
Abstract
A simple, rapid and efficient method has been developed for the extraction, preconcentration and determination of copper, lead and zinc ions in water samples by air-assisted liquid–liquid microextraction coupled with graphite furnace atomic absorption spectrometry (GFAAS). In the proposed method, much less volume of an organic solvent (in the order of some µL) was used as the extraction solvent in the absence of disperser solvent. Fine organic droplets were formed by sucking and injecting of the mixture of aqueous sample solution and extraction solvent with a syringe for several times in a conical test tube. After extraction, phase separation was achieved by centrifugation and the enriched analytes in the sedimented phase were determined by GFAAS. Several variables potentially affecting the extraction efficiency were investigated and optimized. Calibration graphs were linear in the concentration range of 45.0–1100 ng L−1. Detection limits were in the range of 18.0–26.0 ng L−1. The accuracy of the developed procedure was checked by analyzing NRCC-SLRS4 Riverine water as a certified reference material. Finally, the proposed method was successfully applied to determine the selected heavy metals in tap, surface and river water samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Trace metals are widely spread in environment so may enter the food chain. Some trace metals are essential elements and play some important roles in human metabolism. On the other hand, at high concentrations all metals are recognized as potentially toxic [1]. Therefore, the study of heavy metals in the human diet is critical because of these elements’ dual effect as being either essential or toxic. Copper and zinc are typical metal ions in environmental samples, having an important role in many physiological functions and therapeutic applications [2]. However, high concentrations of these metal ions may be toxic leading to side effects. In fact, the human body does not have a good mechanism for the elimination of heavy metals; thus, chronic low-level intake can lead to damaging effects [3]. High amounts of copper in the human body can cause stomach and intestinal illnesses such as nausea, vomiting, diarrhea and stomach cramps [4]. The toxic effects of zinc are well known, and it plays an important role in the progression of several damages to the human body, including disturbances in energy metabolism or increase in oxidative stress, growth retardation, altered immune response, disturbed pregnancy, weight loss and anorexia [5]. Lead is nonessential metal, and it is toxic, even in traces [6, 7]. In spite of great improvements in the sensitivity and selectivity of modern instrumental analysis such as inductively coupled plasma mass spectrometry (ICP-MS), inductively coupled plasma optical emission spectrometry (ICP-OES) and graphite furnace atomic absorption spectrometry (GFAAS), difficulties still lie in the analysis of trace heavy metals because of both their low abundance levels in the samples and the high complexity of the sample matrices [8]. Thus, extraction and preconcentration procedures are required for elimination or at least minimization of matrix effects and concomitants, lowering the detection limit of many metals with different techniques and enhancing the detect ability for many metals [9]. Several sample preparation methods have been developed for the determination of trace heavy metals from various sample matrices, including liquid–liquid extraction (LLE) [10–13], co-precipitation [14–17], cloud point extraction (CPE) [18–22] and solid-phase extraction (SPE) [23–27]. Nevertheless, these methods are time-consuming, tedious and often require large amounts of samples and toxic organic solvents. Recently, much attention is being paid to the development of miniaturized, more efficient and environmentally friendly extraction techniques which could greatly reduce the organic solvent consumption. Therefore, liquid-phase microextraction (LPME) methods [28, 29] such as single-drop microextraction (SDME) [30] and hollow fiber liquid-phase microextraction (HF-LPME) [31, 32] were developed as solvent-minimized sample pretreatment procedures. However, these methods suffer from some disadvantages as follows: fast stirring tends to form air bubbles, extraction is time-consuming, and equilibrium can not be attained after a long time in most cases. To overcome these disadvantages, Rezaee et al. developed a novel liquid-phase microextraction technique termed dispersive liquid–liquid microextraction (DLLME) [33], which is based on a ternary component solvent system. In this extraction method, very large surface area between the fine droplets of an extraction solvent and an aqueous sample is achievable, and the corresponding fast extraction kinetic results in the rapid establishment of equilibrium so high enrichment factors can usually be obtained [34, 35]. DLLME have been developed for the extraction and preconcentration of heavy metals [36–41]. In the conventional DLLME, the extraction solvent is dispersed into the aqueous sample solution with the aid of a disperser solvent. Therefore, the presence of a disperser solvent in aqueous sample solution makes it relatively nonpolar and results in an increased solubility of the target lipophilic analytes into aqueous sample solution leading to relatively low extraction efficiency. Farajzadeh et al. [42] reported a new LPME method in which air was used to assist in dispersion of the extraction solvent into aqueous sample and called “air-assisted liquid–liquid microextraction method” (AALLME).
