
Abstract
A highly sensitive, simple and rapid method is presented for the determination of palladium using graphite furnace atomic absorption spectrometry after its separation and preconcentration by dispersive liquid-liquid microextraction. Ultra traces of Pd were extracted and preconcentrated in acidic water samples by using 2-amino-1-cyclohexene-1-dithiocarboxylic acid as a suitable chelating agent, and carbon tetrachloride and acetone as extraction and disperser solvents, respectively. The experimental parameters were optimized in order to enhance the extraction efficiency. After optimizing the extraction conditions and various instrumental parameters, an enhancement factor of 350 was obtained. The analytical curve absorbance vs. concentration was linear over the range 0.02–0.6 µg L-1 Pd. The detection limit and relative standard deviation were 0.007 µg L-1 and 4.2%, respectively. The method was successfully applied to the determination of palladium in roadside soil and several aqueous samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Palladium is a metal of economic importance due to its extensive use as a hydrogenation catalyst, as a catalyst in various chemical syntheses, as a micro− contactors in electronics, in jewellery manufacture, in production of dental and medicinal devices and, in recent years, as a component in the three-way catalysts in automobile exhaust gas catalytic beads [1]. As a result of such diverse applications, palladium can be distributed and cause damage to the environment, even at low tendency to accumulation in the food chain and low decomposition rates, palladium is grouped within the category of environmental toxins [2].Up to date, the sources of palladium sensitization identified most often for the general population are dental restorations and jewellery. Most case reports refer to palladium sensitivity associated with exposure to palladium-containing dental restorations, its symptoms being contact dermatitis, stomatitis or ucostitis and oral lichen planus. Some palladium compounds have also reported as potent sensitizers of the skin. Because of such issues, the preconcentration and determination of palladium are of great concern.
2-Amino-1-cyclohexene-1-dithiocarboxylic acid (ACDA) and some of its derivatives, first synthesized by Takeshima et al. [3, 4], have already been employed as suitable reagents for the determination of Cu2+, Cd2+, Co2+, and Ni2+ with ACDA in various methods [5-8]. In the present investigation, ACDA was used as an excellent chelating agent for the complexation and extraction of palladium in µg L-1 levels.
There are several literature reports for preconcentration and determination of palladium using liquid-liquid extraction [9-13], solid phase extraction [14-16] and cloud point extraction [17, 18]. Despite the fact that these methods have their own advantages for application to the analysis of Pd in real samples, most of them are associated with one or more of the following disadvantages as high time consumption, solvent loss, and unsatisfactory enrichment factor. Thereby, the development of simple, rapid, selective and efficient separation and preconcentration methods for low level determination of palladium is challenging area of research.
To overcome the above mentioned difficulties, recently a novel microextraction technique, named dispersive liquid-liquid microextraction (DLLME), was developed and successfully applied to the extraction and preconcentration of ultra trace amounts of organic compounds and inorganic elements [19-23]. It is worth mentioning that based on previous literature reports [5-8], among a large number of transition and heavy metal ions it is only palladium(II) ion that can form a stable complex with ACDA in highly acidic solutions.
In DLLME technique, the volume of organic solvent is minimized and sample extraction and preconcentration will be carried out in a single step, faster and simpler than conventional methods. In addition, aqueous to organic phase ratio is higher than 100, thus providing a very high pre-concentration factor. In DLLME, the preconcentrated phase (sedimented phase) can be formed by rapid injection of an appropriate mixture of the extraction and disperser solvents into aqueous phase containing the metal ion complex. Consequently, a cloudy solution is formed and the metal complex will then interacted with the fine droplets of extraction solvent and extracted in it. After centrifuging of fine droplets, the sedimented phase is formed at the bottom of conical tube. Finally, the analyte content of the sedimated phase will be determined by graphite furnace atomic absorption spectrometry (GFAAS).
