
Abstract
A fast, sensitive and simple oil-in-water emulsion (OWE) method was developed for extraction of four phenolic pollutants in environmental water samples followed by gas chromatography and flame ionization detection. In this method, the density of a binary organic solvent (one heavier and one lighter than the sample) was balanced with the density of the sample solution. A stable emulsion was formed at room temperature under vigorous stirring using a Teflon-coated magnetic stirring bar. After addition of 10 µL of the heavier organic solvent and centrifugation, phase separation occurred. The influence of several important parameters on the extraction efficiency of phenolic compounds was evaluated. Under optimized experimental conditions, the calibration graphs were linear in the concentration range 0.025–20 mg L−1 with coefficients of determination more than 0.9994. The limits of detection and quantification were in the range 19.2–76.0 and 64.1–251.0 μg L−1, respectively. Intra-day and inter-day precisions were less than 5.0 %. The procedure was used for the determination of phenolic compounds in spiked water samples with good results. Recoveries range from 96.5 to 103.0%, and relative standard deviations are <2.5% (for n = 3).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phenolic compounds are important contaminants in waste-water due to wide use in many industrial processes, such as pesticides, dyes, pharmaceuticals, and plastics [1–3]. Because of high toxicity, most of phenols and substituted phenols are listed as priority pollutants in many countries.
Several procedures have been reported for measurement of phenolic compounds in environmental water samples, including liquid-liquid extraction (LLE) [4–6] and solid phase extraction (SPE) [1, 7–9] and final analysis by high performance liquid chromatography (HPLC), gas chromatography (GC), or capillary electrophoresis (CE). However, in addition to time consuming and low sensitivity, LLE requires large amount of toxic organic solvents. SPE uses considerably less solvent, but it can be relatively expensive. Hence, a more sensitive, fast, and simple method is necessary for measurement of phenolic compounds.
Dispersion of droplets of a one liquid in another (two immiscible liquids) is defined as an emulsion. The emulsion has been a powerful technology for many separation processes including removal and recovery of various metals such as zinc, silver, cobalt and cadmium [10–12] and successfully used in cosmetic, foods and pharmaceutical industries [13, 14].
The present study described an oil-in-water emulsion (OWE) method for the extraction of four phenolic pollutants in environmental water samples. This method is very fast, flexible in selection of extraction solvents, simple, low cost and high preconcentration factor. In order to show the performances of the proposed method, extraction of phenolic compounds as case study in environmental water samples by gas chromatography and flame ionization detection (GC-FID) were investigated.
Experimental
Reagents
Phenol (Ph), 2,4-dimethylphenol (24DMPh), 2,4,6-trichlorophenol (246TCPh), 1-naphtol (1 Np), salicylaldeyade (internal standard, I.S.), ethanol (HPLC grade), carbon tetrachloride, chloroform, dichloromethane, benzene, toluene, cyclohexane, n-hexane, ethyl acetate, sodium chloride and hydrochloric acid were purchased from Merck (Darmstadt, Germany, www.merck.com). Butyl acetate was acquired from Acros (Geel, Belgium, www.acros.com). Methyl isobutyl keton was supplied from Scharlau (Barcelona, Spain, www.scharlau.com).
Apparatus
The measurements were performed with a gas chromatograph (model GC-17 Shimadzu, www.shimadzu.com) equipped with a flame ionization detector (GC-FID) and a BP21-FFAP capillary column (25-m length and 0.52-mm i.d.). Helium was used as the carrier gas at a total flow rate of 29 mL min−1. After injection of samples, the temperature of the column oven was kept constant for 1 min (100°C), linearly increased to 270°C (30°C min−1) and kept constant at this temperature for the remaining time of analysis. Injector and detector temperatures were set at 280 and 300°C, respectively. Retention times of the peaks were as follows: Ph, 1.69 min; 24DMPh, 2.68 min; 246TCPh, 4.30 min; 1 Np, 5.27 min; I.S., 2.10 min. Quantitative data obtained using the internal standard method based on the peak area of the standard solutions and I.S.