In this work, for the first time AALLME procedure was developed for the extraction and preconcentration of Cu(II), Pb(II) and Zn(II) from water samples. The main goal was to investigate the potential applicability of air (instead of dispersive solvent) as a driving force for better dispersion of the extraction solvent in the aqueous phase in extracting trace amounts of the heavy metals in water samples. In the proposed method, a few microliters of an extraction solvent is transferred into aqueous sample solution containing a chelating agent and then the mixture is repeatedly sucked into a glass syringe and then injected into a tube to achieve a cloudy solutions resulting from dispersion of the extraction solvent into aqueous solutions. After centrifuging the cloudy solution, the extracted heavy metals were settled down in the bottom of the centrifuge tube and used for GFAAS analysis.
Materials and methods
Reagents and solutions
A standard mixed stock solution of Cu(II), Pb(II) and Zn(II) (each 10 mg L−1) was prepared using analytical reagent grade Cu(NO3)2·6H2O, Pb(NO3)2 and ZnSO4.7H2O (Merck, Darmstadt, Germany) by dissolving an appropriate amount of each salt in double-deionized water. Working solution (200 ng L−1) was prepared daily by diluting the mix stock solution with deionized water (Ghazi Company, Tabriz, Iran). Aqueous standard solutions used to construct calibration graphs were obtained by diluting the mix stock standard solution. A mix standard solution with a concentration of 26 µg L−1 was also prepared and injected into GFAAS each day (three times) for quality control, and the obtained signal was also used to calculate enrichment factors (EFs) and extraction recoveries (ERs) of the analytes. Sodium phosphate monobasic (NaH2PO4), sodium phosphate dibasic (Na2HPO4), ammonium chloride, ammonia, acetone, methanol and sodium chloride were also purchased from Merck. Sodium diethyldithiocarbamate (SDDTC) as chelating agent was purchased from Fluka. The tested extraction solvents were obtained from the following sources: chloroform, carbon tetrachloride, 1,2-dibromoethane (1,2-DBE) were from Merck, and 1,1,2-trichloroethane (1,1,2-TCE) as well as 1,1,2,2-tetrachloroethane (1,1,2,2-TCE) was from Janssen Chimica (Belgium).
Real samples
Tap water was collected from the laboratory just before analysis. Surface water was picked up from local area (Tabriz, Iran). River water was collected from Mehranrood River (Tabriz, Iran). They were directly subjected to the extraction by the proposed AALLME method.
Instrumentation
The measurements were performed with a Shimadzu 6800 atomic absorption spectrometer (Japan) equipped with a heated graphite tube atomizer. The instrument settings and furnace programs for determination the extracted amount of each element are described in Table 1. An ASC 6100 autosampler (Shimadzu, Japan) was used to deliver standard solutions and samples from the cup to the graphite tube. The Hettich centrifuge, model D-7200 (Germany), was used for acceleration of phase separation.
AALLME procedure
An amount of 6.0 mL of mix standard solution of Cu(II), Pb(II) and Zn(II) (200 ng L−1) or real sample with pH 7 (adjusted by 100 mM phosphate buffer) was poured into a 10-mL glass centrifuge tube with conical bottom. 1,1,2,2-TCE (115 μL) as extraction solvent and 4 % (w/v) NaCl were added to the above solution. The mixture was sucked into a 10-mL glass syringe and then injected into the tube for eight times via the syringe needle. The mixture was centrifuged at 5000 rpm for 6 min. The extraction solvent droplets were sedimented at the bottom of the conical test tube, and its volume (35 ± 2 µL) was measured using a 50-µL microsyringe (zero dead volume, Hamilton, Switzerland). In order to investigate the extraction amount of the analytes, three 10-µL aliquots of the settled phase were removed and separately injected into GFAAS. The calibration graphs including ten points which were obtained by analyzing aqueous standard solutions containing the analytes at concentrations lying in the range of 45–1100 ng L−1.
