for all tested IgG-SIPs and NIPs the FL model gives an excellent fit, which is confirmed by the high correlation coefficient R2 values (0.990–0.999). The benefit of using the FL fitting model is also supported by the graphs presented in the log plot of the binding isotherms (Fig. S5 in ESM), which show both the linear regions at the low concentrations and the curvature at the high concentrations [33]. The values of m index in the range of 0.457 to 0.78 indicate that the adsorption sites along the IgG-SIPs and NIPs surface are energetically heterogeneous. Nevertheless, all the IgG-SIPs differing in PDA film thickness show somewhat higher binding capacity compared to the corresponding NIPs, that is most likely caused by the presence of the complementary cavities on the surface of the SIP films due to the imprinting phenomenon. Among the IgG-SIPs prepared with different PDA film thicknesses the most pronounced imprinting effect (IF 1.66) was observed for the IgG-SIP with PDA film of 17.7 nm thick. For this case the value of KD, which is considered as a measure of IgG affinity toward the SIP, is found to be approximately one order of magnitude higher for the IgG-SIP than that for the corresponding NIP. Probably, at such PDA film thickness the immobilized IgG molecules are appropriately confined in the polymer and high-affinity binding sites are formed at the polymer surface after the IgG removal procedure. However, binding isotherms observed for the IgG-SIP and NIP pair built-up of the thinnest PDA film (4.4 nm) are clearly inconsistent with the other ones showing the highest difference between the binding affinities of the SIP and NIP while the Bmax value of NIP exceeds Bmax of MIP giving the IF less than unity. This is perhaps due to the non-uniformity of the ultrathin polymer film that may contain significant number of the uncovered areas on the surface, enhancing thus the non-specific adsorption. On the other hand, the lower value of IF was obtained for the IgG-SIP with PDA film of 22 nm thick compared to the IgG-SIPs with other PDA film thicknesses (excl. 4.4 nm thick PDA). This may be explained by the decreasing of the number of the specific cavities on the surface of the polymer film supporting our hypothesis that the polymer film with thickness exceeding the height of the whole immobilized structure (approx.18 nm) may entrap the template more rigorously.
A selectivity test for IgG-SIPs as well as IgA-SIP prepared by the described approach was carried out with respect to IgA and IgG. The preliminary study showed (Fig. S6 in the ESM) that the IgG-SIP binds ca. 2 times more IgG than IgA while the IgA-SIP binds ca. 1.5 times more IgA than IgG.
Conclusion
In summary, we have demonstrated a possibility of preparing the SIP thin films for IgG specific recognition based on a covalent immobilization of IgG through a cleavable cross-linker to a gold electrode of a QCM followed by controlled electropolymerization of the monomer to yield an ultrathin polymeric matrix. The IgG rebinding study performed by the QCM-FIA revealed that the IgG-SIPs built-up of PDA film thickness starting from 8.8 nm and higher demonstrated an apparently enhanced affinity towards IgG when they are compared to the corresponding NIP films. The nanomolar range of KD, estimated by fitting the adsorption isotherms to FL model, indicated strong polymer-template interaction that was expected to result from the multipoint attachment (multiple non-covalent bonds) of the protein to the polymer matrix. IgG-SIPs prepared with PDA thickness of 17.7 nm showed the most pronounced imprinting effect (IF 1.66) compared to other studied IgG-SIPs as well as the nanomolar range binding affinity (KD 296 nM). It was concluded that at such PDA film thickness the immobilized IgG molecules were appropriately confined in the polymer and the more specific binding sites were formed at the polymer surface after the subsequent IgG removal procedure. Although further research is required in this field, we believe that the presented strategy offers a great promise for SIP-based biosensor development demonstrating a way for formation of SIP thin films directly on a sensor transducer surface, which in turn enables convenient real-time label-free detection of a target protein.
