
Abstract
Background
Deregulated autophagy followed by cellular senescence in biliary epithelial cells (BECs) may be closely related to the abnormal expression of mitochondrial antigens and following autoimmune pathogenesis in primary biliary cirrhosis (PBC). We examined an involvement of endoplasmic reticulum (ER) stress in the deregulated autophagy and cellular senescence in PBC.
Methods
We examined the degree of ER stress using markers; glucose-regulated protein 78 (GRP78) and protein disulfide isomerases (PDI), autophagy and cellular senescence in cultured BECs treated with an ER stress inducer, tunicamycin (TM), glycochenodeoxycholic acid (GCDC), and palmitic acid (PA), and the effect of pretreatment with tauroursodeoxycholic acid (TUDCA). We examined the expression of PDI and GRP78 in livers taken from the patients with PBC (n = 43) and 75 control livers.
Results
The expression of ER stress markers was significantly increased in cultured BECs treated with TM, GCDC or PA in BECs (p < 0.05), and pretreatment with TUDCA significantly suppressed the induced ER stress (p < 0.05). Autophagy, deregulated autophagy, and cellular senescence were induced in BECs treated with TM, GCDC, or PA. Pretreatment with TUDCA further increased autophagy in BECs treated with PA and suppressed cellular senescence caused by treatments with TM, GCDC, or PA (p < 0.05). A granular expression of PDI and GRP78 was significantly more extensive in small bile ducts in PBC, compared with control livers (p < 0.05). The expression of GRP78 was seen in senescent BECs in PBC.
Conclusions
ER stress may play a role in the pathogenesis of deregulated autophagy and cellular senescence in biliary epithelial lesions in PBC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary biliary cirrhosis (PBC) is an organ-specific autoimmune disease presenting with chronic, progressive cholestasis and liver failure [1–3]. PBC is characterized by a high frequency of serum anti-mitochondrial antibodies (AMAs) and bile duct lesions such as chronic non-suppurative destructive cholangitis; CNSDC in small bile ducts [1, 4]. Recent progress in this field revealed several susceptible loci for PBC by a genome-wide association study (GWAS) [5, 6]. However, there still remain enigma in the pathogenesis of PBC, for example, a role of AMAs in the occurrence and progression of bile duct lesions [1–4, 7–9].
Macroautophagy (hereafter referred to as autophagy) is a lysosomal pathway that degrades and recycles intracellular organelles such as mitochondria and proteins to maintain energy homeostasis during times of nutrient deprivation and to remove damaged cell components [10, 11]. Deregulated autophagy is caused by blockage of early and late process of autophagosome formation and is accompanied by accumulation of p62/sequastosome-1 [12, 13]. Accumulating data suggest that autophagy plays a role in physiological processes and deregulated autophagy may be involved in a wide variety of disease processes, including neurodegeneration, cancer, infection, and inflammation [10, 14]. Recent progress in this field also revealed a crucial role of the autophagy pathway and proteins in autoimmunity [15, 16]. For example, autophagy has been implicated in intracellular antigen processing for the presentation of MHC class I and II [17–19].
We have recently disclosed a possible involvement of deregulated autophagy in biliary epithelial cells (BECs) in the pathogenesis of bile duct lesions in PBC [12, 20]. Deregulated autophagy in BECs may be closely related to the abnormal expression of mitochondrial antigens and following autoimmune pathogenesis in PBC [21]. Furthermore, we have disclosed that deregulated autophagy may precede cellular senescence in BECs involved in CNSDC showing augmented expression of senescence-associated β-galactosidase (SA-β-gal), p16INK4a, and p21WAF1/Cip1 and telomere shortening in PBC [22–25]. In our previous studies, factors and mechanisms causing deregulated autophagy have remained to be elucidated.
Accumulating data suggest that endoplasmic reticulum (ER) and autophagy are cross-regulated [26, 27]. ER stress is characteristically induced by the accumulation of misfolded proteins in the ER arising from either primary (genetic) or secondary (environmental) factors that affect the folding of proteins within the ER [28]. ER stress is a consequence of maintained and unsuccessful unfolded protein response (UPR) and some mediators of the UPR can directly induce autophagy [29, 30]. Although ER stress can trigger autophagy and thereby foster protein degradation [26, 29, 30], others suggested that defective hepatic autophagy promotes ER stress [27, 31].