The pH measurements were made with a 780 pH meter (Metrohm, Switzerland, www.metrohm.com) equipped with a combine Ag/AgCl glass electrode. The centurion scientific centrifuge (model K280R, UK, www.centurionscientific.co.uk) was used for centrifuging.
OWE preparation
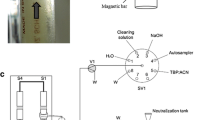
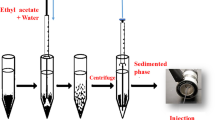
The OWE system components are schematically shown in Fig. 1. An aliquot (5 mL) of solution (pH value 2, adjusted with hydrochloric acid) containing 4 mg L−1 of phenolic compounds and I.S. was placed in a 10-mL volumetric flask. Carbon tetrachloride (20.2 µL), as heavier solvent, and butyl acetate (79.8 µL), as lighter solvent, were added at the bottom and at the top of sample solution using a 250-µL eppendorf micropipette sampler, respectively, (extraction solvents can be mixed preliminarily and add to sample). The mixture was stirred by means of a 19.5 mm × 6 mm stir bar (1 min, 700 rpm), a stable emulsion or a milky solution was formed (it was stable at least for 5 h). In order to break down emulsion, 10 µL heavier solvent was added. The milky solution was transferred to a test tube with conical bottom and separation of the phases was accomplished by centrifuged at 4000 rpm for 3 min. Then the dispersed fine droplets of organic phase were sedimented in the bottom of test tube. The 2 µL of sedimented phase was removed using a 10 µL microsyringe (Hamilton, USA, www.hamiltoncompany.com) and injected onto GC-FID system.

Schematic illustration of OWE device
Calibration curve
Individual solutions of Ph, 24DMPh and I.S. were prepared in double distilled water. Due to the limited solubility of 246TCPh and 1 Np in water, these analytes were initially dissolved in ethanol. Stock standard solutions of phenolic compounds and internal standard were prepared in ethanol at the concentration level of 1000 mg L−1. Working solutions were obtained by appropriate dilution. Calibration standards were made at different concentration ranges. Each one was prepared in five replicates. Equations were obtained by least squares linear regression of the peak area ratio of standard to internal standard versus analyte concentration.
Sample preparation
Tap, mineral, river and waste water samples were collected in glass bottles from Ilam province (Ilam, Iran). The samples were filtered using Whatman No. 42 filter paper before analysis and stored in refrigerator in the dark. The pH of samples was adjusted by hydrochloric acid and the preconcentration procedure was applied based on the above mention method.
Results and discussion
Optimization of the OWE method
In this study the effect of several important parameters influencing the extraction efficiency including the heavier and lighter organic solvents, volume of organic phase and its density, salting-out, pH of aqueous sample, temperature, time of extraction and stirring rate was investigated. Chromatographic peak area of analyte and that of I.S. was used to assess the extraction efficiency under experimental conditions tested. A solution 4 mg L−1 each analyte and I.S. was used for optimization of OWE procedure.
The most important step in the development of the proposed method is the selection of appropriate extraction solvents. The selection of extraction solvents was based on (a) immiscibility with aqueous phase; (b) the density of heavier and lighter solvents must be higher and lower than aqueous phase, respectively (c) good chromatographic analysis. Based on these considerations dichloromethane (CH2Cl2), chloroform (CHCl3) and carbon tetrachloride (CCl4) as heavier extraction solvents and methyl isobutyl ketone (MIBK), cyclohexane, ethyl acetate, butyl acetate, benzene, toluene, and n-hexane as lighter extraction solvents were tested. It was found that except for carbon tetrachloride-butyl acetate system, all other combinations of heavier and lighter solvents did not show stable cloudy solution. Therefore, CCl4 and butyl acetate were selected as the extraction solvents for subsequent experiments.