References
Ye L, Mosbach K (2008) Molecular imprinting: synthetic materials as substitutes for biological antibodies and receptors. Chem Mater 20(3):859–868. doi:10.1021/Cm703190w
Tamayo FG, Turiel E, Martin-Esteban A (2007) Molecularly imprinted polymers for solid-phase extraction and solid-phase microextraction: recent developments and future trends. J Chromatogr A 1152(1-2):32–40. doi:10.1016/j.chroma.2006.08.095
Pichon V, Haupt K (2006) Affinity separations on molecularly imprinted polymers with special emphasis on solid–phase extraction. J Liq Chromatogr Relat Technol 29(7–8):989–1023. doi:10.1080/10826070600574739
Sellergren B, Allender CJ (2005) Molecularly imprinted polymers: a bridge to advanced drug delivery. Adv Drug Deliv Rev 57(12):1733–1741. doi:10.1016/j.addr.2005.07.010
Wulff G (2002) Enzyme-like catalysis by molecularly imprinted polymers. Chem Rev 102(1):1–27. doi:10.1021/Cr980039a
Shi H, Tsai WB, Garrison MD, Ferrari S, Ratner BD (1999) Template-imprinted nanostructured surfaces for protein recognition. Nature 398:593–597. doi:10.1038/19267
Yilmaz E, Haupt K, Mosbach K (2000) The Use of immobilized templates—new approach in molecular imprinting. Angew Chem Int Ed 39:2115–2118
Li Y, Yang HH, You QH, Zhuang ZX, Wang XR (2006) Protein recognition via surface molecularly imprinted polymer nanowires. Anal Chem 78:317–320
Qin L, He X-W, Zhang W, Li W-Y, Zhang Y-K (2009) Surface-modified polystyrene beads as photografting imprinted polymer matrix for chromatographic separation of proteins. J chromatogr A 1216:807–814. doi:10.1016/j.chroma.2008.12.007
Titirici MM, Hall AJ, Sellergren B (2002) Hierarchically imprinted stationary phases: mesoporous polymer beads containing surface-confined binding sites for adenine. Chem Mater 14(1):21–23. doi:10.1021/Cm011207+
Titirici MM, Sellergren B (2004) Peptide recognition via hierarchical imprinting. Anal Bioanal Chem 378:1913–1921. doi:10.1007/s00216-003-2445-5
Nematollahzadeh A, Sun W, Aureliano CSA, Lutkemeyer D, Stute J, Abdekhodaie MJ, Shojaei A, Sellergren B (2011) High-capacity hierarchically imprinted polymer beads for protein recognition and capture. Angew Chem Int Ed 50(2):495–498. doi:10.1002/anie.201004774
Lautner G, Kaev J, Reut J, Öpik A, Rappich J, Syritski V, Gyurcsanyi RE (2011) Selective artificial receptors based on micropatterned surface-imprinted polymers for label-free detection of proteins by SPR imaging. Adv Funct Mater 21(3):591–597. doi:10.1002/adfm.201001753
Menaker A, Syritski V, Reut J, Öpik A, Horváth V, Gyurcsányi RE (2009) Electrosynthesized surface-imprinted conducting polymer microrods for selective protein recognition. Adv Mater 21(22):2271–2275. doi:10.1002/adma.200803597
Schirhagl R, Lieberzeit PA, Blaas D, Dickert FL (2010) Chemosensors for viruses based on artificial immunoglobulin copies. Adv Mater 22:2078–2081. doi:10.1002/adma.200903517
Marx KA (2003) Quartz crystal microbalance: a useful tool for studying thin polymer films and complex biomolecular systems at the solution-surface interface. Biomacromolecules 4:1099–1120
Arce L, Zougagh M, Arce C, Moreno A, Ríos A, Valcárcel M (2007) Self-assembled monolayer-based piezoelectric flow immunosensor for the determination of canine immunoglobulin. Biosens Bioelectron 22(12):3217–3223. doi:10.1016/j.bios.2007.02.014
Zhang Q, Huang Y, Zhao R, Liu G, Chen Y (2008) Determining binding sites of drugs on human serum albumin using FIA-QCM. Biosens Bioelectron 24:48–54. doi:10.1016/j.bios.2008.03.009