We hypothesized that ER stress may be involved in the deregulated autophagy and cellular senescence in biliary epithelial lesions in PBC and assessed the ER stress and its association with autophagy and cellular senescence in cultured BECs and liver tissues taken from patients with PBC. Furthermore, we examined the effect of tauroursodeoxycholic acid (TUDCA), which is known as a chemical chaperone and also is widely used for the treatment of PBC, on ER stress, deregulated autophagy, and cellular senescence in BECs.
Materials and methods
Culture study
Cell culture and treatments
Mouse intrahepatic BECs were isolated from 8-week-old female BALB/c mice and were purified and cultured as described previously [32]. The cell density of the cells was less than 80 % during experiments. In several experiments, BECs were treated with tunicamycin (TM; Sigma-Aldrich, St. Louis, MO, USA; 0.5 μg/ml), a potent inducer of ER stress, glycochenodeoxycholate (GCDC; Sigma-Aldrich, 500 μM), palmitic acid (PA; Sigma-Aldrich, 400 μM) or oleic acid (OA; Sigma-Aldrich, 400 μM) for 6 h or 1 day. PA, a saturated fatty acid, is known to induce ER stress in cultured hepatocytes and other cells [31, 33]. PA and OA were complexed to 0.5 % (wt./vol) bovine serum albumin (BSA; Sigma-Aldrich) according to previous reports [34]. The approximate molar ratio of fatty acids to BSA is 6:1 with 400 μM PA. The addition of BSA or a fatty acid/BSA mixture has not been shown to affect the pH of the media. In some experiments, BECs were treated with serum deprivation for the induction of autophagy, as described previously [20]. In several experiments, the effect of pretreatment with TUDCA (Sigma-Aldrich, 1 mM, 1 h) was also examined. In preliminary experiments, we found that a pretreatment with UDCA was effective for the reduction of ER stress as well as a pretreatment with TUDCA (data not shown). So, we used TUDCA for further experiments in this study.
Real-time quantitative reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cells with a QIAGEN RNeasy Mini kit (QIAGEN) according to the manufacturer’s protocol. After cDNA was synthesized, quantitative real-time PCR was performed to measure a series of ER stress markers, glucose-regulated protein 78 (GRP78)/Bip, C/EBP homologous protein (CHOP), X-box binding protein 1 (XBP-1) (unspliced and spliced) and β-actin mRNA according to a standard protocol using the SYBR Green PCR Master Mix (Toyobo, Tokyo, Japan). Primers used are shown in Table 1. Each experiment was performed twice in triplicate, and the mean was adopted in each experiment.
Immunoblotting
The cell lysate samples (10 μg) were resolved by SDS-PAGE and transferred to a nitrocellulose membrane as described previously [35]. After transfer, the membranes were processed for immunoblotting as described previously [35]. The primary antibodies used are shown in Table 2. Densitometry of the resulting bands was performed using ImageJ software and normalized to the loading control.
Immunofluorescence staining for cultured cells
The BECs growing in a Lab-Tek chamber were fixed and immunostained using the primary antibodies for ER stress markers (PDI and GRP78/Bip), LC3, or p62 (Table 2), as described previously [20]. A similar dilution of the control mouse or rabbit Immunoglobulin G (Dako) was applied instead of the primary antibody as a negative control. The cells were then incubated with Alexa-488-labeled rabbit anti-mouse or rabbit IgG (Molecular Probes, Eugene, OR, USA) for 30 min, counterstained with 10 μg/ml 4′ and 6-diamidino-2-phenylindole (DAPI) and evaluated under a conventional fluorescence microscope (Olympus, Tokyo, Japan).
Assay for cellular senescence
SA-β-gal activity was detected by using the senescence detection kit (BioVision, Mountain View, CA, USA) according to the manufacturer’s protocol [36]. The proportion of senescent cells in each condition was assessed at day 4 by counting the percentage of SA-β-gal-positive cells in at least 1 × 103 total cells using light microscopy.
Double staining for ER stress and cellular senescence
The BECs growing in a Lab-Tek chamber were fixed and SA-β-gal activity was detected as described above. The BECs were then immunostained for ER stress markers as described above.
Human study
Classification of intrahepatic biliary tree
The intrahepatic biliary tree is classified into intrahepatic large and small bile ducts (septal and interlobular bile ducts) by their size and distributions in the portal tracts [37]. Bile ductules, which are characterized by tubular or glandular structures with a poorly defined lumen and located at the periphery of the portal tracts [37, 38], are not included in the small bile ducts.
