
Abstract
A separation/preconcentration procedure based on the coprecipitation of Pb(II), Fe(III), Co(II), Cr(III) and Zn(II) ions with copper(II)-N-benzoyl-N-phenyl-hydroxylamine complex (Cu-BPHA) has been developed. The analytical variables including pH, amount of BPHA, amount of copper(II) as carrier element, and sample volume were investigated for the quantitative recoveries of the elements. No interfering effects were observed from the concomitant ions when present in real samples. The recoveries of the analyte ions were in the range of 95–100%. The detection limits (3 s) for Pb(II), Co(II), Fe(III), Cr(III) and Zn(II) ions were found to be 2.3, 0.7, 0.7, 0.3 and 0.4 µg L−1, respectively. The validation of the procedure was performed by the analysis of CRM (SRM NIST-1547 peach leaves and LGC6019 river water) standard reference materials. The method was applied to the determination of the analytes in real samples including natural waters, hair, urine, soil, sediment and peritoneal fluids samples etc., and good results were obtained (relative standard deviations <4%, recoveries >95%).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heavy metal species in the biotic environment pose not only severe carcinogenic and non-carcinogenic risks to humans, but also potential ecological risks [1]. Therefore, it is important to determine trace elements in environmental samples from the viewpoint of environmental management and/or protection from hazardous infection as well as comprehension of the distribution of trace elements in the environment [2].
Preconcentration of traces of heavy metals is often required prior to the instrumental determination to lower the detection limits and to improve the precision and accuracy of analytical results [3]. Efficient and selective separation of metal ions is gaining more importance because of the increasing demand for high purity products and also for environmental concerns. There are various separation methods such as precipitation, solvent extraction [4], cloud point extraction [5], membrane filtration [6], solid phase extraction [7], ion-exchange [8], solvent sublation [9], electrodeposition [10] are widely used to solve these problems.
The coprecipitation method is useful for the preconcentration of trace metal ions [11] and is one of the most useful ways for the preconcentration/separation of trace elements from sample matrix, and the variety of coprecipitants have been studied [12–14]. This preconcentration purpose is characterised by the formation of insoluble compounds. The coprecipitation is adopted when direct precipitation can not separate the desired metallic species due to its low concentration in sample solution. The coprecipitation phenomenon can be associated with metal adsorption on the precipitate surface or due to metal incorporation onto the precipitate structures. Inorganic or organic substances can be considered as coprecipitation agents. The organic agents usually chosen are those able to originate neutral chelates with metallic species [15]. The carrier element is precipitated to coprecipitate trace elements in sample solutions. The precipitate is separated by using a decantation, short-time centrifugation, and so on; in this process, a part of the precipitate might be lost together along with a portion of the coprecipitated trace elements [2]. The coprecipitation method has an advantage that many elements other than alkali and alkaline earth elements are preconcentrated quite effectively under the same experimental conditions [16]. Concentration factors by coprecipitation using organic complex agents and a carrier ion reach 5,000–30,000 [17].
Many metal ions from water samples have been preconcentrated by coprecipitation with hydroxides of iron(III) [18], indium(III) [19–21], scandium(III) [11], gallium(III) [16], terbium(III) [22] and zirconium [23]. As a carrier element, metals like copper and zinc are popular, because of their limited negative effects for environment. Organic coprecipitants, generally dithiocarbamates of bismuth [24], nickel [25] and copper[26], cobalt [27] have been widely used as efficient collectors of trace elements.
N-Benzoyl-N-phenyl-hydroxylamine (BPHA), a cupferron analogous, is used extensively as an organic precipitant and solvent extractant since it reacts with several metals to form stable chelates that are soluble in organic solvents [28]. It is well known for its excellent thermal stability and can keep unchanged even in strong acid [29, 30]. BPHA and its analogs have been successfully employed as a spectrophotometric, gravimetric, and extracting reagents [31].
In the presented work, simple and rapid coprecipitation procedure by using Cu(II)-BPHA precipitate was used as a new coprecipitation technique in order to preconcentrate Pb(II), Fe(III), Co(II), Cr(III) and Zn(II) ions in environmental samples prior to their flame atomic absorption spectrometric (FAAS) determinations. According to our literature knowledge, there is no study on the coprecipitation of trace elements by using copper(II)-BPHA system. We have investigated a new coprecipitation procedure for trace elements for FAAS determination and found that copper(II)-BPHA system has a good collecting ability for this purpose. Also, Cu(II)-BPHA is a convenient coprecipitant to handle, because it can be dissolved in about 500 µL of conc. nitric acid without heating, which is required for the dissolution of the precipitate.