As previously mentioned, stability of this kind of OWE is based on variation of density of binary extraction solvent to approach density of sample. The effect of density of the binary extraction solvent (CCl4 and butyl acetate) was studied in the range 0.881–1.591 g L−1. The desired density (density of binary organic phase) was calculated as follows [15]:
where ρ l and ρ h represent the density of lighter and heavier organic solvents and v l and v h represent the volume of lighter and heavier organic solvents, respectively. It was found that response gradually increased with an increase in the binary extraction solvent density from 0.881 to 1.024 g L−1. Beyond this point, the response was decreased. Hence, the optimum density of the binary extraction solvent was selected by considering density of aqueous sample.
The effects of ionic strength were extensively evaluated in the traditional liquid-liquid extraction, because salt addition can increase the partition of analytes to the organic phase. The effect of salting-out was investigated by adding NaCl (0–10% w/v). Peak areas of all analytes slightly increase and gradually when amounts of NaCl increase up to 2% and became constant. Hence, a salt concentration of 2% was chosen for further experiments.
The effect of pH in the range from 1 to 7 was evaluated. The analytical signal as a function of pH of aqueous sample is demonstrated in Fig. 2. As it can be observed, the extraction efficiency of analytes at low pH value was superior to that at higher pH value. Based on the results, pH value of 2 was adopted for subsequent tests.

Effect of pH of aqueous solution on the extraction efficiency. Extraction conditions: sample volume: 5 mL; NaCl concentration: 2% (w/v); heavier phase volume: 30.3 µL (CCl4); lighter phase volume: 119.7 µL (butyl acetate); extraction time: 1 min; stirring rate: 800 rpm; extraction temperature: 23°C
The effect of volume of binary extraction solvent (CCl4 and butyl acetate) was examined in the range 40–200 µL. The results of Fig. 3 show that for all analytes, a similar pattern is observed in the volume range between 90 µL and 130 µL. With less than 40 µL of binary extraction solvent no two phase system was observed. However, a further increase in the volume of binary extraction solvent has no significant consequence on the extraction efficiency. This may be because of formation of large droplets of binary extraction solvent [16, 17]. Therefore, all further experiments were carried out at the optimum volume of 100 µL.

Effect of binary organic phase volume on the extraction efficiency. Extraction conditions: sample volume: 5 mL; NaCl concentration: 2% (w/v); pH: 2 (0.01 mol L−1 HCl); heavier phase: CCl4; lighter phase: butyl acetate; extraction time: 1 min; stirring rate: 800 rpm; extraction temperature: 23°C
Stirring rate is one of the most important parameters that can reduce the time required to attain thermodynamic equilibrium and enhanced the extraction efficiency. In order to generate fine and a stable emulsion formation fast agitation was used. The results indicated that the extraction efficiency increased with increasing of stirring rate from 100 to 700 rpm and then approximately remained constant across the range of 800–1000 rpm. Therefore, all further experiments were performed with a stirring rate of 700 rpm.
The extraction efficiency increases with longer extraction time in traditional liquid-liquid extraction (LLE). When the influence of extraction time was studied in the range 0 to 30 min, it was found that this parameter had no influence on the extraction efficiency for the analytes under study. This may be attributed to the quickly equilibrium reaching and independency of proposed method to the time. Thus, in this investigation an extraction time of 1 min was selected for subsequent experiments.
The effect of temperature was investigated in the range 23–65°C. It was found that an increase in the solution temperature reduced the extraction efficiency. This may be attributed to decreases in surface tension of immiscible liquids with increasing temperature [18, 19]. Therefore, a temperature of 23°C was finally selected for subsequent experiments.
Analytical performance of the developed OWE method
Under the above optimum experimental conditions, the proposed method was validated by linearity, precision and limit of detection. The calibration plots were found to be linear in the range of 0.1–20 , 0.025–20, 0.05–20 and 0.05–20 mg L−1, for Ph, 24DMPh, 246TCPh and 1 Np, respectively, with a coefficient of determination (r2) more than 0.9994 (n = 11). For each concentration level, three replicate extractions were performed. The limits of detection (LOD, S/N = 3) and quantification (LOQ, S/N = 10) were in the range 19.2–76.0 and 64.1–251.0 µg L−1, respectively. As it can be seen, the proposed method has low LOD and LOQ and can be used for trace analysis of phenolic pollutants in different water samples. The intra-day and inter-day precisions of the assay were evaluated by analyses of quality control samples at three concentration levels (0.2, 8.0, and 18.0 mg L−1) on the same day (n = 10) and the five consecutive days. It was found that the intra-day and inter-day precisions for all analytes were in the range of 1.2–5.0 and 1.5–5.0%, respectively.
