
Abstract
A highly sensitive electrochemical sensor for the simultaneous determination of catechol (CC) and hydroquinone (HQ) was fabricated by electrodeposition of gold nanoparticles onto carbon nanofiber film pre-cast on an Au electrode. Both CC and HQ cause a pair of quasi-reversible and well-defined redox peaks at the modified electrode in pH 7.0 solution. Simultaneously, the oxidation peak potentials of CC and HQ become separated by 112 mV. When simultaneously changing the concentrations of both CC and HQ, the response is linear between 9.0 μM and 1.50 mM. In the presence of 0.15 mM of the respective isomer, the electrode gives a linear response in the range from 5.0 to 350 μM, and from 9.0 to 500 μM for CC and HQ, respectively, and detection limits are 0.36 and 0.86 μM. The method was successfully examined for real sample analysis with high selectivity and sensitivity.

Highly sensitive and simultaneous determination of catechol and hydroquinone was realized at the GNPs/CNF/Au electrode (d), and its peak currents had nearly two times higher than that of the CNF/Au electrode(c), while only one oxidation peak was observed for both analytes at the bare Au electrode (a) and GNPs/Au electrode (b)
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Catechol (CC, 1,2-dihydroxybenzene) and Hydroquinone (HQ, 1,4-dihydroxybenzene), which are two isomers of dihydroxybenzenes, exist widely in industrial effluents, such as the waste from oil refineries, coal tar, plastic, leather, paint, steel and pharmaceutical industries [1, 2]. They have been included in the lists of priority pollutants to be monitored by international bodies, such as the US Environmental Protection Agency and the European Union [3]. Furthermore, because CC and HQ have similar structures and properties, they usually coexist. Reliable analytical procedures are required for sensitive and simultaneous determination in various matrices. To date, various methods have been exploited for their determination such as high-performance liquid chromatography [4], pH based-flow injection analysis [5], and synchronous fluorescence [6]. However, chromatographic methods are complicated, time-consuming and high cost, and optical methods usually require an additional reagent for the signal generation. Electrochemical methods are relatively simple and provide an easy and fast way in simultaneous determination of CC and HQ. To achieve simultaneous determination, some materials such as carbon nanotubes (CNT) [7–9], poly (glutamic acid) [10], penicillamine [11], aspartic acid [12] and mesoporous Pt [13] were used to modify the electrodes. However, the detection limit was not low and/or the acidic solutions were used for the above modified electrodes, which cannot meet the demand of practical application. The CNT/poly (3-methylthiophene) composite film modified electrodes were used to the simultaneous determination of CC and HQ, while the background current was relatively high and separation of the two peaks (101 mV) was lower [9]. Mesporous carbon-modified electrode was reported with high sensitivity, however, it may suffer from mechanical instability because the electrode was constructed by adsorption of carbon material on substrates [14]. Activated glassy carbon electrode prepared by anodization of glassy carbon was also reported, while anodization damaged the glassy carbon electrode seriously and reduced the lifetime of the electrode [15]. Therefore, the search of a reliable material for the electrode modification is still of considerable interest for the simultaneous determination.
Since the invention of CNT, it has been used as modified material on the electrodes due to its high electrical conductivity, relatively remarkable mechanical and thermal properties, large surface active groups, surface-to-volume ratio, low-ohmic resistance, and easy mass production [16]. Carbon nanofiber (CNF) possesses the similar properties to CNT. Importanly, CNF surpasses CNT in more edge sites on the outer graphite planes and less order, which may lead to more facile electron transfer [17], better dispersion and wettability [18, 19]. CNF was expected to be more suitable to act as modified material for electrochemical sensing and biosensing. However, to our knowledge, only a few reports existed on the development of the CNF modified electrodes [20, 21]. In addition, gold nanoparticles (GNPs) have been widely used to fabricate electrochemical and biological sensors owning to their good conductivity, simplicity of self-assembly through Au–S bond, excellent electrocatalytic ability and biocompatibility [22]. Thus, the nanocomposites of CNF and GNPs were the ideal candidates for electrode modification and expected to acquire the simultaneous and sensitive determination of CC and HQ.