Experimental
Instrument
A Perkin-Elmer AAnalyst 800 Model atomic absorption spectrometer with deuterium background corrector was used for the determination. All measurements were carried out in an air/aceylene flame. The operating parameters for working elements were set as recommended by the manufacturer as given in Table S1 (Supplementary Material). A pH meter Consort C533 model with glass-electrode was employed for measuring pH values in the aqueous phase. An ALC PK 120 model centrifuge was used to centrifuge of solutions. For the digestion of hair samples, a Cem Mars XP-1500 model microwave oven was used.
Reagents and solutions
All the reagents used were of analytical reagent grade. Double distilled deionized water was used throughout the experiments. Laboratory glassware was kept overnight in a dilute HNO3 solution (1:1) and then rinsed with deionized water. The BPHA solution (0.5%, w/v) was prepared daily in ethanol.
Standard stock solutions of the analytes, 1,000 mg L−1, were prepared by dissolving appropriate amounts of their nitrate salts in 2% (w/v) nitric acid. Working standard solutions were prepared fresh daily by stepwise dilution of the stock solutions with double distilled deionized water. The calibration curve was established using the standard solutions prepared in 1 mol L−1 HNO3 by dilution of the stock solutions.
Phosphate buffer solutions were prepared by mixing of appropriate volumes of 1 mol L−1 H3PO4 and NaH2PO4 solutions for pH 2.0 and 3.0, and of 1 mol L−1 NaH2PO4 and Na2HPO4 solutions for pH values of 5.0, 6.0 and 7.0. Acetate buffer solutions were prepared by mixing appropriate volumes of 1 mol L−1 CH3COOH and CH3COONa solutions for pH 4.0. Ammonium buffer solutions were prepared by mixing of appropriate amounts of 3 mol L−1 NH3 and 3 mol L−1 HCl solutions for pH values of 8.0, 9.0 and 10.0.
Analytical procedure
The copper-BPHA coprecipitation method was tested with model solutions prior to its application to real samples. Twenty five millilitres portion of an aqueous solution containing 20 µg of Cr(III), Fe(III) and Pb(II), 10 µg of Co(II) and 2 µg of Zn(II) was placed in a centrifuge tube. The pH of solution was adjusted to 9.0 with ammonia/ammonium chloride buffer solution. Then 250 µL of 1,000 mg L−1 copper(II) as a carrier element was added to this solution. The 0.5 mL of 0.5% (w/v) BPHA solution was poured into the tube. After the formation of the precipitate, the solution was centrifuged at 3,500 rpm for 10 min. The supernatant was removed. The precipitate remained adhering to the tube was dissolved with 500 µL of concentrated HNO3. Then the final volume was completed to 10 mL with double distilled deionized water. The analytes in the final solution were determined by FAAS.
Analyses of standard reference materials
A 100-mg amount of the standard reference material (NIST-1547 peach leaves) sample was dissolved with the mixture of 4 mL of concentrated HNO3 and 2 mL of concentrated H2O2 on a hot plate. After completing the dissolution process, all the sample solutions were clear. The volume of the samples was diluted to approximately 25 mL using distilled deionized water. These sample solutions were analysed by using the proposed preconcentration procedure described above. The final measurement volume of the sample solutions was 5 mL.
The second certified reference material was the LGC6019 River water sample. The pH’s of the samples were adjusted to 9.0 with ammonia/ammonium chloride buffer solution. Then the separation/preconcentration procedure given above was applied to these sample solutions. The concentration of the investigated analyte ions in the final solution was determined by FAAS.
Analysis of peritoneal dialysis fluids
The coprecipitation method was employed for the separation/preconcentration of trace elements in peritoneal dialysis fluid from a hospital at Erciyes University. The electrolyte composition of the peritoneal fluid concentrate (Dianeal 137, Baxter) used in this study was as follows: NaCl 15.7 g L−1, sodium lactate 3.9 g L−1, CaCl2.2H2O 257 mg L−1, MgCl2.6H2O 152 mg L−1, and glucose without water: 13.9 g L−1.
The analysis of peritoneal dialysis concentrate was carried out after dilution 1:4 with the distilled deionized water. First, it was neutralised and then buffered to pH 9.0 with ammonia/ammonium chloride buffer solution. This sample solution was analysed by using the proposed procedure described above.