In the present work, we used this method for selective extraction and pre-concentration of sub-nanogram amounts of Pd (II) ions by ACDA, as a suitable chelating agent, and subsequent determination by GFAAS. Simplicity of the operation, rapidity, low cost, low organic solvent consumption, high recovery, low sample volume and high enrichment factor combined with good detection limit of GFAAS are the advantages of DLLME-GFAAS system for determination of palladium.
Experimental
Reagents
Reagent grade chlorobenzene, carbon tetrachloride, chloroform, as extraction solvent, and ethanol, methanol, acetone and acetonitrile, as disperser solvents, and nitric acid and sulfuric acid were purchased from Merck Chemical Company and used as received. Doubly distilled water was used throughout. Analytical grade palladium chloride and nitrate salts of other cations (all from Merck) were of the highest purity available from Merck and used without further purification except for vacuum drying. ACDA was prepared and purified according to Takeshima et al. [3, 4] and used as a 1.0 × 10-3 M solution in acetone.
Instrumentation
All measurements were carried out using a Shimadzu atomic absorption spectrometer (AA6800G), equipped with a graphite furnace atomizer GFA-6500 and an autosampler ASC6100. Deuterium background correction was employed to correct nonspecific absorbance. All measurements were performed using the peak heights. A palladium hollow cathode lamp (Hamamatsu Photonic Co. Ltd., L233 series) and a pyrolitic coated graphite tube (Shimadzu part no. 206-69984-02) were used. The sample injection volume was 10 µL in all experiments. The experimental parameters and temperature program for the graphite atomizer are listed in Table 1. Argon gas with 99.95% purity was purchased from Roham Gas Co. (Tehran, Iran) and was used as protected and purges gas. A Behdad Universal Centrifuge (Tehran, Iran) was used for centrifugation. All 10 mL screw cap falcon test tubes with conical bottom (extraction vessel) were maintained into 0.1 M HNO3, for cleaning of any inorganic compounds, and washed with doubly distilled water and then with acetone for proper sedimentation of fine droplets of extraction solvent in centrifuging step. The pH measurement on the aqueous phase was performed using a PP-50 Sartorius pH meter equipped with a combined glass electrode.
General procedure
In a calibrated 10 mL volumetric flask was added 1.00 mL of 2 µg L-1 of palladium solution, 0.5 mL of 2.0 M H2SO4, to achieve a pH of 0.9, and 0.25 mL of 1.0 × 10-3 M ACDA solution in acetone and diluted to the mark with distilled water and mixed well. The solution was then removed into a 15 mL screw cap falcon conical test tube. 800 µL of acetone (disperser solvent) containing 40 µL of carbon tetrachloride (extraction solvent) were injected rapidly into the sample solution by using a 1.00 mL Hamilton syringe and the solution was gently shaken. A cloudy water/acetone/carbon tetrachloride solution was formed in the test tube. In this step, the palladium-ACDA complex is extracted into the fine droplets of carbon tetrachloride. The mixture was then centrifuged for 2 min at 4,000 rpm. After this process, the dispersed fine droplets of carbon tetrachloride were sedimented at the bottom of test tube (25 ± 1 µL). After centrifugation, the aqueous phase was carefully removed so that a stable sedimented phase was remained. Afterward, about 20 µL of the sedimented phase was charged into an auto-sampler device, and 10 µL of it was then injected into the graphite tube, and the palladium content was determined by graphite furnace atomic absorption spectrometry. To prevent sample spreading over the atomizer surface, in all experiments, the drying and ashing steps were carried out with a long ramp time [24].
Preparation of roadside soil sample
Palladium of the automobile catalyst origin is introduced into roadside soil in an artificial way. The content of palladium was strongly correlated to the density of traffic. Because of certified reference material was not available, we decided to analyze a roadside soil sample from urban road of Arak, Iran, by the proposed method and validate it by recovery of appropriate amounts of Pd standard solution to the sample, before its digestion.