Keeping this aim in view, this paper described that a GNPs/CNF composite modified electrode fabricated by electrodeposition of GNPs onto the CNF modified Au electrode was used to simultaneous determination of CC and HQ. The electrochemical behaviors of CC and HQ were studied using cyclic voltammograms (CV) and differential pulse voltammetry (DPV) at GNPs/CNF/Au electrode. The influences of GNPs deposition time and the pH of phosphate buffer solution were investigated. At the optimum conditions, the peaks for CC and HQ were separated by about 112 mV in pH 7.0 phosphate buffer. The method had been applied to simultaneous determination of CC and HQ in water sample with high selectivity.
Experimental
Reagents
CC and HQ (Fig. S1, see Supplementary material) were purchased from Da Mao Chemical Reagent Co. Ltd. (Tianjin, China, http://dmchem.cn.alibaba.com) and Zhan Yun Chemical Reagent Co. Ltd. (Shanghai, China, http://www.shzychem.com), respectively. Chloroauric acid (HAuCl4·3H2O) was obtained from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China, http://sinoreagent.cn.alibaba.com). CNF was obtained from Chengdu Organic Chemicals Co. Ltd. (Chengdu, China, http://www.cioc.ac.cn). Phosphate buffer solution was prepared by 0.1 M KH2PO4-Na2HPO4. All chemicals were of analytical grade. All aqueous solutions were prepared with deionized water.
Apparatus and methods
All electrochemical experiments were conducted on a CHI660D electrochemical workstation (Shanghai, China, http://chi.instrument.com.cn). All experiments were performed in a three-electrode cell system with the bare or modified Au electrodes (2 mm in diameter), a Pt wire, and a saturated calomel electrode (SCE) as working electrode, auxiliary electrode, and reference electrode, respectively. The electrochemical parameters for DPV were optimized at 50 mV amplitude and 0.2 s pulse width. For CV measurements, potential ranges from −0.3 to +0.6 V at the scan rate of 50 mV s−1. The surface morphologies of the modified Au electrode were evaluated by scanning electron microscope (SEM, JSM-6700F, Japan) images. The electrochemical impedance spectroscopy (EIS) measurements were performed in the presence of 2 mM [Fe(CN) 3−/4−6 ] solution containing 0.1 M KCl and plotted in the form of complex plane diagrams (Nyquist plots). They were recorded with a frequency range of 0.1–100 kHz. The amplitude of the applied sine wave potential was 5 mV, with a formal potential 0.22 V.
Preparation of the GNPs/CNF/Au electrode
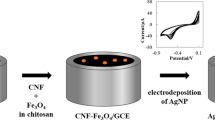
The GNPs/CNF/Au electrode was prepared in three steps. Firstly, the bare Au electrode was polished to mirror smooth with 0.3 and 0.05 μm Al2O3 slurry, in turn, followed by sonicating in acetone, ethanol and deionized water, and then cleaned by cycling in 0.5 M H2SO4 in potential range from −0.3 to +1.5 V for 12 cycles at scan rate of 50 mV s−1. Secondly, CNF was sonicated for 6 h in [V(HNO3):V(H2SO4) = 1:3]. It was then rinsed with deionized water and evaporated to dryness. 5 μL CNF suspension (1 mg mL−1), which was prepared by dispersing the desired amount of acid CNF in N,N-dimethylformamide, was dropped on the fresh Au electrode surface. Then the electrode was placed under an infrared lamp to remove the solvent. Thirdly, the CNF/Au electrode was immersed in 0.5 M KNO3 solution containing 5 mM HAuCl4. The electrochemical deposition of GNPs was conducted for 4 min at −0.2 V. After that the GNPs/CNF/Au electrode was obtained. The electrodes were rinsed by deionized water and stored in phosphate buffer solution before use.
Results and discussion
Characterization of the GNPs/CNF/Au electrode
Electrochemical impedance spectroscopy (EIS) is an efficient tool for studying the interface properties of surface-modified electrodes. The electron-transfer resistance (R et) at the electrode surface is equal to the semicircle diameter of the Nyquist plots and can be used to describe the interface properties of the electrode. Figure 1 showed the results for the Niquist plots on bare Au electrode, CNF/Au electrode and GNPs/CNF/Au electrode. For the bare Au electrode, the R et was 21.6 ± 0.5 Ω. After modifying CNF, the R et (66.7 ± 1.0 Ω), which was reflected by the appearance of substantial increase in the semicircular part of the spectrum, increase due to the blocking effect of CNF in the charge transfer process. This is probably because the negative charges of carboxylic groups on the purified CNF surface limit the access of [Fe(CN) 3−/4−6 ] to the electrode surface [23]. For the GNPs/CNF/Au electrode, the R et decreased to 9.2 ± 0.5 Ω, revealing a very low electron-transfer resistance on the GNPs/CNF/Au electrode. The results demonstrated that the GNPs/CNF composite film was successfully immobilized onto the Au electrode surface, and GNPs were excellent electric conducting material and accelerated the electron transfer.