Water samples
The presented method was applied to the different natural water samples for the preconcentration/separation of the analyte ions. The water samples were collected from different regions of Turkey. The water samples were stored in polyethylene bottles (pre-washed before with detergent, doubly distilled deionized water, dilute HNO3 and doubly distilled deionized water, respectively) and acidified to pH about 2.0 with 5 mL of concentrated nitric acid per liter of the sample and were subsequently stored at 4 °C in a refrigerator. The water samples were filtered through a cellulose membrane filter with a pore size of 0.45 µm. Prior to trace metal analysis, the acidified water samples were neutralised, and then the proposed procedure was applied to the samples. The initial volume of the water samples was 750 mL, and the final volume was 5 mL. The levels of the investigated analyte ions in the final solutions were determined by FAAS.
Application to real samples
A 1.0-g aliquots of each of soil and sediment samples were digested with 15 mL of aqua regia and then heated until obtaining a clear solution. After the mixture was evaporated almost to dryness, 10 mL of aqua regia was again added to the residue. Then the mixture was again evaporated to dryness. After the evaporation, 5 mL of double distilled deionized water was added to the residue and then dissolved. The resulting solution was filtered through a cellulose filter paper. The residue on the filter paper was washed with 1–2 mL of 1 mol L−1 HNO3. The filtrate was diluted to 25 mL of double distilled deionized water. Then the proposed procedure given above was applied to this sample solution. A blank digest was carried out in the same way.
For the microwave digestion of hair samples, to a 0.25-g aliquot of hair sample in a digestion vessel 10 mL of concentrated HNO3 was added. The working conditions of the microwave digestion system were as follows; ramp time: 20 min, pressure: 150 psi, temperature: 210 °C, hold time: 10 min, power: 1,200 W (100%), cool down time: 15 min. After the digestion completed, the proposed preconcentration procedure was applied to the hair sample solution.
Urine samples were digested as follows: An aliquot of 10 mL of urine sample was treated with a mixture of 5 mL of concentrated H2O2 and 2.5 mL of concentrated HNO3 on a hot plate at 80 °C. The system was moderately heated up to the disappearance of the amber color. Then, the sample was evaporated almost to dryness. Thereafter, fresh portions of 2.5 mL of concentrated HNO3 were added to the dark residue and again heated to dryness [32]. Then the proposed procedure was applied to the resulting sample solutions. A blank digest was carried out in the same way.
Results and discussion
The optimised conditions for the preconcentration/separation procedure were established using 25 mL of deionised water containing 2–20 µg of each element and submitting these solutions to the preconcentration procedure.
Effects of pH on the coprecipitation
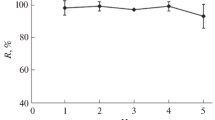
For the quantitative coprecipitation efficiency of the analyte ions, the working pH of the media is one of the best important factors. The influences of pH on the recoveries of the analytes for the copper-BPHA precipitate system were studied in the pH range of 2.0–10.0 by using model solutions containing the analyte ions. The pH values were adjusted by using the related buffer solutions given before. The results are shown in Fig. 1. The recoveries of all the analyte ions were not quantitative, except for iron(III), at acidic pH values. Therefore, the pH 9.0 was selected as working pH for the subsequent studies, and at this pH the ammonia/ammonium chloride buffer system shows a good buffering capacity. Quantitative recovery values (>95%) were obtained at pH 9.0 for the working metals ions.

Influences of pH on the recoveries of the analyte ions (N = 3)
Effect of the amount of BPHA
The effects of amount of BPHA reagent on the recoveries (%) of the analyte ions were investigated. The results are given in Fig. 2. When the experiments were performed without BPHA at pH 9.0, the recovery values were not quantitative for the studying metal ions. The recoveries of the analyte ions increased with the increasing amounts of BPHA. These results show that for the quantitative recoveries of the studied metal ions, BPHA is necessary. For all the subsequent works 500 µL of 0.5% (w/v) BPHA solution was used.

Effect of the amount of BPHA (N = 3)
Amount of copper as carrier element
The influences of amount of copper(II) as carrier element on the recoveries of the analyte ions were also investigated. Without copper, the recoveries for the analyte ions were below 60%. Quantitative recovery values (>95%) for the analyte ions were obtained for 0.25 mg of copper(II). All the further works were performed with 250 µL of 1,000 mg L−1 of copper(II) solution. The results are given in Fig. 3.