1.0000 g of the soil sample was weighed into a PTFE beaker. In order to decompose it, the sample was heated at 100˚C in the presence of 2 × 10 mL of aqua regia for 1 h; afterwards, 1 mL concentrated HF was added and the solution was heated to near dryness. Now, 1 mL of concentrated HCl was added and the solution made up to 25 mL with distillated water and, finally, 1.00 mL of the solution was taken and the recommended procedure was followed.
Results and discussion
In order to reach the optimized conditions for quantitative extraction and high enrichment factor of palladium by DLLME method, the influence of different parameters such as volume and nature of both disperser and extraction solvents, pH of aqueous solution, concentration of chelating agent, salt effect and extraction time were investigated. The enhancement factor (EF) was calculated from the slope ratio of calibration curves obtained after and before DLLME.
Influence of type and volume of extraction solvent
The selection of an appropriate solvent as a key parameter is an important task in DLLME process. The organic extraction solvents should possess densities lower than water as well as low water solubilities, and are selected based on their highest extraction efficiencies for the palladium-ACDA complex. Thus, the ability of chlorobenzene, carbon tetrachloride and chloroform, with respective densities of 1.107, 1.590 and 1.492 g cm-3, were compared for the extraction of palladium-ACDA by the proposed method. A series of sample solutions was studied by using 800 µL of acetone, as dispersing solvent, containing different volumes of extraction solvent to achieve a final 25 ± 1 µL volume of the sedimented phase. Thereby, 43, 40 and 45 µL volumes of chlorobenzene, carbon tetrachloride, and chloroform were selected, respectively, and used for the DLLME and GFAAS determination of 0.3 µg L-1 palladium. The results revealed that percent recovery by using chlorobenzene, carbon tetrachloride and chloroform is 81 ± 5, 104 ± 3 and 78 ± 4, respectively. Therefore, carbon tetrachloride, with the highest extraction efficiency, was selected as the best solvent for further experiments.
In order to evaluate the optimal volume of extraction solvent, solutions containing different volumes of carbon tetrachloride were examined with the same DLLME procedures. The experimental conditions were fixed and included the use of 800 µL acetone containing different volume of carbon tetrachloride (i.e., 40, 47, 55, 65, 75, 85 µL). According to the results obtained, by increasing the volume of carbon tetrachloride, the volume of sedimented phase increased from 25 to 77 µL. Meanwhile, the extraction recovery was found to be almost quantitative in the case of all solvent volumes examined, which emphasizes the high distribution coefficient of palladium-ACDA complex in carbon tetrachloride under the experimental conditions used. However, as expected, the enrichment factor found to decrease significantly with increasing volume of carbon tetrachloride. Subsequently, at lower volume of CCl4, higher enrichment factor and quantitative recovery are reachable. On the other hand, our experience revealed that in carbon tetrachloride volumes lower than 40 µl, picking up of 10 µL of the resulting sedimented phase by autosampler was practically difficult. Thus, 40 µL of carbon tetrachloride was selected as the optimum volume of extraction solvent.
Influence of nature and volume of disperser solvent
The most important point for the selection of a suitable disperser solvent is its mutual miscibility in organic phase (extraction solvent) and aqueous phase (sample solution) is. Thereby, acetone, methanol, ethanol, and acetonitrile which possess these abilities were tested as potential disperser solvents. Thus, under the same experimental conditions, a series of sample solutions were prepared by using 800 µL of each disperser solvent containing 40 µL of carbon tetrachloride and the recommended procedure was followed. The results showed that, in all cases, the recovery is almost quantitative, and variations in percent recovery are not remarkable. Thus, among the three candidate solvents, acetone was selected as disperser solvent for further studies, due to its low toxicity and cost.
Since the change in volume of acetone resulted in a change in the volume of sedimented phase, it was necessary to optimize the volume of disperser solvent. It was found that, at low volume of acetone, the cloudy solution was not formed completely while, at high acetone volume, the solubility of carbon tetrachloride in aqueous solution was increased. Therefore, it was important to consider the effect of acetone volume on the extraction efficiency of the system.