Liver tissue preparation
A total of 118 liver tissue specimens (all were biopsied or surgically resected) were collected from the liver disease file of our laboratory and affiliated hospitals. The Ethics Committee of Kanazawa University approved this study. The liver specimens in this study were 43 PBC, ten primary sclerosing cholangitis (PSC), 43 chronic viral hepatitis (CVH), ten livers with extrahepatic biliary obstruction (EBO), and 12 “histologically normal” livers. All PBC were from patients fulfilling the clinical, serological, and histological characteristics consistent with the diagnosis of PBC [1]. PBC livers were staged histologically [1], and 25 and 18 of PBC were stages 1, 2 (early PBC) and stages 3, 4 (advanced PBC), respectively. Twenty-four CVH were regarded as F0-2 and 19 as F3, 4, respectively [39]. Nine and 34 of CVH cases were serologically positive for hepatitis B surface antigen (HBsAg) and anti-hepatitis C viral antibody (HCVAb), respectively. Causes of EBO were obstruction of the bile duct at the hepatic hilum or the extrahepatic bile ducts, because of carcinoma or stones, and the duration of jaundice was less than 1 month. “Histologically normal” livers were obtained from surgically resected livers for traumatic hepatic rupture or metastatic liver tumor. Normal liver tissues were obtained from an area apart from the tumor, and carcinoma tissues were not evaluated. Chemotherapy was not performed before liver resection in all patients with histologically normal liver.
Liver tissue samples were fixed in 10 % neutral-buffered formalin, and embedded in paraffin. More than 20 serial sections, 4 μm thick, were cut from each block. Several were processed routinely for histologic study, and the remainder was processed for the subsequent immunohistochemistry.
Immunohistochemistry
We examined immunohistochemically the expression of ER stress-related proteins: PDI and GRP78 using the primary antibodies are shown in Table 2, as described previously [23]. A similar dilution of the control mouse or rabbit Immunoglobulin G (Dako) was applied instead of the primary antibody as a negative control. Positive and negative controls were routinely included. Histological analysis was performed in a blinded manner. For the assessment of PDI and GRP78 expression in small bile ducts, the extent of expression was evaluated as follows: 1+ (focal, positive cells detected in less than a half of small bile ducts) and 2+ (extensive, positive cells detected in more than a half of small bile ducts).
Double immunostaining
We also performed double immunostaining for GRP78 with senescence markers (p16INK4a and p21WAF1/Cip1). In brief, GRP78 detected as described above, followed by second staining for either of p16INK4a or p21WAF1/Cip1 using Vector Blue Alkaline Phosphatase Substrate Kit (Vector Labs, Burlingame, CA).
Statistical analysis
Statistical analysis of differences was performed using the Kruskal–Wallis test with Dunn’s post test. When the p value was less than 0.05, the difference was regarded as significant.
Results
Culture study
Treatment with TM, GCDC, and PA induces ER stress in BECs and pretreatment with TUDCA reduces the ER stress
Real-time PCR
We examined the mRNA expression level of ER stress markers, GRP78, CHOP, and XBP-1 (spliced and unspliced form), in cultured BECs treated with TM, GCDC, or PA. Figure 1a summarizes the data. The expression of ER stress markers was significantly increased in BECs treated with TM, GCDC, or PA (p < 0.05). TM and PA upregulated all three of ER markers, whereas GCDC did only CHOP. It is known that some drugs and factors activated only a part of three major ER stress sensors, PKR-like ER stress kinase (PERK), activating transcription factor (ATF6) and inositol-requiring transmembrane kinase and endonuclease 1 (IRE1). Although there was no report describing the sensor of ER stress activated by GCDC, to our knowledge, GCDC may activate only PERK-signaling and consequently enhance CHOP. Pretreatment with TUDCA significantly suppressed the expression of ER stress markers in BECs treated with TM, GCDC, or PA (p < 0.05).