Calculation of EF and ER
EF is defined as the ratio between the analyte concentration in the sedimented phase (Csed) and the initial concentration of analyte (C0) in the sample [43]:
Csed is obtained from a calibration graph. ER is defined as the percentage of the total analyte amount (n0) which is extracted to the sedimented phase (nsed).
where Vsed and Vaq are the volumes of sedimented phase and aqueous solution, respectively.
Results and discussion
The parameters being effective on the extraction process, namely amount of chelating agent, nature and level of extraction solvent, extraction numbers, salting-out effect, sample volume, pH, centrifuge rate and time and coexisting ions effect, were investigated and optimized.
Optimization of the furnace temperature program
Drying, ashing and atomization temperatures had an important effect on the determination of Cu(II), Pb(II) and Zn(II). So, temperature effects on the signals of the analytes were investigated and the results are shown in Fig. S1 in Electronic Supplementary Material (ESM). The optimum conditions are listed in Table 1.
Effect of the concentration of SDDTC
SDDTC is a suitable chelating reagent that can react with many metallic ions to form stable complexes. Because of the multielement capability of the analysis, SDDTC was selected as a nonspecific chelating agent [44, 45] for the simultaneous determination of Cu(II), Pb(II) and Zn(II) (Fig. 1) using the proposed AALLME preconcentration technique. The amount of SDDTC can markedly effect on the extraction efficiency. Therefore, the influence of SDDTC concentration on the AALLME of Cu(II), Pb(II) and Zn(II) was evaluated in the concentration range of 2.0 × 10−4–2.2 × 10−3 mol L−1, and the results are shown in Fig. S2 in ESM. According to the results, ER increases up to 1.4 × 10−3 mol L−1 and then slightly decreases. It can be assumed that this slight decrease in ER at high concentrations of SDDTC is due to the competitive extraction of free SDDTC (i.e., the portion not participated in complex formation) into the extraction solvent which can easily saturate the low-volume extraction solvent and decrease its capability for extracting the complexes.

Reaction of the selected heavy metals with diethyldithiocarbamate; M = Cu, Pb and Zn
Effect of type and volume of extraction solvent
The selection of an appropriate extraction solvent is of great importance in the optimization of an AALLME process. The extraction solvent has to fulfill some requirements: heavier or lighter than water, low volatility, low solubility in water and high extraction efficiency toward the analytes. For this purpose, several extracting solvents including chloroform, carbon tetrachloride, 1,2-DBE, 1,1,2-TCE and 1,1,2,2-TCE were investigated. To achieve the same volume of precipitated phase (35 ± 2 µL), different volumes of the selected extraction solvents were used. The obtained volumes were 186, 179, 135, 159 and 115 µL for chloroform, carbon tetrachloride, 1,2-DBE, 1,1,2-TCE and 1,1,2,2-TCE, respectively. The results (Fig. 2) showed that 1,1,2,2-TCE and 1,2-DBE are the most effective extraction solvents giving the highest extraction efficiency for the target analytes among the five solvents investigated. Due to low consumption of 1,1,2,2-TCE compared to 1,2-DBE (115 vs. 135 µL), it was selected as the extraction solvent for the further experiments. In microextraction procedures, the volume of extraction solvent is another important factor that affects the extraction efficiency. Increasing the extraction solvent volume could increase the extracted amount of analyte, whereas its concentration in the sedimented phase will be diluted. To evaluate the effect of extraction solvent volume on the extraction performance, different volumes of 1,1,2,2-TCE (115–165 μL) were tested. Analytical signals decreased by increasing volume of 1,1,2,2-TCE due to dilution effect. But ERs of the analytes remain the same for various extractant volumes in the range of 115–165 µL (Fig. S3 in ESM). It is noted that based on Eq. 3, by increasing Vorg while Vaq is constant, ER% should be increased.

Effect of the chemical identity of extraction solvent in ERs of the selected elements. Extraction conditions: sample, 6 mL deionized water containing 200 ng L−1 of Cu2+, Pb2+ and Zn2+; SDDTC concentration, 1.4 × 10−3 mol L−1; extraction numbers, 7 times; centrifuge rate, 5000 rpm; and centrifuge time, 5 min. The error bars represent standard deviations (n = 3)
[D: partition coefficient of the analyte; Vaq: volume of aqueous phase; Vorg: volume of organic phase]However, when D is very high compared with \(\frac{{V_{aq} }}{{V_{org} }}\), increasing Vorg does not affect the ER%.
