
Abstract
A full-length cDNA clone (LeST3), encoding a putative tomato sugar transporter, was isolated from mycorrhizal roots by using a PCR-based approach. Based on sequence similarity, conserved motifs and predicted membrane topology, LeST3 was classified as a putative monosaccharide transporter of the sugar transporter subgroup of the major facilitator superfamily. Southern blot analysis showed that LeST3 represents a single-copy gene in tomato. To investigate its function, LeST3 was expressed in a hexose transport-deficient mutant of Saccharomyces cerevisiae. Although LeST3 was correctly transcribed in yeast, it did not restore growth on hexoses of the S. cerevisiae mutant. LeST3 gene expression was increased in the leaves of plants colonised by the arbuscular mycorrhizal (AM) fungi Glomus mosseae or Glomus intraradices and in those of plants infected with the root pathogen Phytophthora parasitica. These data suggest that LeST3 plays a role in the transport of sugars into the sink tissues and responds to the increased demand for carbohydrates exerted by two AM fungi and by a root pathogen to cope with the increased metabolic activity of the colonised/infected tissues or to supply carbohydrates to the AM fungus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The first evidence for carbon transfer from the plant to the fungus in arbuscular mycorrhizal (AM) symbiosis was provided by Ho and Trappe (1973). Nowadays, there is enough evidence that as a consequence of mycorrhizal establishment, a higher proportion of assimilates is directed toward the root, and the rate of net photosynthesis of the host increases (Douds et al. 1988; Graham 2000; Tinker et al. 1994). Measurements of carbon flux indicate that up to 20% of the photosynthetically fixed carbon may be allocated to the roots during a mycorrhizal association (Graham 2000). Sucrose, the long-distance transport carbohydrate in higher plants, should play an important role in carbon transfer in AM symbioses, and mechanisms must exist to ensure that mycorrhizal roots receive an adequate supply of sugars for formation, maintenance and function of mycorrhizal structures. Accumulation of sucrose synthase and vacuolar invertase transcripts, enzymes responsible for sucrose catabolism, in arbuscule-colonised root cortical cells has led to the hypothesis that these enzymes provide metabolites and generate sink strength during the active phase of the mycorrhizal symbiosis (Blee and Anderson 2002; Hohnjec et al. 2003).
Studies using isotopic labelling with nuclear magnetic resonance spectrometry in AM roots and radiorespirometry measurements on isolated intraradical hyphae have shown that the internal mycelium of the fungus can take up and use hexoses, mainly glucose, but not sucrose (Shachar-Hill et al. 1995; Solaiman and Saito 1997). Therefore, it is assumed that sucrose allocated to the symbiotic interfaces is hydrolysed to glucose and fructose by a cell-wall-bound invertase and that monosaccharides are taken up by the fungus via ATP- and proton-potential-dependent mechanisms at the symbiotic interface (Ferrol et al. 2002a; Smith et al. 2001). However, experimental evidence for this hypothesis is still lacking.
Sugar transporters play a pivotal role in sugar distribution throughout the plant. Therefore, it is likely that these transporters are regulated by the development of the AM symbiosis. A gene encoding a Medicago hexose transporter, likely to be involved in sugar uptake, has been shown to be up-regulated in Glomus versiforme colonised roots, specifically in highly colonised regions of the root (Harrison 1996). Plants have several monosaccharide and disaccharide transporters to coordinate sugar movement in diverse tissues at different developmental stages and under varying environmental conditions (for a review, see Büttner and Sauer 2000; Lemoine 2000; Williams et al. 2000). These transporters are members of the major facilitator superfamily, which is characterised by a common structural motif consisting of 12 transmembrane-spanning domains and the presence of several other conserved amino acid motifs (Saier et al. 1999). Whereas disaccharide transporters appear to be specific for plants, numerous genes that are homologous to monosaccharide transporters have been found in bacteria, fungi and mammals. Monosaccharide transporters have been cloned from different fungi such as yeasts (Heiland et al. 2000), Aspergillus (Yu et al. 2000), Neurospora (Madi et al. 1997), the ectomycorrhizal fungus Amanita muscaria (Nehls et al. 1998) and the pathogen Uromyces fabae (Voegele et al. 2001).
In the present article, as a first step to study mechanisms underlying carbon transfer in arbuscular mycorrhizas, we have followed a PCR-based approach to identify genes showing increased expression or specifically active in mycorrhizal plants. A gene encoding a putative tomato sugar transporter, whose expression increased in leaves but not in roots of mycorrhizal plants, has been identified. Regulation of this gene in plants infected with the root pathogen Phytophthora parasitica has been also assessed to determine whether expression of this gene was specifically regulated by the AM symbiosis.