Nyquist plots of 2 mM [Fe(CN) 3−/4−6 ] containing 0.1 M KCl solution at bare Au electrode (a), CNF/Au electrode (b) and GNPs/CNF/Au electrode (c). The frequency range is from 0.1 Hz to 100 kHz
The surface morphology of GNPs/CNF was characterized by SEM. As shown in Fig. 2, a network-like structure of CNF with the diameter about 150 nm was observed, which indicated that CNF without aggregation was immobilized on the Au electrode surface. Treatment of the CNF/Au electrode with the HAuCl4 solution was carried out by electrochemical deposition. The number and radius of GNPs increase with the increase of deposition time. The dense and uniform GNPs with the average diameter about 80 nm were obtained on CNF for 4 min deposition, while the aggregation occurred for 6 min deposition. The edges of outer wall on CNF serve as suitable sites for the stabilization of GNPs and the acid functional groups can improve the metal–support interaction, then, GNPs on CNF surface exhibit high ratio surface and stability. The above nanostructures of GNPs/CNF are expected to have an excellent potential in electrochemistry.

SEM images of GNPs/CNF/Au electrodes with different deposition time of GNPs. 2 (a), 4 (b) and 6 (c) minutes
Electrochemical behaviors of CC and HQ at the electrodes
The electrochemical behaviors of CC and HQ at bare Au electrode (a), GNPs/Au electrode (b), CNF/Au electrode (c) and GNPs/CNF/Au electrode (d) were studied by CV and DPV. Figure S2, S3 showed CVs obtained from 4 mM CC or 4 mM HQ in 0.1 M phosphate buffer solution.
Compared with bare Au electrode, there was an increase in current response at modified electrodes and the increase order was: \( I_{{{\left( {{\text{bare}}\;{\text{Au}}\;{\text{electrode}}} \right)}}} < I_{{{\left( {{\text{CNF}}/{\text{Au}}\;{\text{electrode}}} \right)}}} < I_{{{\left( {{\text{GNPs}}/{\text{Au}}\;{\text{electrode}}} \right)}}} < I_{{{\left( {{\text{GNPs}}/{\text{CNF}}/{\text{Au}}\;{\text{electrode}}} \right)}}} \). Meanwhile, a decrease in the separation of peak to peak was observed and the decrease order was: \( \Delta {E_{{({\rm{bare}}\;{\rm{Au}}\;{\rm{electrode}})}}} < \Delta {E_{{({\rm{CNF}}/{\rm{Au}}\;{\rm{electrode}})}}} < \Delta {E_{{({\rm{GNPs}}/{\rm{Au}}\;{\rm{electrode}})}}} < \Delta {E_{{({\rm{GNPs}}/{\rm{CNF}}/{\rm{Au}}\;{\rm{electrode}})}}} \), indicating nearly reversible or quasi-reversible electron transfer kinetics for GNPs/CNF/Au electrode interfaces. It may be ascribed to the high surface area of CNF and efficient catalytic effect of GNPs.
Figure 3 showed the CV and DPV obtained for CC and HQ coexisting at the above four electrodes. At the bare Au electrode and GNPs/Au electrode, only one oxidation peak was observed for both analytes, which indicated the impossiblility for the simultaneous determination, but gladly current response was found to enhance greatly at the GNPs/Au electrode. At the CNF/Au electrode, the oxidation waves of CC and HQ were clearly resolved with peak potentials of 167 and 55 mV in DPV, respectively. The increased separation can ascribe partly to the degree of the electrostatic interaction between the CNF surface and these phenolic compounds with different pKa values. Both HQ (pKa = 10.0) and CC (pKa = 9.4) exist in their protonated forms at pH 7.0 [24]. The fraction of positively charged HQ was increased more than CC at the surface of pretreated CNF, which was negatively charged [25]. This leaded to the more beneficial conditions for electron transfer to HQ at the acid CNF besides the more preferable structure of HQ, capable of resonance stabilization of the intermediate, for redox reaction. Moreover, after deposition the GNPs, the peak currents had nearly two times higher than the response currents at CNF/Au electrode, indicating that the GNPs effectively catalyze the oxidation process of CC and HQ. This may be that GNPs can effectively increase surface area and active sites of electrode. Thus, such a large increase in the separation of oxidation peak potentials associated with an increase in the peak currents reflected GNPs/CNF composite film acted as an efficient promoter to enhance the kinetics of the electrochemical process. These results indicated that the simultaneous or selective determination was feasible at the GNPs/CNF/Au electrode.