Influences of amount of copper(II) on the coprecipitation efficiency of the analyte ions (N = 3)
Effect of standing time
Firstly, the effects of the rate of centrifugation were examined in the range of 1,500–3,500 rpm under the optimal experimental conditions with model solutions. All the analytes were recovered quantitatively at the centrifugation rate of 3,500 rpm. The influences of the standing time on the recoveries of the analyte ions were investigated in the range of 5–40 min at 3,500 rpm. Quantitative recoveries (>95%) were obtained for all the analytes for 10 min of standing time and for the centrifugation rate of 3,500 rpm.
Dissolution of the precipitate
For the dissolution of the copper(II)-BPHA precipitate, several common concentrated mineral acids were examined, i.e., HNO3 and HCl. As a result, it was found that the copper(II)-BPHA precipitate has been easily dissolved in 500 µL of concentrated nitric acid. Then the resulting clear solution was diluted to 5 mL with distilled water.
Effect of sample volume
The influences of the sample volume on the coprecipitation efficiencies of the analytes were examined. The analyte ions were quantitatively recovered for the sample volumes in the range of 25–750 mL. The copper-BPHA precipitate including the analyte ions was successfully dissolved in 500 µL of concentrated nitric acid. The final volume was completed to 5 mL with double distilled deionized water. The recoveries (%) were quantitative for all the metal ions. The preconcentration factor is calculated as the ratio of the highest sample volume (750 mL) to the final measurement volume (5 mL). So, the preconcentration factor was 150. The results are given in Fig. S1 (Supplementary Material).
Matrix effects
As pointed out in “Introduction” section, one of the main problems in atomic absorption spectrometric determination of metals is matrix interferences originated from the sample. For this purpose, the influences of the some ions and substances were investigated. The results are given in Table S2 (Supplementary Material). The tolerance limit of foreign ions and/or materials was taken as the value which caused an error of not more than ±5% in the absorbance measurements. Therefore, all of the investigated foreign ions and/or subtances have no serious interfering effect on the recommended coprecipitation procedure. The recoveries were generally satisfactory and changed from 91 to 103%.
Analytical performance
The limit of detection (LOD) of the proposed method for the determination of investigated elements were studied under the optimum experimental conditions by applying the procedure for blank solutions. The detection limits was established by analyzing 20 blank solutions (3 s). The detection limits of Pb(II), Co(II), Fe(III), Cr(III) and Zn(II) were found to be 2.3, 0.7, 0.7, 0.3 and 0.4 µg L−1, respectively. The relative standard deviations of the proposed method for the analyte ions were < 4%. A common misconception is that the LOD is the smallest concentration that can be measured. Instead, quantitation is generally agreed to begin at a concentration equal to 10 standard deviations of the blank. This is called the limit of quantitation (LOQ) or limit of determination. Therefore, LOQ is equal to about 3.3 LOD [33]. So, LOQ values for Pb(II), Co(II), Fe(III), Cr(III) and Zn(II) ions were found to be 7.7, 2.3, 2.3, 1.0 and 1.3 µg L−1, respectively.
Accuracy of the method
In order to estimate the accuracy of the procedure, different amounts of the studied metal ions were added to sea water and soil samples. The resulting solutions were submitted to the proposed preconcentration/separation procedure. The obtained results are shown in Table 1. A good agreement was obtained between the added and the measured analyte amounts. The recovery values calculated for the added analytes were always higher than 95%, thus confirming the accuracy of the procedure and its independence from the matrix effects. These results confirm the validity of the proposed method.
The method presented here was also checked with the two certified reference materials, LGC6019 river water and NIST-1547 peach leaves, for the determination of Co(II), Pb(II), Fe(III), Cr(III) and Zn(II) ions. The acquired results are given in Table 2. The results were in good agreement with the certified values of the certified reference materials.
Application to real samples
The procedure presented here was applied to various real samples including hair, peritoneal fluids, urine, soil, sediment and water samples. The results are given in Table 3. Also, the results obtained from the application of the proposed method to the different natural water samples can be seen in Table 4.
Comparison with other coprecipitation methods
Comparative data from some recent papers based on the coprecipitation procedures were summarised in Table S3 (Supplementary Material). The method presented in this study is most promising for the trace elements with a preconcentration factor of 150. The preconcentration factor achieved with presented procedure is superior with respect to the coprecipitation methods given in the literature. The precipitate could be easily dissolved with 500 µL HNO3. The detection limits for the trace elements were generally lower than those of the methods given in Table S3. Also, the method is relatively rapid as compared with previously reported procedures for the enrichment of traces metal ions.