Materials and methods
Biological material, growth conditions and plant harvest
Tomato seeds (Lycopersicon esculentum Mill, cv 76R; Peto Seed Company, CA) were surface-sterilized and sown in wet autoclaved vermiculite. Plantlets were transplanted when the first true leaf was expanded. Plants were grown in 500 ml pots containing a sterile mixture of quartz sand and soil (1:1, v/v). Four different treatments were applied: uninoculated control plants (C), plants inoculated with the mycorrhizal fungi Glomus mosseae (Nicol. and Gerd.), Gerd. and Trappe BEG119 (Gm) or Glomus intraradices Smith and Schenck BEG121 (Gi) and plants inoculated with the root pathogen P. parasitica var. nicotianae (Phy). Mycorrhizal inoculation was performed as described by Benabdellah et al. (1999) using a soil-sand-based inoculum of the AM fungi. Control plants received a filtrate (<20 μm) of the AM inoculum containing the microbial populations without the AM propagules. Plants infected by the root pathogen P. parasitica were prepared by watering 4 week-old control plants with a suspension of P. parasitica mycelium as previously described by Pozo et al. (1999).
Plants were grown in a constant environment (25/18°C day/night temperature, 60% relative humidity, 16 h photoperiod, 400 μmol photons m−2 s−1), watered three times per week with a low-phosphorus-content (25%) Long Ashton nutrient solution (Hewitt 1952) and harvested 6 weeks after inoculation with the AM fungi and 2 weeks after inoculation with P. parasitica. Mycorrhiza development was estimated in root samples after trypan blue staining (Phillips and Hayman 1970) using the gridline intersect method (Giovannetti and Mosse 1980). The spread of P. parasitica was estimated by evaluation of the necrotic lesions on the roots (Pozo et al. 1999).
Nucleic acid isolation
Total RNA was isolated from frozen roots and leaves of plants of all treatments according to Logemann et al. (1987). Tomato genomic DNA was extracted from frozen tomato leaves as previously described by Murray and Thompson (1980). G. mosseae genomic DNA was extracted from 1,000 spores using the DNeasy Plant Mini Extraction Kit (QIAGEN, Germany) according to the manufacturer’s specifications, except that the liquid nitrogen grinding step was substituted by crushing the spores in the lysis buffer using a micropestle.
Degenerate RT-PCR
A 2 μg RNA sample of G. mosseae mycorrhizal roots was DNase treated and used to synthesize first-strand cDNA in a 20 μl reaction containing 4 U Avian myeloblastosis virus reverse transcriptase (Finnzymes Oy, Finland), 200 ng random hexamer primers, 1 mM each dNTP, 20 U RNase inhibitor and 1X reverse transcription buffer. Degenerate sense primer sug1 [5′-CCIGA(AG)(AT)C(ACT)CCI(CA)GIT-3′] and antisense primer sug2 [5′-A(AG)ICC(CT)TTIGT(CT)TGIGG-3′], designed in base to conserved motifs present in all hexose transporters, were used for PCR amplification of first-strand cDNA. The PCR reaction mixture (50 μl) contained 5% of the RT reaction described above, 200 mM dNTPs, 3 mM MgCl2, 2 mM each primer and 0.3 U Taq polymerase. Amplifications were performed in an automated thermal cycler (Gene Amp PCR System 2400, Perkin Elmer, Foster City, CA) with an initial denaturation for 5 min at 94°C, followed by 35 cycles with denaturation for 30 s at 94°C, annealing for 1 min at 45°C and extension for 1 min at 72°C, followed by a final extension for 7 min at 72°C.
Rapid amplification of cDNA ends
Five micrograms of total RNA was used for first-strand cDNA synthesis using the sug3 primer (5′-CAAATAATATGGACAGTG-3′) for 5′ rapid amplification of cDNA ends (RACE) and an oligo(dT)-adapter primer for 3′-RACE according to the instructions of the supplier of the RACE systems (Gibco-BRL, Germany). For 5′-RACE, one third of the single-stranded cDNA (sscDNA) was tailed with dCTP using terminal transferase, and one fifth of the tailed cDNA was PCR-amplified with sug4 primer (5′-TGCAGCATACAGAACGAACGT-3′) and the dCTP-binding anchor primer (Gibco-BRL). For 3′-RACE, one tenth of the sscDNA was directly used for PCR with the UAP and sug5 (5′-GGTCTATATCGGTTCCTTTTCA-3′) primers. All conditions were as described by Gibco-BRL.
Subcloning and sequencing
Polymerase chain reaction products obtained by RT-PCR or by RACE were electrophoretically separated on 1.6% agarose, and bands of the expected sizes were purified from the gels using the QIAEX II gel extraction kit (QIAGEN) and cloned into the pGEM-T Easy vector (Promega, Madison, WI), following the manufacturer’s protocols. Nucleotide sequences were determined by using Taq polymerase cycle sequencing and an automated DNA sequencer (Perkin-Elmer ABI Prism 373). All clones were sequenced in both strands using universal forward and reverse primers.