ER stress induced by TM, GCDC, and PA and effect of TUDCA. a The expression of mRNA in biliary epithelial cells treated with tunicamycin (TM, 0.5 μg/ml), glycochenodeoxycholate (GCDC, 500 μM) or palmitic acid (PA, 400 μM) or oleic acid (OA, 400 μM) for 6 h with or without pretreatment with tauroursodeoxycholic acid (TU, 1 mM) for 1 h was quantified with real-time PCR and normalized as a ratio using β-actin as the housekeeping gene. Data are expressed as the mean ± SD. * p < 0.05 compared to the control; # p < 0.05 compared to the TU pretreatment; n = 6 for each group. b Immunofluorescent staining for ER stress marker, protein disulfide isomerase, (shown in green) in biliary epithelial cells treated with tunicamycin (TM, 0.5 μg/ml), glycochenodeoxycholate (GCDC, 500 μM) or palmitic acid (PA, 400 μM) or oleic acid (OA, 400 μM) for 6 h with or without pretreatment with tauroursodeoxycholic acid (TU, 1 mM). The expression of PDI was absent in control (no treatment) and biliary epithelial cells treated by OA, whereas PDI-positive dots were increased in the cytoplasm of biliary epithelial cells treated by TM, GCDC, and PA. The upregulated expression of PDI by treatment with TM, GCDC, and PA was inhibited by pre-treatment with TU
Immunofluorescence
The expression of ER stress marker PDI was assessed by immunofluorescent staining in BECs treated with GCDC, PA, or OA for 6 h with or without pretreatment with TUDCA (Fig. 1b). The expression of PDI was absent in control (no treatment) and BECs treated by OA, whereas PDI-positive dots were increased in the cytoplasm of BECs treated by GCDC or PA (Fig. 1b). The increased expression of PDI by treatment with GCDC or PA was inhibited by pre-treatment with TUDCA (Fig. 1b, Supplemental Fig. 1).
TM, GCDC, or PA induced autophagy and the pretreatment with TUDCA further increased autophagy induced by PA
Immunoblotting showed that LC3-II/LC3-I ratio, which indicates the level of autophagy, was upregulated by the treatment with TM, GCDC, or PA (Fig. 2a). The LC3-II/LC3-I ratio was further increased by pre-treatment with TUDCA in BECs treated with PA, whereas pre-treatment with TUDCA did not affect the LC3-II/LC3-I ratio in BECs treated with TM or GCDC (Fig. 2a).

Autophagy and deregulated induced by TM, GCDC, and PA and effect of TUDCA. a Immunoblotting for microtubule-associated proteins-light chain 3β (LC3). LC3-II/LC3-I ratio, which indicates the level of autophagy, was upregulated by the treatment with glycochenodeoxycholate (GCDC, 500 μM) or palmitic acid (PA, 400 μM) or tunicamycin (TM, 0.5 μg/ml), for 6 h with or without pretreatment with tauroursodeoxycholic acid (TU, 1 mM). LC3-II/LC3-I ratio was not increased by the treatment with oleic acid (OA, 400 μM). Increased LC3-II/LC3-I ratio induced by PA was further upregulated by pretreatment with TU. * p < 0.05 vs. control (Cont); ** p < 0.05. n = 4 for each group. b Immunofluorescent staining for a deregulated autophagy marker p62 (shown in green) in biliary epithelial cells treated with tunicamycin (TM, 0.5 μg/ml), glycochenodeoxycholate (GCDC, 500 μM) or palmitic acid (PA, 400 μM) or oleic acid (OA, 400 μM) for 6 h with or without pretreatment with tauroursodeoxycholic acid (TU, 1 mM). The expression of p62 was absent in control (no treatment) or biliary epithelial cells treated with OA, whereas a granular expression of p62 was increased in the cytoplasm of biliary epithelial cells treated by GCDC and PA. The upregulated expression of p62 by treatment with GCDC and PA was inhibited by pre-treatment with TU
ER stress was induced by increased autophagy
The expression of ER stress marker PDI was assessed by immunofluorescent staining in BECs treated with serum deprivation for the induction of autophagy. PDI-positive dots were increased in the cytoplasm of BECs treated with serum deprivation (Supplemental Fig. 2).
TM, GCDC, or PA induced deregulated autophagy with p62 accumulation and pretreatment with TUDCA suppressed deregulated autophagy
Immunofluorescent staining disclosed that p62 expression suggesting a deregulated autophagy was observed as cytoplasmic dots in BECs treated with GCDC and PA (Fig. 2b, Supplemental Fig. 3). The p62-positive cytoplasmic dots induced by GCDC and PA were decreased by pretreatment with TUDCA (Fig. 2b, Supplemental Fig. 3).
TM, GCDC, and PA induced cellular senescence and pretreatment with TUDCA suppressed stress-induced cellular senescence
Cellular senescence was assessed by percentage of cells positive for SA-β-gal. Percentage of cells positive for SA-β-gal was significantly increased in cells treated with TM (SA-β-gal labeling index, 8.9 ± 2.7), GCDC (10.4 ± 4.1), and PA (11.5 ± 3.5), when compared with control (0.4 ± 0.5) (p < 0.01) (Fig. 3). The stress-induced cellular senescence was significantly inhibited by a pre-treatment with TUDCA (TM+TU, 3.3 ± 1.5; GCDC+TU, 2.3 ± 1.5; PA+TU, 1.5 ± 1.7) (p < 0.01) (Fig. 3).