Application to real sample analysis
In order to investigate the recovery of the proposed method, four water samples were collected, spiked with standards at three concentration levels (0.1, 5.0 and 15.0 mg L−1) and analyzed. The results are listed in Table 1. The recoveries of the samples ranged from 96.0–102.0% and relative standard deviations (RSDs) were in the range of 1.0 to 2.5%, respectively. The preceding results demonstrated that the proposed method was feasible for quantitative the determination of phenolic pollutions in environmental water samples.
The proposed method was applied to determination of phenolic pollutions in tap, mineral, river and waste water samples. Representative chromatograms of different water sample extracts are shown in Fig. 4. Average concentrations of phenolic compounds are summarized in Table 1. As it can be seen, tap, river and mineral water samples were free of phenolic compounds and the phenolic pollutions (except 24DMPh) were found in waste water sample.

GC-FID chromatograms of OWE extracts of unspiked a standard, b waste water, c tap water, d river water, e mineral water
Table 2 indicates the limit of detection (LOD), relative standard deviation (RSD), coefficient of determination (r2), linear range, extraction time and recovery using solid-phase microextraction-gas chromatography-flam ionization detection (SPME-GC-FID) [8], phase transfer catalysis-gas chromatography-mass spectrometric detection (PTC-GC-MS) [20], micro-liquid chromatography-ultraviolet absorption spectrometry (µLC-UV) [21], single-drop microextraction-gas chromatography-mass spectrometric detection (SDME-GC-MS) [3], solvent microextraction-gas chromatography-mass spectrometric detection (SME-GC-MS) [22] and OWE methods for the determination of phenolic compounds in water samples. The proposed method provides similar quantification extraction efficiency, with advantages of being faster and using smaller volume of organic solvents.
Conclusions
In this research work a simple OWE method was developed and applied for the determination for the determination of phenolic compounds in water samples. The method was based on ternary component solvents emulsion formation. The present method is attractive due to its simplicity, analytical precision, considerable saving in terms of time of analysis, and organic solvent used. The results from validation indicate the proposed method can be used for the determination of phenolic pollutants in water samples.
References
Liu X, Ji Y, Zhang Y, Liu H (2007) Oxidized multiwalled carbon nanotubes as a novel solid-phase microextraction fiber for determination of phenols in aqueous samples. J Chromatogr A 1165:10
Shen S, Chang Z, Liu H (2006) Three-liquid-phase extraction system for separation of phenol and p-nitrophenol from wastewater. Sep Pur Technol 49:17
Saraji M, Bakhshi M (2005) Determination of phenols in water samples by single-drop microextraction followed by in-syringe derivatization and gas chromatography-mass spectrometric detection. J Chromatogr A 1098:30
Andres MPS, Leon-Gonzalez ME, Perez-Arribas LV, Polo-Diez LM (2000) Determination of pollutant phenols by capillary high-performance liquid chromatography with UV-detection. J High Resol Chromatogr 23:367
Almeda S, Nozal L, Arce L, Valcarcel M (2007) Direct determination of chlorophenols present in liquid samples by using a supported liquid membrane coupled in-line with capillary electrophoresis equipment. Anal Chim Acta 587:97
Reis MTA, Freitas OMF, Ismael MRC, Carvalho JMR (2007) Recovery of phenol from aqueous solutions using liquid membranes with Cyanex 923. J Membr Sci 305:313
Sirvent G, Sanchez JM, Salvado V (2004) Preconcentration and determination of priority pollutant phenols in waters at trace levels using a polymeric solid-phase extraction cartridge. J Sep Sci 27:1524