Sequence analysis
The sequences were analysed using the Genetic Computer Group program (Madison, WI). The sequence data were compared to gene libraries (EMBL and GenBank) with BLAST (Altschul et al. 1990) and FASTA (Pearson and Lipman 1988) programs. Amino acid sequence comparisons were made with the BESTFIT program. Multiple sequence alignments of translated gene sequences were carried out with the program CLUSTALW (version 1.5; Thompson et al. 1994). Genetic distances were estimated by using the Kimura two-parameter method employed by PHYLIP (Felstein 1993). The tree was displayed with the help of the TREEVIEW program (Page 1996). Protein structure was predicted by the program TopPred2, calculated with a window size of 10 amino acids (Kyte and Doolitle 1982).
Southern blot
Tomato genomic DNA (15 μg) was digested overnight with the restriction enzymes BamHI and XbaI and fully digested DNA was separated on a 0.8% (w/v) agarose gel. Digested DNA was then transferred to positively charged nylon membrane (Hoffmann-La Roche Ltd, Switzerland) by downward capillary blot in 10XSSC (1XSSC is 0.15 M NaCl and 0.015 M sodium citrate). The sug4–sug5 region of LeST3 was labelled with [α-32P]dCTP by PCR and used as a probe. Hybridization of immobilized nucleic acids to the 32P-labelled probe was performed overnight in 7% SDS (w/v), 500 mM NaPi (pH 7.2) and 1 mM EDTA (Church and Gilbert 1984) at 65°C. Nonspecifically bound probe was removed by washing the filter twice with 6XSSC and 0. 1% SDS for 15 min at 65°C, followed by two subsequent stringent washes with 0.1XSSC and 0.1% SDS at 65°C for 15 min each. Hybridization signals were visualised using a Phosphor imager (Bio-Rad, Hercules, CA).
Analysis of LeST3 expression
LeST3 gene expression was studied by real-time RT-PCR (iCycler, BioRad). cDNAs were obtained from 1 μg of DNase-treated total RNA from the different treatments in a 20 μl reaction containing 200 U SuperScript II reverse transcriptase (Invitrogen, Groningen, the Netherlands), 200 ng random hexamer primers, 0.5 mM each dNTP, 20 U RNase inhibitor and 1Xreverse transcription buffer. The primer set used to amplify LeST3 in the synthesized cDNAs were sug4 and sug5. Specificity of this primer set was confirmed by cloning the sug4–sug5 PCR products into the pGEMT-Easy vector (Promega) and sequencing of 10 positive clones. Each 25 μl reaction contained 0.1 μl of the synthesized cDNA, 200 mM dNTPs, 200 nM each primer, 3 mM MgCl2, 2.5 μl 1× SyBR Green (Molecular Probes, Eugene, OR) and 0.5 U Platinum Taq DNA polymerase (Invitrogen, Groningen, the Netherlands) in 1XPCR buffer [20 mM Tris–HCl (pH 8.4), 50 mM KCl].
The PCR program consisted of a 5 min incubation at 95°C to activate the hot-start recombinant Taq DNA polymerase, followed by 35 cycles of 30 sec at 95°C, 45 s at 58°C and 45 s at 70°C, where the fluorescence signal was measured. The specificity of the PCR amplification procedure was checked with a heat dissociation protocol (from 70°C to 100°) after the final cycle of the PCR. The efficiency of the primer set was evaluated by performing real-time PCR on several dilutions of plasmid DNA. The results obtained on the different treatments were standardized to the 18S rRNA levels, which were amplified with the tomato-specific primers R1 (5′-AAAAGGTCGACGCGGGCT-3′) and R2 (5′-CGACAGAAGGGACGAGAC-3′) (Ferrol et al. 2002b). Real-time PCR experiments were repeated three times, with the threshold cycle (ct) determined in triplicate. The average for the triplicate of one representative experiment was used in all subsequent analyses. The relative levels of transcription were calculated by using the \( 2^{{ - \Delta \Delta c_{{\text{t}}} }} \)method (Livak and Schmittgen 2001).
In all RT-PCR reactions a non-RT control was used to detect any possible DNA contamination.
Heterologous expression of LeST3 in Saccharomyces cerevisiae
The entire sequence of LeST3 was obtained by PCR using tomato cDNA as DNA template and a set of primers flanking the complete open reading frame. The full-length LeST3 was ligated into the NotI site of the E. coli/S. cerevisiae shuttle vector pFL61 (Minet et al. 1992). S. cerevisiae strain EBY.VW4000 (Δhxt1-17 Δgal2 Δstl1 Δagt1 Δmph2 Δmph3) (Wieczorke et al. 1999) was transformed with the plasmid pFL61/LeST3 and pFL61 only (empty vector) using the LiAc method. Ura+ transformants were identified by growth at 30°C on 2% agar plates consisting of 0.67% yeast nitrogen base with ammonium sulfate supplemented with leucine, uracil, tryptophan, histidine and 2% maltose. Two of the selected transformants were streaked on plates supplemented with glucose, fructose, mannose or galactose as carbon sources. RNA from S. cerevisiae was isolated with the RNeasy Plant Mini kit (QIAGEN) following the manufacturer’s instructions.