In order to achieve a constant volume of sedimented phase throughout the experiments, the volume of acetone and carbon tetrachloride were simultaneously changed, so that the volume of the sedimented phase remained nearly constant at 25 ± 1 µL. In this respect, several experiments were carried out by using varying volume ratios of acetone (mL)/carbon tetrachloride (µL) of 0.25/37, 0.50/38, 0.75/39, 1.0/40, 1.25/42, 1.5/43.5 . The results showed that the extraction efficiency is increased with increasing volume of acetone up to 0.75 mL, and remained more or less constant up to 1.0 mL of acetone, and then decreased significantly upon further increase in volume of acetone used. The observed decrease in the extraction efficiency at acetone volumes lower than 0.75 mL is most possibly due to incomplete formation of cloudy state, while at volumes higher than 1.0 mL acetone, the decreased extraction efficiency could be due to the increased solubility of palladium-ACDA complex in aqueous phase, which results in diminished distribution coefficient and extraction recovery of palladium. According to the results, a volume of 0.8 mL of acetone was chosen as optimum disperser volume.
Effect of extraction time
The extraction time in DLLME process is considered as the time interval between the injection moment of the mixture of disperser/extraction solvent, and the moment of starting the centrifugation process. In order to study the influence of extraction time, the extraction procedure was carried out at different time intervals, in the range of 0–20 min, under the same experimental conditions. The obtained results clearly showed that the proposed extraction method is very fast so that the extraction time has no considerable effect on the signal of palladium. This is mainly due to an infinitely large surface area between extraction solvent and aqueous phase, which results in rapid transfer of palladium-ACDA complex from aqueous phase into the organic solvent is. Such a short extraction time can be considered as one of the main advantages of the DLLME method, as reported before [19-23].
Influence of pH of test solution
For successful extraction of palladium ion from the aqueous phase to the organic solvent by DLLME method, it is necessary to optimize the pH of test solution for the selective formation of a neutral Pd-ACDA complex with sufficient hydrophobicity and quantitative extraction into the extraction solvent. The effect of pH on extraction of palladium ion from water sample was studied in the range of 0.6–8.5 and the results are shown in Fig. 1. As it is obvious, the absorbance signal was increased by decreasing of pH from 6.4 to 1.25 and remained more or less constant at lower pH values. Therefore, a pH of 0.9 maintained by adding an appropriate amount of 2.0 M sulfuric acid was selected for further studies.

Effect of pH of test solution on the absorbance of palladium after DLLME. Extraction condition: sample volume, 10.0 mL; concentration of Pd2+, 0.4 µg L-1; sedimented phase volume, 25 ± 1 µL; pH, 0.9; concentration of ACDA, 2.5 × 10-5 M. volume of extraction solvent, 40 µL. Error bars correspond to the standard deviations of three replicate measurements.
Influence of ACDA concentration
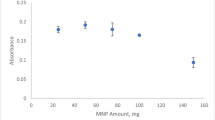
The influence of ACDA concentration, as an excellent chelating agent for Pd2+ ion, on the efficiency of DLLME was investigated under the same experimental conditions, and the results are shown in Fig. 2. As seen, the extraction efficiency increased with increasing concentration of the ACDA until a reagent concentration of about 4.0 × 10-4 M is reached. Further increase in concentration of ACDA resulted in a gradual decrease in extraction efficiency of the system for palladium ion, most probably due to the extraction of excess of ACDA, which in turn decreases the limited capacity of the organic solvent for the quantitative extraction of the palladium-ACDA complex. Thus, an ACDA concentration of 3.75 × 10-4 M was selected for subsequent experiments.

Effect of ACDA concentration on recovery of palladium ion in DLLME. Extraction conditions: same as Fig. 2. Error bars correspond to the standard deviation of three replicate measurements.