Zhou F, Li X, Zeng Z (2005) Determination of phenolic compounds in wastewater samples using a novel fiber by solid-phase microextraction coupled to gas chromatography. Anal Chim Acta 538:63
Penalver A, Pocurull E, Borrull F, Marce RM (2002) Solid-phase microextraction coupled to high-performance liquid chromatography to determine phenolic compounds in water samples. J Chromatogr A 953:79
Fouad EA, Bart HJ (2008) Emulsion liquid membrane extraction of zinc by a hallow-fiber contactor. J Membr Sci 307:156
Othman N, Mat H, Goto M (2006) Separation of silver from photographic wastes by emulsion liquid membrane system. J Membr Sci 282:171
Kumbasar RA (2009) Selective extraction and concentration of cobalt from acidic leach solution containing cobalt and nickel through emulsion liquid membrane using PC-88A as extractant. Sep Pur Technol 64:273
Mirhosseini H, Tan CP, Hamid NSA, Yusof S, Chern BH (2009) Characterization of the influence of main emulsion components on the physicochemical properties of orange beverage emulsion using response surface methodology. Food Hydrocolloids 23:271
Domenico GA, Massimo MA, Sanzo RDA, Rossi COB, Silverstro A, Ruffolo B, Bruno de Cindio A (2009) Characterization of dairy emulsions by NMR and rheological techniques. Food Hydrocolloids 23:619
Meloan CE (1999) Chemical separations: principles, techniques, and experiments, John Wiley & Sons, INC, p. 451
Ashby NP, Binks BP (2000) Pickering emulsion stabilised by laponite clay particles. Phys Chem Chem Phys 2:5640
Tadros T, Izquierdo P, Esquena J, Solans C (2004) Formation and stability of nano-emulsion. Adv Colloid Interface Sci 108–109:303
Hoang TKN, La VB, Deriemaeker L, Finsy R (2004) Ostwald ripening and solubilization in alkane in water emulsions stabilized by different surfactants. Phys Chem Chem Phys 6:1413
Chen G, Tao D (2005) An experimental study of stability of oil-water emulsion. Fuel Process Technol 86:499
Fiamegos YC, Nanos CG, Pilidis GA, Stalikas CD (2003) Phase-transfer catalytic determination of phenols as methylated derivatives by gas chromatography with flame ionization and mass-selective detection. J Chromatogr A 983:215
Ruiz-Jimenez J, Luque de Castro MD (2007) In-column micro-high-performance liquid chromatographic concentration-separation prior to ultraviolet detection for the determination of chlorophenols in water samples. J Chromatogr A 1174:78
Bagheri H, Saber A, Mousavi SR (2004) Immersed solvent microextraction of phenol and chlorophenols from water samples followed by gas chromatography-mass spectrometry. J Chromatogr A 1046:27
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Characteristic parameters of the calibration equations for the proposed oil-in-water emulsion method for simultaneous analysis of phenolic compounds. (DOC 23 kb)
Table S2
Results from determination of within-day and between-day precision of phenolic compounds. (DOC 41 kb)
Fig. S1
Effect of stirring speed on the extraction efficiency of phenolic compounds. Extraction conditions: sample volume = 5 mL aqueous solution containing 2% NaCl and 0.01 mol L−1 HCl (pH 2); heavier phase volume = 20.2 µL CCl4; lighter phase volume = 79.8 µL of butyl acetate; extraction time = 1 min; temperature = 23°C. (DOC 184 kb)
Fig. S2
Effect of temperature on the extraction efficiency of phenolic compounds. Extraction conditions: sample volume = 5 mL aqueous solution containing 2% NaCl and 0.01 mol L−1 HCl (pH 2); heavier phase volume = 20.2 µL CCl4; lighter phase volume = 79.8 µL of butyl acetate; stirring rate = 700 rpm; extraction time = 1 min. (DOC 181 kb)
Rights and permissions
About this article
Cite this article
Daneshfar, A., Khezeli, T. Extraction of phenolic compounds from environmental water samples using oil-in-water emulsions. Microchim Acta 167, 211–216 (2009). https://doi.org/10.1007/s00604-009-0242-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-009-0242-3