Conclusion
Coprecipitation with Cu(II)-BPHA system offers a useful multielement preconcentration technique for the environmental samples. The procedure has been successfully applied to the analyte ions with acceptable accuracy and precision. The time required for the coprecipitation and determination was about 20 min. The coprecipitated analyte ions can be sensitively determined by FAAS without any influence of copper and BPHA.
The coprecipitation method is rapid and has good reproducibility. The usefulness of the method is shown by the control analyses of the standard reference materials. The procedure offers a useful multielement enrichment technique in various samples including water analysis, hair, urine etc. with acceptable accuracy and precision. The detection limits of the analytes are superior compared to those of the preconcentration techniques given in the literature [22, 34].
References
Zou AM, Chen ML, Shu Y, Yang M, Wang JH (2007) Biological cell-sorption for separation/preconcentration of ultra-trace cadmium in a sequential injection system with detection by electrothermal atomic absorption spectrometry. J Anal At Spectrom 22:392–398
Kagaya S, Miwa S, Mizuno T, Tohda K (2007) Rapid coprecipitation technique using yttrium hydroxide for the preconcentration and separation of trace elements in saline water prior to their ICP-AES determination. Anal Sci 23:1021–1023
Hiraide M, Hommi H, Kawaguchi H (1991) Coprecipitation of copper(II)-humic complexes with indium hydroxide. Anal Sci 7:169–171
Agrawal YK, Vora SB, Shah G (2005) Solvent extraction, separation and recovery of lanthanum(III) and cerium(IV) from monazite sand by N-phenylbenzo-18-crown-6 hydroxamic acid. Indian J Chem A 44:497–503
Manzoori JL, Karim-Nezhad G (2004) Development of a cloud point extraction and preconcentration method for Cd and Ni prior to flame atomic absorption spectrometric determination. Anal Chim Acta 521:173–177
Ihara K, Hasegawa SI, Naito K (2003) Collection of iron(III) from homogeneous aqueous solutions on membrane filters using chromazurol B with triton X-100. Anal Sci 19:265–268
Tokalıoğlu Ş, Kartal Ş, Elçi L (2002) Determination of trace metals in waters by FAAS after enrichment as metal-HMDTC complexes using solid phase extraction. Bull Korean Chem Soc 23:693–698
Shinotsukai K, Suzuki K (2007) Simultaneous determination of platinum group elements and rhenium in rock samples using isotope dilution inductively coupled plasma mass spectrometry after cation exchange separation followed by solvent extraction. Anal Chim Acta 603:129–139
Kim YS, Shin JH, Choi YS, Lee W, Lee YI (2001) Solvent sublation using 8-hydroxyquinoline as a ligand for determination of trace elements in water samples. Microchem J 68:99–107
Najafi NM, Eidizadeh M, Seidi S, Ghasemi E, Alizadeh R (2009) Developing electrodeposition techniques for preconcentration of ultra-traces of Ni, Cr and Pb prior to arc-atomic emission spectrometry determination. Microchem J 93:159–163
Minami T, Atsumi K, Ueda J (2003) Determination of cobalt and nickel by graphite-furnace atomic absorption spectrometry after coprecipitation with scandium hydroxide. Anal Sci 19:313–315
Ueda J, Mizui C (1988) Preconcentration of gallium(III) and indium(III) by coprecipitation with hafnium hydroxide for electrothermal atomic absorption spectrometry. Anal Sci 4:417–421
Minamisawa H, Ilzima A, Minamisawa M, Tanaka S, Arai N, Shibukawa M (2004) Preconcentration of gallium by coprecipitation with synthetic zeolites prior to determination by electrothermal atomic absorption spectrometry. Anal Sci 20:683–687
Zolotov YA, Kuz’min NM (1990) Preconcentration of trace elements. Elsevier, Amsterdam, p 81
Pereira MG, Arruda MAZ (2003) Trends in preconcentration procedures for metal determination using atomic spectrometry techniques. Microchim Acta 141:115–131
Sawatari H, Fujimori E, Haraguchi H (1995) Multi-element determination of trace elements in seawater by gallium coprecipitation and inductively coupled plasma mass spectrometry. Anal Sci 11:369–374
Zhang Q, Minami H, Inonue S, Atsuya I (2001) Preconcentration by coprecipitation of arsenic and tin in natural waters with a Ni-pyrrolidine dithiocarbamate complex and their direct determination by solid-sampling atomic-absorption spectrometry. Fresenius J Anal Chem 370:860–864