It was observed that by increasing volume of the extraction solvent from 115 to 165 µL, volume of the sedimented phase increased from 35 to 77 µL. Therefore, use of low volume of the extractant leads to high EFs and low detection limits. In the case of ˂115 μL of 1,1,2,2-TCE, the volume of sedimented phase was less than 35 µL, by which the analysis of the analytes was impossible (10 µL was required for analysis of each analyte). Thereby, 115 µL of 1,1,2,2-TCE was used as the extraction solvent in the subsequent experiments to obtain 35 µL sedimented phase volume.
Effect of salt addition
In most cases, addition of a salt plays an important role in the conventional extraction procedures. By increasing ionic strength, the solubility of many analytes in aqueous solutions decreases due to salting-out effect and this improves the extraction efficiency of various extraction methods. So ER is often increased in the presence of a salt. It was observed that by increasing the concentration of NaCl, the volume of the sedimented phase diminished. This decrease in volume is most probably due to increase in the density of the aqueous phase with respect to the organic phase which leads to transfer of a slight portion of the organic phase to surface of the aqueous phase. Therefore, the experiments were performed using different volumes of extraction solvent to achieve 35 µL of the sedimented phase volume (115, 112, 109, 107, 105 and 101 µL for 0, 2, 4, 6, 8 and 12 %, w/v, NaCl concentration, respectively), while other experimental parameters were constant. The extraction efficiencies relating to the extracted amount of the analytes against NaCl concentration are presented in Fig. 3. The results showed that with increasing the concentration of NaCl up to 4 %, ER increased and then reached a plateau after that. Regarding the extraction efficiency increasing with NaCl concentration increasing, the salting-out effect is thought to be responsible. Addition of salt increases the ionic strength of the samples and makes complexes (element-SDDTC) less soluble and forces it to migrate into the organic phase. Therefore, 4 % (w/v) NaCl was used to ensure obtaining analytes responses of the highest possible level.

Effect of NaCl addition on the ERs of Cu2+, Pb2+ and Zn2+. NaCl %, w/v, (1,1,2,2-TCE volume, µL); 0 (115), 2 (112), 4 (109), 6 (107), 8 (105), 12 (101); other conditions are the same as used in Fig. 2. The error bars represent standard deviations (n = 3)
Effect of sample solution volume
In order to study the effect of sample solution volume on the extraction efficiency, it was investigated in the range of 3–8 mL. The results (Fig. 4) proved that analytical signals increased by increasing the sample volume up to 6 mL and then decreased at higher volumes. This is due to the increase in EF of the analytes in the extract. However, more increasing of sample size is not reasonable, because it prevents the formation of organic phase drops. The volume of sedimented phase was also decreased from 44 to 32 µL by increasing the volume of aqueous phase from 3 to 8 mL. Due to high ER obtained, 6 mL was selected for the future studies.

Effects of sample volume on the ER of Cu2+, Pb2+ and Zn2+. Extraction conditions: NaCl, 4 % (w/v); other conditions are the same as used in Fig. 3. The error bars represent standard deviations (n = 3)
Effect of sample solution pH
The pH value of sample solution plays an important role in the proposed procedure because it determines the chemical form of analytes and functional groups of chelating groups that can occur, and thus the extraction efficiency of the target compounds can be influenced by pH. Extraction of metallic ions by AALLME involves prior complex formation with sufficient hydrophobicity to be extracted into the small volume of the extraction solvent. Therefore, the pH of aqueous phase is one of the most important factors in extraction of metallic ions from various media in the view of formation of the related complex. The influence of the pH on the ER of the selected heavy metals was investigated in the pH range of 2.0–12.0, while the other experimental conditions kept constant. The results are depicted in Fig. 5. At low value of pH, the extraction efficiency of the analytes is low. It may be attributed to the interaction of DDTC− with hydronium ions rather than with the analytes [46]:
ER increased with increasing solution pH from 2.0 to 6.0 and was effective in pHs 6.0 and 7.0. Further increasing pH of aqueous solution led to decrease in ER of the analytes probably due to hydrolysis of analytes [47–49]:
Therefore, the further works were performed at pH 7.0.