Results
Molecular cloning and sequence analysis of LeST3
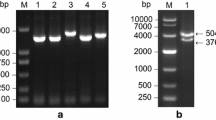
PCR amplification of sscDNA from mycorrhizal roots with a pair of degenerated primers designed on the base of the conserved amino acid domains PESPR and PETKG present in all sugar transporters produced a band of ∼850 bp. Cloning of the amplified DNA and sequencing of several clones allowed identification of a partial clone encoding a putative tomato sugar transporter. By 5′ and 3′-RACE-PCR, a full-length 1699-bp sugar transporter cDNA was obtained (LeST3, accession number AJ278765), with only a few nucleotides upstream the putative ATG start codon and a tail of 210 bp. The plant origin of the cloned gene was demonstrated by PCR amplification of tomato and G. mosseae genomic DNA with the gene-specific primers sug4 and sug5 (Fig. 1) and by Southern blot analysis of tomato genomic DNA (Fig. 2). Moreover, the existence of a gene showing 99.79% identity to LeST3 in the recently released TIGR tomato gene index (TC number 164118) provides further evidence for the plant origin of the identified gene. Southern blot analysis also revealed that the LeST3 probe hybridised to a single band in each of the digested genomic DNAs, which suggests the presence of only one LeST3 copy in the tomato genome.

Ethidium bromide-stained gel of the PCR products obtained after amplification of tomato (lane 1) and G. mosseae (lane 2) genomic DNA with the LeST3 specific primers sug4 and sug5. Lanes 3 and 4 are the negative PCR control and the DNA markers, respectively

Genomic Southern blot analysis of LeST3. Tomato genomic DNA (10 μg) was digested with the restriction enzymes indicated, electrophoresed and transferred onto a nylon membrane. The membrane was hybridised with the 32P-labelled LeST3 cDNA region sug4–sug5 and washed under high-stringency conditions
The open reading frame of LeST3 encoded a polypeptide of 480 amino acids with a calculated molecular mass of 52 kDa. Hydropathy analysis using the Kyte–Doolitle algorithm (Kyte and Doolitle 1982) revealed that the protein encoded by the cloned gene possesses 12 putative transmembrane domains arranged in two groups of six each separated by a hydrophilic loop of 62 amino acids. This hydrophobicity profile is characteristic of proteins belonging to the major facilitator superfamily. A comparison of the LeST3 sequence with entries of the GenBank revealed definitive similarity to a cDNA AFLP fragment from a tobacco gene induced by hydrogen peroxide (92%) and to a number of known sugar transporters from different organisms, with the highest levels of identity to putative and unpublished sugar transporters from Arabidopsis (81–68%), to the dehydration- and cold-induced Arabidopsis ERD6 gene (60%) and to the beet vacuole-localized putative sugar transporter (59%). The predicted amino acid sequence of LeST3 is more similar to sugar transporters from other plants and to glucose transporters from mammals than to sugar transporters from tomato. All these genes belong to the monosaccharide transport family of the sugar porter family of the major facilitator superfamily.
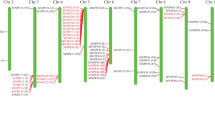
The deduced amino acid sequence of LeST3 was aligned with those of different higher plant monosaccharide transporters and a phylogenetic tree was constructed (Fig. 3). This analysis shows the existence of three major gene subfamilies within the monosaccharide plant transporters: the plasma membrane transporters, the plastidic transporters and a third subfamily clustering the LeST3 gene, the beet vacuole-localized transporter and some uncharacterised transporters from Arabidopsis.

Unrooted phylogenetic tree showing the evolutionary relationships of plant hexose transporters. Transporters belonging to the plasma membrane and plastid families, and the vacuolar and other uncharacterised hexose transporters are encircled. Sequences were obtained from the GenBank database with the following accession numbers: Arabidopsis thaliana (At2: NM_130369, At5: NM_121889, At3: NM_111387; ERD6: D89051, pGlcT: AF215855, SFP1: NM_122617; SFP2: NM_122618, STP1: X55350, STP3: AJ002399, STP4: X66857), B. vulgaris (U43629), L. esculentum (HT2: AJ132224, HT: AJ010942; ST3: AJ278765), M. truncatula (MST1: U38651), Nicotiana tabacum (pGlcT: AF215852, MST1: X66856), O. europaea (pGlcT: AY036055), Oryza. sativa (pGlcT: AC007858), P. armeniaca (AF215853), Petunia x hybrida (MT1: AF061106), Saccharum hybrida (SGT2: L21753), Spinacia oleracea (pGlcT: AF215851), Solanum tuberosum (pGlcT: AF215853), Vicia faba (STP1: Z93775), Vitis vinifera (HT: AJ001061), Zea mays (pGlcT: AF215854)
Heterologous expression of LeST3 in yeast
A hexose import-deficient mutant of S. cerevisiae was transformed with the yeast expression vector pFL61 and its derivative pFL61-LeST3 containing the LeST3 cDNA downstream of the constitutive S. cerevisiae phosphoglycerate kinase promoter to check whether the LeST3 protein functions as a monosaccharide transporter. However, selected transformants were not able to grow in the presence of either glucose, fructose, mannose or galactose as carbon source. To determine whether the construct was accepted by the S. cerevisiae transcription machinery we isolated RNA from two of the transformed yeasts and expression of LeST3 transcript was assessed by RT-PCR using gene-specific primers. Figure 4 shows that the LeST3 gene was strongly expressed in the mutant yeast transformed with the pFL61 vector carrying the LeST3 gene; however, no signal was detected in the mutant yeast transformed with the empty vector.