Kashiwagi Y, Kokufuta E (2000) Selective determination of selenite and selenate in wastewater by graphite furnace AAS after iron(III) hyroxide coprecipitation and reductive coprecipitation on palladium collector using hydrazinium sulfate. Anal Sci 16:1215–1219
Hiraide M, Chen ZS, Kawaguchi H (1991) Coprecipitation of traces of heavy metals with Indium hydroxide for graphite-furnace atomic absorption spectrometry. Anal Sci 7:65–68
Hiraide M, Ozaki N, Pak YN, Tanaka T, Kawaguchi H (1993) Coprecipitation with indium hyroxide followed by flotation in a flow system for monitoring copper(II) ions in water. Anal Sci 9:367–370
Kagaya S, Kosumi S, Ueda J (1994) Differential pulse polarographic determination of lead using a rapid coprecipitation technique with indium hyroxide. Anal Sci 10:83–87
Minami T, Sohrin Y, Ueda J (2005) Determination of chromium, copper and lead in river water by graphite-furnace atomic absorption spectrometry after coprecipitation with terbium hydroxide. Anal Sci 21:1519–1521
Tamari Y, Hirai R, Tsuji H, Kusaka Y (1987) Zirconium coprecipitation method for fluorometric determination of ppt level selenium(IV) and selenium(VI) in groundwaters. Anal Sci 3:313–317
Sato H, Ueda J (2001) Coprecipitation of trace metal ions in water with bismuth(III) diethyldithiocarbamate for an electrothermal atomic absorption spectrometric determination. Anal Sci 17:461–463
Atsuya I, Itoh K, Ariu K (1991) Preconcentration by coprecipitation of lead and selenium with Ni/pyrrolidine dithiocarbamate complex and their simultaneous determination by internal standard atomic absorption spectrometry with the solid sampling technique. Pure Appl Chem 63:1221–1226
Tokalıoğlu Ş, Oymak T, Kartal Ş (2007) Coprecipitation of lead and cadmium using copper(II) mercaptobenzothiazole prior to flame atomic absorption spectrometric determination. Microchim Acta 159:133–139
Elçi L, Şahin U, Öztaş S (1997) Determination of trace amounts of some metals in samples with high salt content by atomic absorption spectrometry after cobalt-diethyldithiocarbamate coprecipitation. Talanta 44:1017–1023
Vita OA, Levier WA, Litteral E (1968) Solvent extraction separations with BPHA, applications to the microanalysis of niobium and zirconium in uranium. Anal Chim Acta 42:87–94
Nan Z (2000) Reaction mechanism of N-benzoyl-N-phenylhydroxylamine with vanadium (IV) in the weakly acidic medium. Talanta 52:785–789
Xiong C, Jiang Z, Hu B (2006) Speciation of dissolved Fe(II) and Fe(III) in environmental water samples by micro-column packed with N-benzoyl-N-phenylhydroxylamine loaded on microcrystalline naphthalene and determination by electrothermal vaporization inductively coupled plasma-optical emission spectrometry. Anal Chim Acta 559:113–119
Inoue S, Zhang Q, Uto M (2004) Solvent extraction of lanthanides(III) with N-p-phenylbenzoyl-N-phenylhydroxylamine. Solvent Extr Ion Exc 22:121–133
Lemos VA, Novaes GDS, de Carvalho AL, Gama EM, Santos AG (2009) Determination of copper in biological samples by flame atomic absorption spectrometry after precipitation with Me-BTAP. Environ Monit Assess 148:245–253
Thomsen V, Schatzlein D, Mercuro D (2003) Limits of detection in spectroscopy. Spectroscopy 18:112–114
Soylak M, Kars A, Narin I (2008) Coprecipitation of Ni2+, Cd2+ and Pb2+ for preconcentration in environmental samples prior to flame atomic absorption spectrometric determinations. J Hazard Mater 159:435–439
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Influences of the sample volume on the recoveries of the analyte ions (N = 3) (DOC 202 kb)
Table S1
Instrumental conditions for the measurement of the analytes by FAAS (DOC 29 kb)
Table S2
Effect of matrix ions and/or subtances on coprecipitation efficiency of the trace elements (DOC 47 kb)
Table S3
Comparative data from some recent studies on coprecipitation of heavy metal ions (DOC 33 kb)
Rights and permissions
About this article
Cite this article
Saçmacı, Ş., Kartal, Ş. Determination of some trace metal ions in various samples by FAAS after separation/preconcentration by copper(II)-BPHA coprecipitation method. Microchim Acta 170, 75–82 (2010). https://doi.org/10.1007/s00604-010-0391-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-010-0391-4