Effect of sample pH on the enrichment performances. The conditions are the same as used in Fig. 4. The error bars represent standard deviations (n = 3)
Optimization of numbers of extraction
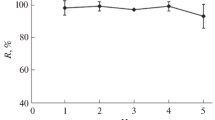
In this study, the numbers of extraction is defined as the numbers of repeatedly sucking extraction solvent and sample solution mixture into a 10-mL glass syringe and then its injecting into the test tube. To some extent similar to multiple batch extraction, it is predictable that by increasing extraction numbers, recoveries should be increased, too. Therefore, to reach the equilibrium status, the numbers of extraction was studied in the range of 2–12 times. According to the results (Fig. 6), by increasing extraction numbers, ER is also increased till eighth extraction and then remains almost constant. Hence, eight times of extraction was selected as the optimum for the further studies. It is mentioned that this step is very fast and consumes less than 1 min.

Effect of extraction numbers on the ERs of Cu2+, Pb2+ and Zn2+. Extraction conditions: sample pH, 7.0; other conditions are the same as used in Fig. 5. The error bars represent standard deviations (n = 3)
Optimization of centrifuging rate and time
Other parameters that may affect the extraction efficiency are centrifuging rate and time. They were studied in the ranges of 1000–8000 rpm and 2–8 min, respectively. Therefore, two series of experiments were carried out. In one set of experiments, a constant centrifugation time (5 min) was selected, while its speed varied in the range of 1000–8000 rpm. Another set of experiments were performed at a constant centrifugation speed (5000 rpm), while the run time varied (2–8 min). According to the results, 5000 rpm and 6 min were selected as centrifuge rate and time, respectively.
Effect of coexisting ions
The effect of common coexisting ions in natural water samples on the ER of the analytes was studied. In these experiments, 6.0 mL of solution containing 200 ng L−1 of the analytes and various amounts of coexisting ions was treated according to the recommended procedure. A given species was considered to interfere if it resulted in a ±5 % variation in the absorbance signal. The tolerable concentration ratios of the coexisting ions to the analytes were found to be as follows: 5000 for Al3+, Cr3+, Ca2+, Mg2+, K+; 2200 for Hg2+; 650 for Sn4+, Fe3+, Cd2+, Sn2+; 400 for CO3 2−, PO4 3−; 200 for SO4 2−; and 150 for Co2+, Ni2+.
Analytical figures of merit
Under the optimized conditions, quantitative characteristics, namely linear range (LR), squared determination coefficient (r2), precision, limit of determination (LOD) and limit of quantification (LOQ) of the analytes were evaluated and are summarized in Table 2. The LODs values were 20.0, 26.0 and 18.0 ng L−1 for Cu, Pb and Zn, respectively. To evaluate repeatability of the method, extractions were carried out on the six separate solutions at a concentration of 200 ng L−1. High EFs, low LODs and LOQs are main advantages of the proposed method. The accuracy of the proposed method was assessed with the measurement of the analytes in NRCC-SLRS-4 Riverine water as certified references material. For analysis of Cu and Zn, the certified reference material was diluted till 2.5-fold and then adjusted to the proposed procedure. The certified and observed values are given in Table 3. It was found that the analyzed results were in good agreement with the certified values.
Application of the proposed method to analysis of real samples
To demonstrate the performance of the present method, it was utilized to determine Cu, Pb and Zn concentration in different real water samples. Recovery studies were also carried out after the samples were spiked with known concentrations of the analytes at levels of 150, 250 and 350 ng L−1. The recoveries in comparison with those obtained for deionized water at the same concentrations are summarized in Table 4. The recovery values were ranged from 89 to 97 %, which indicated that the matrix effect was negligible.
Comparison of the proposed method with other methods
Table 5 summarizes the analytical characteristics of some other analytical methods along with those of the proposed AALLME-GFAAS method for the extraction and determination of Cu, Pb and Zn in different real samples. Most of the analytical characteristics of the proposed method were good and comparable with those of the other reported methods. The repeatability of the method is good, and RSD for the proposed method is lower than those of some of the others. The most spectral methods have LRs in the range of 2–3 orders of magnitude. As it can be seen from the Table 5, except the second method (SPME-HPLC-UV) which is a separation method, other methods (spectral methods) have the similar LRs. Also, the proposed method has the LRs in the anticipation ranges. Therefore, the LRs of the method are comparable with those of the other reported related methods. Other advantages of the method compared to others are: low LOD and LOQ.