Expression analysis of LeST3 in S. cerevisiae. Gene expression was analysed by RT-PCR. RNA isolated from two of the yeasts transformed with pFL61 (lanes 1 and 2) and its derivative pFL61-LeST3 (lanes 3 and 4) was reverse transcribed and PCR amplified with the LeST3 specific primers sug4 and sug5. 18S rRNA was used as a control of gene expression
Expression analysis of LeST3
LeST3 expression was assessed in leaves and roots of non-mycorrhizal and G. mosseae- or G. intraradices-colonised tomato plants showing 40% mycorrhizal colonisation. Surprisingly, mycorrhizal colonisation did not affect root LeST3 transcript levels; however, a clear up-regulation of LeST3 mRNA was observed in the leaves of the plants colonised by either G. mosseae or G. intraradices (Fig. 5). To determine whether the observed effect was a symbiosis-specific response, expression of LeST3 was also analysed in plants inoculated with the root pathogen P. parasitica. Roots of the P. parasitica-inoculated plants clearly showed necrotic areas (average rating 2.0, medium level of symptoms). As shown in Fig. 6, LeST3 was also up-regulated in the leaves of the P. parasitica-infected plants, whereas it was not affected in the roots.

Effect of mycorrhizal colonisation on LeST3 gene expression in tomato root and leaf tissues. RNAs were extracted from leaves and roots of control (C), G. intraradices (Gi) and G. mosseae (Gm) inoculated tomato plants. RNAs were reverse transcribed and expression was assayed by quantitative real-time RT-PCR using gene-specific primers for LeST3 and 18S rRNA. The fold change in LeST3 gene expression induced in each tissue by the mycorrhizal colonisation was calculated using the \( 2^{{ - \Delta \Delta c_{{\text{t}}} }} \)method. Data represent the means of three replicates from a representative experiment. Error bars, SD

Effect of P. parasitica on LeST3 gene expression. RNAs were extracted from leaves and roots of control (C) and P. parasitica (Phy) inoculated tomato plants. RNAs were reverse transcribed and expression was assayed by quantitative real-time RT-PCR using gene-specific primers for LeST3 and 18S rRNA. The fold change in LeST3 gene expression induced by P. parasitica in each tissue was calculated using the \( 2^{{ - \Delta \Delta c_{{\text{t}}} }} \)method. Data represent the means of three replicates from a representative experiment. Error bars, SD
Discussion
Based on sequence similarity, conserved motifs and predicted membrane topology, the tomato gene LeST3 has been classified as a putative monosaccharide transporter of the sugar porter family included in the major facilitator superfamily (Saier et al. 1999). However, the phylogenetic analysis suggests that although homologous, LeST3 is clearly different from the functionally characterised plant plasma membrane monosaccharide transporters.
The attempt to study its function in vivo through heterologous expression in a hexose transport-deficient mutant of S. cerevisiae was not successful. In this regard, it is interesting that the LeST3 protein is closely related to the ERD6 protein of Arabidopsis and to a sugar beet putative sugar transporter whose activity was also reported to be undetectable in yeast cells (Chiou and Bush 1996; Kiyosue et al. 1998). Different possibilities can be argued to explain these facts: (1) the native structure of the LeST3 protein in yeast cells may be different from that in tomato, (2) the protein encoded by LeST3 is involved in sugar efflux rather than in import or (3) the protein may be targeted to an intracellular membrane. Although no intracellular targeting signals have been found in the deduced amino acid sequences of LeST3, the observation that this gene shows higher homology to the tonoplast-localized sugar transporter of sugar beet and to the intracellular mammalian glucose transporters than to the plant plasma membrane transporters suggests that the protein encoded by LeST3 might be localized in the tonoplast. Transport systems for vacuolar sugar uptake and efflux in plant cells still await identification and characterisation (Lalonde et al. 2004). It has been proposed that sucrose stored in the vacuoles is broken down by a vacuolar invertase to generate hexoses, which are slowly released to the cytosol, where they constitute an easily accessible carbon source (Sturm and Tang 1999). Hence, the role of a tonoplast hexose transporter could be to provide an adequate supply of carbon to the cytosol when there is an increased demand for carbohydrates by cells. If the protein encoded by LeST3 is localized in the tonoplast, as the phylogenetic analysis suggests, it might be involved in the efflux of glucose from the vacuole. Further experiments need to be done to localize the encoded protein.