Syritski V, Reut J, Menaker A, Gyurcsányi RE, Öpik A (2008) Electrosynthesized molecularly imprinted polypyrrole films for enantioselective recognition of L-aspartic acid. Electrochim Acta 53(6):2729–2736. doi:10.1016/j.electacta.2007.10.032
Malitesta C, Losito I, Zambonin PG (1999) Molecularly imprinted electrosynthesized polymers: new materials for biomimetic sensors. Anal Chem 71:1366–1370
Liu K, Zeng J-X, Wei W-Z, Liu X-Y, Gao Y-P (2006) Application of a novel electrosynthesized polydopamine-imprinted film to the capacitive sensing of nicotine. Anal Bioanal Chem 385:724–729. doi:10.1007/s00216-006-0489-z
Ouyang R, Lei J, Ju H (2008) Surface molecularly imprinted nanowire for protein specific recognition. Chemical Communications. 5761–5763. doi:10.1039/b810248a
Zhou W-H, Tang S-F, Yao Q-H, Chen F-R, Yang H-H, Wang X-R (2010) A quartz crystal microbalance sensor based on mussel-inspired molecularly imprinted polymer. Biosens Bioelectron 26:585–589. doi:10.1016/j.bios.2010.07.024
Orata D, Buttry DA (1987) Determination of ion populations and solvent content as functions of redox state and pH in polyaniline. J Am Chem Soc 109(12):3574–3581. doi:10.1021/ja00246a013
Valério E, Abrantes LM, Viana AS (2008) 4-Aminothiophenol self-assembled monolayer for the development of a DNA biosensor aiming the detection of cylindrospermopsin producing cyanobacteria. Electroanalysis 20:2467–2474. doi:10.1002/elan.200804350
Sabatani E, Cohen-Boulakia J, Bruening M, Rubinstein I (1993) Thioaromatic monolayers on gold: a new family of self—assembling monolayers. Langmuir 9:2974–2981
Buttry DA, Ward MD (1992) Measurement of interfacial processes at electrode surfaces with the electrochemical quartz crystal microbalance. Chem Rev 92(6):1355–1379. doi:10.1021/cr00014a006
Langmuir I (1918) The adsorption of gases on plane surfaces of glass, mica and platinum. J Am Chem Soc 40(9):1361–1403. doi:10.1021/ja02242a004
Shea KJ, Spivak DA, Sellergren B (1993) Polymer complements to nucleotide bases—selective binding of adenine-derivatives to imprinted polymers. J Am Chem Soc 115(8):3368–3369
Freundlich H (1906) Over the adsorption in solution. Z Phys Chem 57:385–471
Sips R (1948) On the structure of a catalyst surface. J Chem Phys 16(5):490–495
Umpleby RJ, Baxter SC, Chen YZ, Shah RN, Shimizu KD (2001) Characterization of molecularly imprinted polymers with the Langmuir-Freundlich isotherm. Anal Chem 73(19):4584–4591
Umpleby RJ, Baxter SC, Rampey AM, Rushton GT, Chen YZ, Shimizu KD (2004) Characterization of the heterogeneous binding site affinity distributions in molecularly imprinted polymers. J Chromatogr B 804(1):141–149. doi:10.1016/j.jchromb.2004.01.064
Muhammad T, Nur Z, Piletska EV, Yimit O, Piletsky SA (2012) Rational design of molecularly imprinted polymer: the choice of cross-linker. Analyst 137(11):2623–2628
Acknowledgements
This work has been supported by the Estonian Ministry of Education and Research (grant PUT150) and the Estonian Science Foundation (grant ETF8249).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1.38 MB)
Rights and permissions
About this article
Cite this article
Tretjakov, A., Syritski, V., Reut, J. et al. Surface molecularly imprinted polydopamine films for recognition of immunoglobulin G. Microchim Acta 180, 1433–1442 (2013). https://doi.org/10.1007/s00604-013-1039-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-013-1039-y