Conclusions
In this study, for the first time a developed microextraction technique, namely AALLME coupled with GFAAS‚ was used for preconcentration and determination of Cu, Pb and Zn in aqueous samples. The method is rapid, precise, efficient and sensitive. In the proposed method, much less volume of an organic solvent (at µL order) was used as the extraction solvent in the absence of disperser solvent. Finally, the proposed method was successfully applied for the determination of the target analytes in different real aqueous sample. The results indicated that this extraction procedure was noticeable due to its outstanding advantages including minimum organic solvent consumption, simplicity, low cost, rapidness, high efficiency and environmentally friendly.
Abbreviations
- AALLME:
-
Air-assisted liquid–liquid microextraction
- EF:
-
Enrichment factor
- ER:
-
Extraction recovery
- GFAAS:
-
Graphite furnace atomic absorption spectrometry
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- RSD:
-
Relative standard deviation
- SDDTC:
-
Sodium diethyldithiocarbamate
References
S. Baytak, A.R. Turker, Talanta 65, 938 (2005)
T.M. Statham, K.A. Mumford, S.C. Stark, D.B. Gore, G.W. Stevens, Sep. Sci. Technol. 50, 2427 (2015)
S. Verbych, N. Hilal, G. Sorokin, M. Leaper, Sep. Sci. Technol. 39, 2031 (2005)
E.J. Underwood, Trace Elements in Human and Animal Nutrition (Academia, London, 1971)
P. Bratter, P. Schramel, Trace elements in Analytical Chemistry (Medicine and Biology, New York, 1980)
D.L. Tsalev, Z.K. Zaprianov, Atomic Absorption in Occupational and Environmental Health Practice, Analytical Aspects and Health Significance, vol. 11 (CRC Press, Boca Raton, 1983)
V.P. Kudesia, Water Pollution (Pregati Prakashan Publications, Meerut, 1990)
H.G. Seiler, A. Siegel, H. Siegel, Handbook on Metals in Clinical and Analytical Chemistry (Marcel Dekker, New York, 1994)
J. Shao, Sh Qin, W. Li, Y. He, Sep. Sci. Technol. 48, 2735 (2013)
M.R. Jamali, Y. Assadi, F. Shemirani, Sep. Sci. Technol. 42, 3503 (2007)
N.W. Ockwig, T.M. Nenoff, Chem. Rev. 107, 4078 (2007)
M. Bustelo, B. Fernandez, J. Pisonero, R. Pereiro, N. Bordel, V. Vega, V.M. Prida, A. Sanz-Medel, Anal. Chem. 83, 329 (2011)
A. Oliva, A. Molinari, F. Zuniga, P. Ponce, Microchim. Acta 140, 201 (2002)
H. Sato, J. Ueda, Anal. Sci. 17, 461 (2001)
E.L. Chruscinska, A.S. Zając, D. Olejnik, Food Addit. Contam. Part B 5, 251 (2012)
H.Y. Lee, Sep. Sci. Technol. 48, 1602 (2013)
S. Kagaya, Y. Araki, N. Hirai, K. Hasegawa, Talanta 67, 90 (2005)
Naeemullah, T.G. Kazi, M. Tuzen, J. Ind. Eng. Chem. 35, 93 (2016)
M.A. Bezerra, M.A.Z. Arruda, S.L.C. Ferreira, Appl. Spectrosc. Rev. 40, 269 (2005)
Sh. Yang, X. Fang, L. Duan, Sh Yang, Z. Lei, X. Wen, Spectrochim. Acta A 148, 72 (2015)
J. Chen, K.C. Teo, Anal. Chim. Acta 434, 325 (2001)
J.L. Manzoori, A. Bavili-Tabrizi, Microchim. Acta 141, 201 (2003)
M. Amjadi, A. Samadi, Colloid Surf. A 434, 171 (2013)