Analysis of LeST3 regulation during colonisation by two different AM fungi has shown that development of the symbiosis increases LeST3 expression levels in leaves but not in roots of the tomato plants. AM fungi, as obligate symbionts, depend for their growth and activity on the supply of carbon compounds by the photosynthetic partner (Jennings 1995), and it has been previously observed that carbon allocation to the roots increases during the mycorrhizal association (Douds et al. 1988; Wright et al. 1998). Therefore, up-regulation of LeST3 by development of the symbiosis might be related to the increased carbon partitioning that occurs in mycorrhizal associations. In a previous work, up-regulation of two H+-ATPase isoforms in leaves of mycorrhizal plants was also associated with their increased assimilate partitioning activity (Ferrol et al. 2002b). As far as we know, this is the second report of a plant sugar transporter gene that is regulated by mycorrhizal colonisation. Harrison (1996) observed that colonisation of Medicago truncatula roots by the mycorrhizal fungus G. versiforme induces expression of the monosaccharide transporter gene MtST1 in the zones of the root cortex that were highly colonised by the mycorrhizal fungus. It was proposed that this induction could be a consequence of the need of the colonised host cells to acquire more hexoses to support their increased metabolism. That LeST3 is not regulated in the roots by the symbiosis is not surprising since sugar transporters are present in multigene families in higher plants and there is increasing evidence that the different isoforms are expressed in a cell-, tissue- and developmental stage-specific manner, thus providing potential mechanisms for local regulation of sugar transport (Barker et al. 2000; Gear et al. 2000).
Up-regulation of LeST3 in the leaves of the plants infected with P. parasitica indicates that regulation of this gene could be a general response in plant–fungus interactions. There is evidence that some sugar transporters play a role in plant–microorganism interactions. When plants are subjected to pathogen infection, usually plant metabolism responds by increasing respiration, synthesizing wound-sealing compounds, strengthening the cell wall and producing defence compounds (Baker et al. 2000; Moerschbacher and Mendgen 2000). This increased metabolic activity is probably fed by an enhanced flow of sugars to the infection sites. In Arabidopsis leaves infected with the pathogen Erysiphe cichoracearum, it has been reported that glucose uptake in host tissues is enhanced after fungal infection and this coincides with the induction of expression of the monosaccharide transporter gene AtSTP4 (Fotopoulus et al. 2003). The sink-specific, wound-induced monosaccharide transporter AtSTP4 is also induced approximately fourfold in Arabidopsis seedlings infected with Alternaria brassicicola, Fusarium oxysporum and in suspension-cultured cells of Arabidopsis treated with bacterial or fungal elicitors, and it has been proposed that this transporter regulates monosaccharide import into sink tissues to meet the increased demand of cells responding to environmental stresses (Truernit et al. 1996). Our data suggest that LeST3 responds to an increased demand for carbohydrates by the roots colonised by G. mosseae or infected by P. parasitica to cope with the increased metabolic activity of the colonised/infected cells or to supply carbohydrates to the AM fungus.
Sugars have also been described to play an important role in signalling (Koch 2004). An alternative hypothesis would be to consider LeST3 as a component of the signal transduction pathway that triggers the defence response in both a successful mycorrhizal association (Pozo et al. 1998) and in a plant–pathogen interaction (Herbers et al. 1996). Although AM fungi are able to induce defence responses in roots of their host plants, it is widely accepted that these reactions are transient, localized and weaker than those induced by pathogenic fungi (Pozo et al. 2002). Up-regulation of LeST3 by the symbiosis was detected in shoots 6 weeks after mycorrhizal inoculation, but the role of the symbiosis in inducing resistance in aerial parts of tomato is unknown.