Effect of ionic strength
In order to investigate of influence of the ionic strength of test solution on the extraction efficiency of the system, different amounts of NaCl (0-5 %w⁄v) were tested, while the other experimental conditions were kept constant. The results revealed that the volume of sedimented phase increases slightly (less than 2%) with increasing concentration of NaCl, which results in some decreased enrichment factor of the system. Meanwhile, as expected, the salting out effect increases the enrichment factor. Therefore, the combination of these opposite phenomena practically resulted in no significant effect on recovery and enrichment factor of the DLLME.
Effect of diverse ions
In order to study the effect of various cations and anions on the determination of Pd2+, a fixed amount of palladium ion (0.3 µg L-1) was taken with different amounts of a large number of potentially interfering ions and the recommended procedure was followed. A relative error of 5% was considered tolerable. The results are summarized in Table 2. As is obvious, under the optimized conditions used and, especially, at an acidic solution of pH 0.9, none of the ionic species examined interfere in extraction and determination of palladium, even at concentrations of 1,000 µg L-1 and higher.
Figures of merit
The figures of merit of the proposed DLLME-GFAAS method, including linear range, limit of detection, reproducibility and enhancement factor are listed in Table 3. The results clearly demonstrate a distinct improvement in the figures of merit of GFASS brought about upon its combination with DLLME technique. Under the optimum experimental conditions, the linear range (LR) of the calibration curve found to be 0.02–0.60 µg L-1, with a regression coefficient of 0.995. The enhancement factor for the proposed method found to be 350, as obtained from the slope ratio of calibration curve after (1.70) and before (0.0049) of extraction. The limit of detection (LOD) was calculated as the ratio of three times of standard deviation of blank signal and the slop of calibration curve after preconcentration. The LOD was achieved 0.007 µg L-1. The high sensitivity presented by GFAAS is a decisive factor when analytes have to be determined at very low concentration [24]. The relative standard deviation (RSD) for the eight replicate measurements of 0.30 µgL-1 palladium from 10 mL of standard solutions was found to be 4.2%.
Comparison of DLLME-GFAAS with other methods
In Table 4 are compared the main analytical characteristics (i.e., LR, LOD, RSD, EF, extraction time, and sample consumption) of the proposed DLLME-GFAAS method for the determination of palladium with those of some of the best previously methods for this purpose. These reported methods include liquid-liquid extraction (LLE)-spectrophotometry [10], online pre-concentration-graphite furnace atomic absorption spectrometry (GFAAS) [12], adsorptive pre-concentration-flame atomic absorption spectrometry (FAAS) [14], cloud point extraction (CPE)-flame atomic absorption spectrometry (FAAS) [17], cloud point extraction (CPE)-spectrophotomtry [18], solid phase extraction (SPE)-flame atomic absorption spectrometry (FAAS) [25], solid phase extraction (SPE)-inductively coupled plasma-atomic emission spectrometry (ICP-AES) [26].
As it can be concluded from Table 4, although the proposed method possess some improved features including the low LOD, high enhancement factor, short extraction time (less than 3 min) and low sample consumption (10 mL), some of the previously reported methods have also several advantageous properties such as better RSD, low sample consumption, and on line capability. The results clearly revealed that DLLME-GFAAS is a sensitive, rapid, and simple technique that can be used for preconcentration and determination of ultra trace amounts of palladium from real samples.
Analytical applications
The proposed DLLME-GFAAS method was validated by extraction and determination of palladium from tap, mineral and waste water and synthetic samples and the results are summarized in Table 5. For this purpose, 10 mL of the water and synthetic samples were spiked with 0.2 and 0.4 µg L-1 palladium standard solution and the recommended procedure was followed to assess the matrix effects. The method was also applied to the determination of palladium content of, as well as recovery of some added Pd to, a roadside soil sample and the results are also included in Table 5. As it is obvious from Table 5, in all cases, the extraction efficiency of palladium was excellent, and showed no serious matrix effects.