Y. Ye, A. Ali, X. Yin, Talanta 57, 945 (2002)
A.N. Anthemidis, G. Giakisikli, G.A. Zachariadis, Anal. Methods 3, 2108 (2011)
Q. Zhou, A. Xing, K. Zhao, J. Chromatogr. A 1360, 76 (2014)
V.N. Bulut, A. Gundogdu, C. Duran, H.B. Senturk, M. Soylak, L. Elci, M. Tufekci, J. Hazard. Mater. 146, 155 (2007)
A. Sarafraz-Yazdi, A. Amiri, TrAC Trends Anal. Chem. 29, 1 (2010)
M.A. Farajzadeh, S.M. Sorouraddin, M.R. Afshar Mogaddam, Microchim. Acta 81, 829 (2014)
X. Wen, Q. Deng, J. Guo, Spectrochim. Acta A 79, 1941 (2011)
C.J. Zeng, X.D. Wen, Z.Q. Tan, P.Y. Cai, X.D. Hou, Microchem. J. 96, 238 (2010)
J. Abulhassani, J.L. Manzoori, M. Amjadi, J. Hazard. Mater. 176, 481 (2010)
M. Rezaee, Y. Assadi, M.R. Milani Hosseini, E. Aghaee, F. Ahmadi, S. Berijani, J. Chromatogr. A 1116, 1 (2006)
C.B. Ojeda, F.S. Rojas, Chromatographia 69, 1149 (2009)
M. Rezaee, Y. Yamini, M. Faraji, J. Chromatogr. A 1217, 2342 (2010)
Y. Zhang, J. Duan, M. He, B. Chen, B. Hu, Talanta 115, 730 (2013)
E.S. Silva, L.O. Correia, L.O. Santos, E.V.S. Vieira, V.A. Lemos, Microchim. Acta 178, 269 (2012)
K. Kocot, B. Zawisza, R. Sitko, Spectrochim. Acta B 73, 79 (2012)
E.Z. Jahromi, A. Bidari, Y. Assadi, M.R.M. Hosseini, M.R. Jamali, Anal. Chim. Acta 585, 305 (2007)
P.X. Baliza, L.S.G. Teixeira, V.A. Lemos, Microchem. J. 93, 220 (2009)
H. Sereshti, V. Khojeh, S. Samadi, Talanta 83, 885 (2011)
M.A. Farajzadeh, M.R. Afshar Mogaddam, Anal. Chim. Acta 728, 31 (2012)
S.M. Sorouraddin, S. Nouri, Anal. Methods 8, 1396 (2016)
X. Jia, Y. Han, X. Liu, T. Duan, H. Chen, Spectrochim. Acta Part B 66, 88 (2011)
X. Wen, Q. Yang, Z. Yan, Q. Deng, Microchem. J. 97, 249 (2011)
G.B. Raju, W. Forsling, Colloids Surf. 60, 53 (1991)
A. Huchicki, Talanta 14, 1371 (1967)
M.M. Hassanien, A.M. Hassan, W.I. Mortada, A.A. El-Asmy, Am. J. Anal. Chem. 2, 697 (2011)
A.A. Baba, F.A. Adekola, J. King Saud Univ. Sci. 25, 297 (2013)
M. Tuzen, M. Soylak, L. Elci, Anal. Chim. Acta 548, 101 (2005)
V. Kaur, J.S. Aulakh, A.K. Malik, Anal. Chim. Acta 603, 44 (2007)
Y. Yamini, M. Rezaee, A. Khnchi, M. Faraji, A. Saleh, J. Chromatogr. A 1217, 2358 (2010)
K. Bakkali, N.R. Martos, B. Souhail, E. Ballesteros, Food Chem. 116, 590 (2009)
M.V. Reboucas, D. Domingos, A.S.O. Santos, L. Sampaio, Fuel Process. Technol. 91, 1702 (2010)
A. Niazi, S. Habibi, M. Ramezani, Arab. J. Chem. 8, 706 (2015)
M. Mirzaei, M. Behzadi, N. Mahmoud Abadia, A. Beizaeia, J. Hazard. Mater. 186, 1739 (2011)
Acknowledgments
The author would like to thank the Research Office at the University of Tabriz for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sorouraddin, S.M. Simultaneous separation and preconcentration of ultra-trace of Cu(II), Pb(II) and Zn(II) in water samples by air-assisted liquid–liquid microextraction prior to determination by graphite furnace atomic absorption spectrometry. J IRAN CHEM SOC 13, 2219–2227 (2016). https://doi.org/10.1007/s13738-016-0940-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-016-0940-9