CV (A) and DPV (B) of 0.4 mM CC and 0.4 mM HQ in phosphate buffer solution (pH 7.0) at bare Au electrode (a), GNPs/Au electrode (b), CNF/Au electrode (c) and GNPs/CNF/Au electrode (d)
Optimization of the general procedures
The pH of the solution and GNPs deposition time were important influence factors for the electrochemical reaction. Voltammetry was carried out to characterize effect of solution pH on electrochemical behaviors of CC and HQ in 0.1 M phosphate buffer solution as shown in Fig. 4A. The results showed that the oxidation currents of CC and HQ increased as the pH changing from 5.0 to 7.0, and then decreased gradually from 7.0 to 10.0. This value of 7.0, at which the currents reached the maximum, was chosen in this work, and importantly, neutral environment can meet the demand of practical application better.

Effects of pH (A) and GNPs deposition time (B) on the peak currents of 0.4 mM CC (a) and 0.4 mM HQ (b)
The effect of the deposition time of GNPs was investigated by means of DPV. The oxidation peak currents of 0.4 mM CC and HQ on the GNPs/CNF/Au electrode increased with increasing deposition time from 1 to 4 min. From Fig. 4B we can see that the peak currents of CC and HQ decreased slightly after 4 min deposition. This may be associated with the decrease of the real surface area of the electrode resulting from the aggregation of GNPs on the electrode surface. Therefore, 4 min was selected in the following investigation.
Simultaneous determination of HQ and CC
The utilization of the GNPs/CNF/Au electrode for simultaneous determination of CC and HQ was demonstrated by changing the both concentrations. As shown in Fig. S4, the DPV results indicated that the simultaneous determination with well-distinguished two peaks at potentials, corresponding to the oxidation of CC and HQ. The oxidation peak currents of CC and HQ increased linearly with the concentration of their own in the range of 9.0 μM to 1.5 mM (I = 0.4055 + 0.0877 C, C in μM, I in μA, R = 0.9980) for CC and (I = 0.4527 + 0.0471 C, C in μM, I in μA, R = 0.9993) for HQ. Thus, the selective and sensitive determination of HQ and CC was achieved simultaneously at this modified electrode.
With the dihydroxybenzene isomer concentration increasing, the peak current increased at GNPs/CNF/Au electrode. In the presence of 0.15 mM isomer, the oxidation peak current of CC was linear to its concentration in the range of 5.0 μM to 0.35 mM, and the linear regression equation was I = 0.3051 + 0.0845 C (C in μM, I in μA, R = 0.9993) with detection limit of 0.36 μM (Fig. 5A). As shown in Fig. 5B, the oxidation peak current of HQ was linear to its concentration in the range of 9.0 μM to 0.50 mM, and the regression equation was I = 0.5057 + 0.0517 C (C in μM, I in μA, R = 0.9987) with detection limit of 0.86 μM. The detection limits of both CC and HQ are lower than the modified electrode with Zn/Al hydroxide film [26] and the glassy carbon electrode modified with similar carbon material composite film (PASA/MWNTs) [8] due to the electrocatalytic property of CNF and GNPs described above.

(A) DPV of CC at (a) 5.0, (b) 10, (c) 20, (d) 40, (e) 60, (f) 120, (g) 170, (h) 200, (i) 250, (j) 350 μM in the presence of 150 μM HQ. (B) DPV of HQ at (a) 9.0, (b) 40, (c) 100, (d) 160, (e) 200, (f) 250, (g) 350, (h) 500 μM in the presence of 150 μM CC
Stability, reproducibility of GNPs/CNF/Au electrode and interference
The stability and reproducibility of the GNPs/CNF/Au electrode were investigated by the measurement of the response to the mixed sample containing 0.4 mM CC and 0.4 mM HQ. The relative standard deviation (RSD) of the oxidation peak current by 8 successive measurements was 1.53% and 1.72% for CC and HQ, respectively. The fabrication reproducibility was estimated at 5 modified electrodes that were prepared under the same condition, and the RSD was 1.86% and 1.94% for CC and HQ, respectively. When the electrode was kept at 4 °C for 1 week, the peak currents remained more than 93.5% of their initial values. The above results revealed the good stability and reproducibility of GNPs/CNF/Au electrode due to the strong adsorption between CNF on the electrode surface and the firm combination of GNPs with CNF.