The determination of palladium in a waste water sample obtained from ET unit of Arak Petrochemical Company, which contained a large amount of copper ion, was also carried out by the proposed method. Here, a 10.0 mL portion of the waste water was transferred into a 500 mL volumetric flask and diluted to the mark with doubly distilled water. The copper content of the resulting solution was 30 mgL-1. The extraction and determination of palladium content in this solution which was carried out via standard addition method found to be 448 ± 30 µg L-1 by the recommended procedure. The result revealed a satisfactory agreement with that obtained ICP-AES (i.e., 466 ± 20 µg L-1).
Conclusions
In this work we introduced DLLME-GFAAS method for the analysis of ultra trace amounts of palladium in tap, mineral and waste waters, and synthetic and roadside soil samples. The proposed DLLME method takes advantages from low cost, use of minimized toxic organic solvents, simplicity of operation, rapidity, high enhancement factor and high sensitivity and selectivity. The limit of detection of the proposed DLLME-GFAAS method (0.007 µg L-1) is significantly improved over that of the best of previously reported methods for palladium determination.
References
Precious Metal Division, Johnson Matthey Chemical Company (web page http://www.matthey.com /divisions/precious).
Environmental Health Criteria of 226-Palladium (web page http://www.who.int/ipcs//publications/ehc/en/ehc 226.pdf)
Yokoyama M, Takeshima T (1968) Spectrophotometric determination of nickel with 2-amino-1-cyclopentene-1-dithiocarboxylic acid. Anal Chem 40:1344–1345
Takeshima T, Yokoyama M, Imamoto T, Akano M, Asaba H (1969) Reaction of active methylene compounds with carbon disulfide in the presence of ammonia. III. Reaction of cyclopentanone and cycloheptanone. J Org Chem 34:730–732
Ensafi AA, Bakhshi M (2003) New stable optical film sensor based on immobilization of 2-amino-1-cyclopentene-1-dithiocarboxylic acid on acetyl cellulose membrane for Ni(II) determination. Sens Actuators B 96:435–440
Shamsipur M, Hashemi OR, Safavi A (2005) Flotation-separation and ICP-AES determination of ultra trace amounts of copper, cadmium, nickel and cobalt using 2-aminocyclopentene-1-dithiocarboxylic acid. Anal Sci 21:1063–1066
Gholivand MB, Khorsandipoor S (2000) Selective and efficient uphill transport of Cu(II) through bulk liquid membrane using N-ethyl-2-aminocyclopentene-1-dithiocarboxylie acid as carrier. J Membr Sci 180:115–120
Safavi A, Abdollahi H, Hormozinezhad MR, Kamali R (2004) Cloud point extraction, preconcentration and simultaneous spectrophotometric determination of nickel and cobalt in water samples. Spectochim Acta A 60:2897–2901
Mateeva N, Arpadjan S, Deligeorgiev T, Mitewa M (1992) Extraction systems for the flame atomic absorption spectrometric determination of trace amounts of mercury and palladium. Analyst 117:1599–1561
Gholivand MB, Nozari N (2000) Extraction and spectrophotometric determination of trace amount of Pd(II) with 2, 2’-dithiodianiline. Talanta 52:1055–1060
Ensafi AA, Eskandari H (1999) Highly selective liquid—liquid extraction from sulfuric acid medium and spectrophotometric determination of palladium(II) with α-benzilmonoxime. Microchem J 63:266–275
Limbeck A, Rendl J, Puxbaum H (2003) TAAS determination of palladium in environmental samples with on-line preconcentration and matrix separation. J Anal At Spectrom 18:161–165
Alizadeh N, Salimi S, Jabbari A (2002) Liquid-liquid extraction of palladium(II) from hydrobromic acid media by hexadecylpyridinium bromide. Anal Sci 18:307–312