In conclusion, data presented in this paper indicate that the tomato gene LeST3 encodes a putative sugar transporter whose expression increases in the leaves of a plant colonised by an AM fungus or by a root pathogen. Regulation of this gene by the interaction between the plant root and both types of fungi in the leaves of the plant suggests that this gene might be involved in the allocation of carbon compounds from source to sink tissues. Localization of this transporter and expression analysis in other plant–microbe interactions is required to understand its precise role.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Baker CJ, Orlandi EW, Deahl KL (2000) Oxygen metabolism in plant/bacteria interactions: characterization of oxygen uptake response of plant suspension cells. Physiol Mol Plant Pathol 57:159–167
Barker L, Kühn C, Weise A, Schulz A, Gebhardt C, Hirner B, Hellmann H, Schulze W, Ward JM, Frommer WB (2000) SUT2, a putative sucrose sensor in sieve elements. Plant Cell 12:1153–1164
Benabdellah K, Azcón-Aguilar C, Ferrol N (1999) Plasma membrane ATPase and H+ transport activities in microsomal membranes from mycorrhizal tomato roots. J Exp Bot 337:1343–1349
Blee KA, Anderson AJ (2002) Transcripts for genes encoding soluble acid invertase and sucrose synthase accumulate in root tip and cortical cells containing mycorrhizal arbuscules. Plant Mol Biol 50:197–211
Büttner M, Sauer N (2000) Monosaccharide transporters in plants: structure, function and physiology. Biochim Biophys Acta 1465:263–274
Chiou T-J, Bush DR (1996) Molecular cloning, immunochemical localization to the vacuole, and expression in transgenic yeast and tobacco of a putative sugar transporter from sugar beet. Plant Physiol 110:511–520
Church GM, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci U S A 81:1991–1995
Douds DD Jr, Johnson CR, Koch KE (1988) Carbon cost of the fungal symbiont relative to net leaf P accumulation in a split-root VA mycorrhizal symbiosis. Plant Physiol 86:491–496
Felstein J (1993) PHYLIP, Version 3.5. Department of Genetics, University of Washington, Seattle
Ferrol N, Gianinazzi S, Gianinazzi-Pearson V (2002a) Arbuscular mycorrhiza induced ATPases and membrane nutrient transport mechanisms. In: Gianinazzi S, Schüepp H, Barea JM, Haselwandter K (eds) Mycorrhizal technology in agriculture: from genes to bioproducts. Birkhäuser, Basel, pp 113–122
Ferrol N, Pozo MJ, Antelo M, Azcón-Aguilar C (2002b) Arbuscular mycorrhizal symbiosis regulates plasma membrane H+-ATPase gene expression in tomato plants. J Exp Bot 53:1683–1687
Fotopoulos V, Gilbert MJ, Pittman JK, Marvier AC, Buchanan AJ, Sauer N, Hall JL, Williams LE (2003) The monosaccharide transporter gene, AtSTP4, and the cell-wall invertase, Atbetafruct1, are induced in Arabidopsis during infection with the fungal biotroph Erysiphe cichoracearum. Plant Physiol 132:821–829
Gear ML, McPhillips ML, Patrick JW, McCurdy DW (2000) Hexose transporters of tomato: molecular cloning, expression analysis and functional characterization. Plant Mol Biol 44:687–697
Giovannetti M, Mosse B (1980) An evaluation of techniques for measuring vesicular–arbuscular infection in roots. New Phytol 84:489–500
Graham JH (2000) Assessing costs of arbuscular mycorrhizal symbiosis agroecosystems fungi. In: Podila GK, Douds DD Jr (eds) Current advances in mycorrhizae research. APS, St. Paul, pp 127–140
Harrison MJ (1996) A sugar transporter from Medicago truncatula: altered expression pattern in roots during vesicular–arbuscular (VA) mycorrhizal associations. Plant J 9:491–503
Heiland S, Radovanovic N, Höfer M, Winderickx J, Lichtenberg H (2000) Multiple hexose transporters of Schizosaccharomyces pombe. J Bacteriol 182:2153–2162
Herbers K, Meuwly P, Frommer WB, Metraux JP, Sonnewald U (1996) Systemic acquired resistance mediated by the ectopic expression of invertase: possible hexose sensing in the secretory pathway. Plant Cell 8:793–803
Hewitt EJ (1952) Sand and water culture methods used in the study of plant nutrition. In: Technical communication, vol. 22. Commonwealth Agricultural Bureaux, Farnham Royal, Bucks, UK
Ho I, Trappe JM (1973) Translocation of 14C from Festuca plants to their endomycorrhizal fungi. Nature 244:311–327
Hohnjec N, Perlick AM, Pühler A, Küster H (2003) The Medicago truncatula sucrose synthase gene MtSucS1 is activated both in infected regions of root nodules and in the cortex of roots colonized by arbuscular mycorrhizal fungi. Mol Plant-Microb Interact 16:903–915
Jennings DH (1995) The physiology of fungal nutrition. Cambridge University Press, Cambridge, UK
Kiyosue T, Abe H, Yamaguchi-Shinozake K, Shinozaki K (1998) ERD6, a cDNA clone for an early dehydration-induced gene of Arabidopsis, encodes a putative sugar transporter. Biochim Biophys Acta 1370:187–191
Koch K (2004) Sucrose metabolism: regulatory mechanisms and pivotal roles in sugar sensing and plant development. Curr Opin Plant Biol 7:235–246
Kyte J, Doolitle RF (1982) A simple method for displaying the hydrophobic character of a protein. J Mol Biol 157:105–132