The interference of some common ions, small biomolecules was evaluated. It was found that for 0.15 mM CC and 0.15 mM HQ, 2.0 mM K+, Na+, Mg2+, Ca2+, NH +4 , NO −3 , Cl−, Br−, glucose, cane sugar, uric acid, and citric acid did not interfere with the determination (signal change below 5%).
Analytical applications
In order to assess the possible applications of this method in direct simultaneous determination of CC and HQ, synthetic samples consisting of CC and HQ in local tap water were tested. The amounts of CC and HQ in the tap water sample were then determined by calibration method and they were summarized in Table 1. When known amount of CC and HQ were added to tap water samples, quantitative recoveries of 95.7–103.5% were obtained. All the results suggested the feasibility of the electrode in simultaneous determination of CC and HQ in water samples.
Conclusions
Combining the advantageous features of GNPs and CNF, GNPs/CNF composite film was constructed on the surface of Au electrode conveniently and firmly. Under the optimum condition, GNPs/CNF/Au electrode showed excellent sensitivity and selectivity properties and can separate oxidation peaks towards CC and HQ, which were indistinguishable at the bare Au electrode. The GNPs/CNF film is expected to be an ideal electrode material for the study of electrochemistry of the redox molecules. Besides electroanalytical application, the prepared electrode with the outstanding electrochemical properties and inherent biocompatibility should have the potential to be used to amperometric biosensors.
References
Terashima C, Rao TN, Sarada BV, Tryk DA, Fujishima A (2002) Electrochemical oxidation of chlorphenols at a boron-doped diamond electrode and their determination by high-performance liquid chromatography with amperometric detection. Anal Chem 74:895–902
Xiao WX, Dan X (2007) Aminopyrene functionalized mesoporous silica for the selective determination of resorcinol. Talanta 72:1288–1292
Xie TY, Liu QW, Shi YR, Liu QY (2006) Simultaneous determination of positional isomers of benzenediols by capilary zone electrophoresis with square wave amperometric detection. J Chromatogr A 1109:317–321
Cui H, He CX, Zhao GW (1999) Determination of polyphenols by high-performance liquid chromatography with inhibited chemiluminescence detection. J Chromatogr A 855:171–179
Garcia-Mesa JA, Mateos R (2007) Direct automatic determination of bitterness and total phenolic compounds in virgin olive oil using a pH-based flow-injection analysis system. J Agric Food Chem 55:3863–3868
Pistonesi MF, Nezio MSD, Centurión ME, Palomeque ME, Lista AG, Band BSF (2006) Determination of phenol, resorcinol and hydroquinone in air samples by synchronous fluorescence using partial least-squares (PLS). Talanta 69:1265–1268
Qi HL, Zhang CX (2005) Simultaneous determination of hydroquinone and catechol at a glassy carbon electrode modified with multiwall carbon nanotubes. Electroanalysis 17:832–838
Zhao DM, Zhang XH, Feng LJ, Li J, Wang SF (2009) Simultaneous determination of hydroquinone and catechol at PASA/MWNTs composite film modified glassy carbon electrode. Colloids Surf, B 74:317–321
Zhang H, Zhao JS, Liu HT, Liu RM, Wang HS, Liu JF (2010) Electrochemical determinationof diphenols and their mixtures at the multiwall carbon nanotubes/poly (3-methylthiophene) modified glassy carbon electrode. Microchim Acta 169:277–282
Wang L, Huang P, Bai J, Wang H, Zhang L, Zhao Y (2007) Direct simultaneous electrochemical determination of hydroquinone and catechol at a poly(glutamic acid) modified glassy carbon electrode. Int J Electrochem Sci 2:123–132
Wang L, Huang PF, Bai JY, Wang HJ, Zhang LY, Zhao YQ (2007) Covalent modification of a glassy carbon electrode with penicillamine for simultaneous determination of hydroquinone and catechol. Microchim Acta 158:151–157