Tokahoghi S, Oymak T, Kartal S (2004) Determination of palladium in various samples by atomic absorption spectrometry after preconcentration with dimethylglyoxime on silica gel. Anal Chim Acta 511:255–260
Dong Y, Gai K (2005) Spectrophotometric determination of palladium after solid-liquid extraction with 4-(2-pyridylazo)-resorcinol at 90°C. Bull Korean Chem Soc 26:943–946
Daniel S, Praveen RS, Rao TP (2006) Ternary ion-association complex based ion imprinted polymers (IIPs) for trace determination of palladium(II) in environmental samples. Anal Chim Acta 570:79–87
Priya BK, Nayam PS, Uvardhan KS, Kumar KS, Rekha D, Rao AV, Rao GC, Chiranjeevi P (2007) RETRACTED: Cloud point extraction of palladium in water samples and alloy mixtures using new synthesized reagent with flame atomic absorption spectrometry (FAAS). J Hazard Mat 144:152–158
Shemirani F, Rahnama Kozani R, Jamali MR, Assadi Y, Milani Hosseini M (2006) Cloud-point extraction, preconcentration, and spectrophotometric determination of palladium in water samples. Intern J Environ Anal Chem 86:1105–1112
Rezaei M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A 1116:1–9
Berijani S, Assadi Y, Anbia M, Milani Hosseini MR, Aghaee E (2006) Dispersive liquid—liquid microextraction combined with gas chromatography-flame photometric detection: Very simple, rapid and sensitive method for the determination of organophosphorus pesticides in water. J. Chromatogr A 1123:1–9
Zeini Jahromi E, Bidari A, Assadi Y, Milani Hosseini MR, Jamali MR (2007) Dispersive liquid—liquid microextraction combined with graphite furnace atomic absorption spectrometry: Ultra trace determination of cadmium in water samples. Anal Chim Acta 585:305–11
Kozani RR, Assadi Y, Shemirani F, Milani Hosseini MR, Jamali MR (2007) Part-per-trillion determination of chlorobenzenes in water using dispersive liquid—liquid microextraction combined gas chromatography—electron capture detection. Talanta 72:387–393
Rezaee M, Yamini Y, Shariati S, Esrafili A, Shamsipur M (2009) Dispersive liquid—liquid microextraction combined with high-performance liquid chromatography-UV detection as a very simple, rapid and sensitive method for the determination of bisphenol A in water samples. J Chromatogr A 1216:1511–1514
Volynsky AB, BYa S, YuA Z (1984) Solvent extraction-electrothermal atomic-absorption analysis. Talanta 31:449–458
Stalikas CD (2002) Micelle-mediated extraction as a tool for separation and preconcentration in metal analysis. Trends Anal Chem 21:343–355
Farhadi K, Teimouri G (2005) Flame atomic absorption determination of palladium in solutions after preconcentration using octadecyl silica membrane disks modified by thioridazine·HCl. Talanta 65:925–929
Jamali MR, Assadi Y, Shemirani F, Salsvati-Niasari M (2007) Application of thiophene-2-carbaldehyde-modified mesoporous silica as a new sorbent for separation and preconcentration of palladium prior to inductively coupled plasma atomic emission spectrometric determination. Talanta 71:1524–1529
Liang P, Zhao E, Li F (2009) Dispersive liquid—liquid microextraction preconcentration of palladium in water samples and determination by graphite furnace atomic absorption spectrometry. Talanta 77:1854–1857
Acknowledgements
The support of this work by Iran National Science Foundation (INSF) and TWAS-Iran Chapter is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shamsipur, M., Ramezani, M. & Sadeghi, M. Preconcentration and determination of ultra trace amounts of palladium in water samples by dispersive liquid-liquid microextraction and graphite furnace atomic absorption spectrometry. Microchim Acta 166, 235–242 (2009). https://doi.org/10.1007/s00604-009-0186-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-009-0186-7