Lalonde S, Wipf D, Frommer WB (2004) Transport mechanisms for organic forms of carbon and nitrogen between source and sink. Annu Rev Plant Biol 55:341–372
Lemoine R (2000) Sucrose transporters in plants: update on function and structure. Biochim Biophys Acta 1465:246–262
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the \( 2^{{ - \Delta \Delta c_{{\text{t}}} }} \)method. Methods 25:402–408
Logemann J, Schell J, Willmitzer L (1987) Improved method for the isolation of RNA from plant tissues. Anal Biochem 163:16–20
Madi L, McBride SA, Bailey LA, Ebbole DJ (1997) rco-3, a gene involved in glucose transport and conidation in Neurospora crassa. Genetics 146:499–508
Minet M, Dufour M-E, Lacroute F (1992) Complementation of Sacharomyces cerevisiae auxotrophic mutants by Arabidopsis thaliana cDNAs. Plant J 2:417–422
Moerschbacher B, Mendgen K (2000) Structural aspects of defense. In: Slusarenko AJ, Fraser RS, van Loon LC (eds) Mechanisms of resistance to plant diseases. Kluwer, Dordrecht, pp 231–277
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Nehls U, Wiese J, Guttengerger M, Hampp R (1998) Carbon allocation in ectomycorrhizas: identification and expression analysis of an Amanita muscaria monosaccharide transporter. Mol Plant-Microb Interact 11:167–176
Page RDM (1996) Treeview: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A 85:2444–2448
Phillips JM, Hayman DS (1970) Improved procedure for clearing roots and staining parasitic and vesicular–arbuscular mycorrhizal fungi for rapid assessment of infection. Trans Br Mycol Soc 55:158–160
Pozo MJ, Azcón-Aguilar C, Dumas-Gaudot E, Barea JM (1998) Chitosanase and chitinase activities in tomato roots during interactions with arbuscular mycorrhizal fungi or Phytophthora parasitica. J Exp Bot 49:1729–1739
Pozo MJ, Azcón-Aguilar C, Dumas-Gaudot E, Barea JM (1999) β-1,3 glucanase activities in tomato roots inoculated with arbuscular mycorrhizal fungi and/or Phytophthora parasitica and their possible involvement in bioprotection. Plant Sci 141:149–157
Pozo MJ, Slezack-Deschaumes S, Dumas-Gaudot E, Gianinazzi S, Azcón-Aguilar C (2002) Plant defense responses induced by arbuscular mycorrhizal fungi. In: Gianinazzi S, Schüepp H, Barea JM, Haselwandter K (eds) Mycorrhizal technology in agriculture: from genes to bioproducts. Birkhäuser, Basel, pp 103–112
Saier MH Jr, Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang SC, Jack DL, Jahn PS, Lew K, Liu J, Pao SS, Paulsen IT, Tseng TT, Virk PS (1999) The major facilitator superfamily. J Mol Microbiol Biotechnol 1:257–279
Shachar-Hill Y, Pfeffer PE, Douds D, Osman SF, Doner LW, Ratcliffe RG (1995) Partitioning of intermediate carbon metabolism in vesicular–arbuscular leek. Plant Physiol 108:2979–2995
Smith SE, Dickson S, Smith FA (2001) Nutrient transfer in arbuscular mycorrhizas: how are fungal and plant processes integrated? Aust J Plant Physiol 28:683–694
Solaiman MD, Saito M (1997) Use of sugars by intraradical hyphae of arbuscular mycorrhizal fungi revealed by radiorespirometry. New Phytol 136:533–538
Sturm A, Tang G-Q (1999) The sucrose-cleaving enzymes of plants are crucial for development, growth and carbon partitioning. Trends Plant Sci 4:401–497
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tinker PB, Durall DM, Jones MD (1994) Carbon use in mycorrhizas: theory and sample calculations. New Phytol 128:115–122
Truernit E, Schmid J, Epple P, Illig J, Sauer N (1996) The sink-specific and stress-regulated Arabidopsis STP4 gene: enhanced expression of a gene encoding a monosaccharide transporter by wounding, elicitors, and pathogen challenge. Plant Cell 8:2169–2182
Voegele RT, Struck C, Hahn M, Mendgen K (2001) The role of haustoria in sugar supply during infection of broad bean by the rust fungus Uromyces fabae. Proc Natl Acad Sci U S A 98:8133–8138
Wieczorke R, Krampe S, Weierstall T, Friedel K, Hollenberg CP, Boles E (1999) Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett 464:123–128
Williams LE, Lemoine R, Sauer N (2000) Sugar transporters in higher plants—a diversity of roles and complex regulation. Trends Plant Sci 5:283–290
Wright DP, Read DJ, Scholes JD (1998) Mycorrhizal sink strength influences whole plant carbon balance of Trifolium repens L. Plant Cell Environ 21:881–891
Yu J, Chang P-K, Bhatnagar D, Cleveland TE (2000) Cloning of a sugar utilization gene cluster in Aspergillus parasiticus. Biochim Biophys Acta 1493:211–214
Acknowledgements
We thank Dr. Boles (University of Heinrich-Heine, Düsseldorf, Germany) for providing the S. cerevisiae strain EBY.VW4000 and Dr. Barea for helpful comments. We are grateful to Ms. Custodia Cano for excellent technical assistance. This research was supported by CICyT (AGL2003-01551), Spain.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
García-Rodríguez, S., Pozo, M.J., Azcón-Aguilar, C. et al. Expression of a tomato sugar transporter is increased in leaves of mycorrhizal or Phytophthora parasitica-infected plants. Mycorrhiza 15, 489–496 (2005). https://doi.org/10.1007/s00572-005-0354-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00572-005-0354-5