Wang L, Huang PF, Wang HJ, Bai JY, Zhang LY, Zhao YQ (2007) Covalent modification of glassy carbon electrode with aspartic acid for simultaneous determination of hydroquinone and catechol. Ann Chim 97:395–404
Ghanem MA (2007) Electrocatalytic activity and simultaneous determination of catechol and hydroquinone at mesoporous platinum electrode. Electrochem Commun 9:2501–2506
Yu JJ, Du W, Zhao FQ, Zeng BZ (2009) High sensitive simultaneous determination of catechol and hydroquinone at mesoporous carbon CMK-3 electrode in comparison with multi-walled carbon nanotubes and Vulcan XC-72 carbon electrodes. Electrochim Acta 54:984–988
Ahammad AJS, Sarker S, Rahman MA, Leea J-J (2010) Simultaneous determination of hydroquinone and catechol at an activated glassy carbon electrode. Electroanalysis 22:694–700
Zhang MG, Gorski W (2005) Electrochemical sensing based on redox mediation at carbon nanotubes. Anal Chem 77:3960–3965
Banks CE, Compton RG (2005) Exploring the electrocatalytic sites of carbon nanotubes for NADH detection: an edge plane pyrolytic graphite electrode study. Analyst 130:1232–1239
Weeks ML, Rahman T, Frymier PD, Islam SK, McKnight TE (2008) A reagentless enzymatic amperometric biosensor using vertically aligned carbon nanofibers (VACNF). Sensor Actuator B 133:53–59
Werner P, Verdejo R, Wöllecke F, Altstädt V, Sandler JKW, Shaffer MSP (2005) Carbon nanofibers allow foaming of semicrystalline poly(ether ether ketone). Adv Mater 17:2864–2869
Arvinte A, Valentini F, Radoi A, Arduini F, Tamburri E, Rotariu L, Palleschi G, Bala C (2007) The NADH electrochemical detection performed at carbon nanofibers modified glassy carbon electrode. Electroanalysis 19:1455–1459
Rassaei L, Sillanpaa M, Bonne MJ, Marken F (2007) Carbon nanofiber–polystyrene composite electrodes for electroanalytical processes. Electroanalysis 19:1461–1466
Daniel MC, Astruc D (2004) Gold nanoparticles:assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem Rev 104:293–346
Li HQ, Liu RL, Zhao DY, Xia YY (2007) Electrochemical properties of an ordered mesoporous carbon prepared by direct tri-constituent co-assembly. Carbon 45:2628–2635
Harvey D (2000) Modern analytical chemistry, 1st edn. Mc Graw-Hill, New York, p 151
Ding YP, Liu WL, Wu QS, Wang XG (2005) Direct simultaneous determination of dihydroxybenzene isomers at C-nanotube-modified electrodes by derivative voltammetry. J Electroanal Chem 575:275–280
Li MG, Ni F, Wang YL, Xu SD, Zhang DD, Chen SH, Wang L (2009) Sensitive and facile determination of catechol and hydroquinone simultaneously under coexistence of resorcinol with a Zn/Al layered double hydroxide film modified glassy carbon electrode. Electroanalysis 21:1521–1526
Acknowledgments
We greatly appreciate the supports of the National Natural Science Foundation of China (No. 20775047).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
The molecule structures of catechol and hydroquinone). (DOC 30.5 kb)
Fig. S2
CV of CC in 0.1 M phosphate buffer solution (pH 7.0) containing 4 mM analyte at bare Au electrode (a), GNPs/Au electrode (b), CNF/Au electrode (c) and GNPs/CNF/Au electrode (d). (DOC 95 kb)
Fig. S3
CV of HQ in 0.1 M phosphate buffer solution (pH 7.0) containing 4 mM analyte at bare Au electrode (a), GNPs/Au electrode (b), CNF/Au electrode (c) and GNPs/CNF/Au electrode (d). (DOC 93 kb)
Fig. S4
DPV of increasing concentrations of CC and HQ in phosphate buffer solution (pH 7.0): (a) 9.0, (b) 50, (c) 150, (d) 250, (e) 550, (f) 820, (g) 1500 μM. (DOC 111 kb)
Rights and permissions
About this article
Cite this article
Huo, Z., Zhou, Y., Liu, Q. et al. Sensitive simultaneous determination of catechol and hydroquinone using a gold electrode modified with carbon nanofibers and gold nanoparticles. Microchim Acta 173, 119–125 (2011). https://doi.org/10.1007/s00604-010-0530-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-010-0530-y