Cellular senescence induced by TM, GCDC, and PA and effect of TUDCA. a Cellular senescence was assessed by senescence-associated β-galactosidase activity (SA-β-gal) on day 4 after a treatment with tunicamycin (TM, 0.5 μg/ml, for 2 h), glycochenodeoxycholate (GCDC, 500 μM) or palmitic acid (PA, 400 μM) or oleic acid (OA, 400 μM) for 6 h with or without pretreatment with tauroursodeoxycholic acid (TU, 1 mM). Pretreatment with TUDCA suppressed the stress-induced cellular senescence in cultured BECs. b Percentage of cells positive for SA-β-gal was significantly increased in cells treated with TM (SA-β-gal labeling index, 8.9 ± 2.7), GCDC (10.4 ± 4.1), and PA (11.5 ± 3.5), when compared with control (0.4 ± 0.5). The stress-induced cellular senescence was significantly inhibited by a pre-treatment with TUDCA (TM+TU, 3.3 ± 1.5; GCDC+TU, 2.3 ± 1.5; PA+TU, 1.5 ± 1.7). Data was expressed as mean ± SD. * p < 0.01 compared to control, ** p < 0.01. c Expression of ER stress markers in senescent BECs. Expression of ER stress marker protein disulfide isomerase (PDI, shown in green) in senescent BECs treated with TM and GCDC assessed by SA-β-gal (SA) on day 4 after a treatment with TM (0.5 μg/ml, for 2 h) or GCDC (500 μM). Expression of ER stress marker PDI was co-localized in senescent BECs treated with TM or GCDC
Expression of ER stress markers in senescent BECs
Expression of ER stress marker protein PDI was detected in senescent BECs treated with TM and GCDC, whereas it was not observed in BECs in control BECs (Fig. 3c).
Human study
Increased expression of ER stress markers in damaged BECs in small bile ducts in PBC
PDI
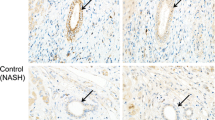
The expression of an ER stress marker PDI was absent or faint in control livers (Fig. 4a), whereas an intense granular and vesicular expression of PDI was seen in small bile ducts (SBDs), especially those involved in CNSDC, in PBC (Fig. 4a). The expression of PDI was significantly more extensive in SBDs in PBC, st1/2, compared to control PSC, CVH, st1/2 and NL (p < 0.01) (Fig. 4b). There was no significant difference between PBC, st1/2 and PBC, st3/4.

Increased expression of ER stress markers protein disulfide isomerase (PDI) in damaged small bile ducts in primary biliary cirrhosis (PBC). a The expression of PDI was absent in biliary epithelial cells in the small bile duct (arrow) in normal liver (NL). Intense granular and vesicular expression of PDI was seen in damaged small bile ducts (arrows) in PBC (PBC #1 and #2). Immunostaining for PDI. Original magnification, ×400. b Semiquantitative evaluation of PDI expression in biliary epithelial cells in small bile ducts in PBC and control livers. Grey column, score 1 (mild) and black, score 2 (extensive). * p < 0.01 compared to PSC, CVH F1/2 and NL; # p < 0.05 compared to PSC
GRP78
The expression of an ER stress marker GRP78 was absent or faint in control livers (Fig. 5a), whereas an intense granular and vesicular expression of GRP78 was seen in small bile ducts (SBDs), especially those involved in CNSDC, in PBC (Fig. 5a). The granular expression of GRP78 was heterogeneous in part of the damaged bile ducts in PBC (Fig. 5a). The expression of GRP78 was significantly more extensive in SBDs in PBC, st1/2, compared to control PSC and CVH, st1/2 (p < 0.01) (Fig. 5b).

Increased expression of ER stress markers glucose-regulated protein 78 GRP78 in damaged small bile ducts in primary biliary cirrhosis (PBC). a The expression of GRP78 was absent in biliary epithelial cells in the small bile duct (arrow) in control liver, primary sclerosing cholangitis (PSC). Intense granular expression of GRP78 was seen diffusely (PBC #1 and 2) in the damaged small bile duct in PBC. There were several round cells, probably macrophages, showing granular expression. PBC cases #1 and #2 (stage 2). Immunostaining for GRP78. Original magnification, ×400. b Semiquantitative evaluation of GRP78 expression in biliary epithelial cells in small bile ducts in PBC and control livers. Grey column, score 1 (mild) and black, score 2 (extensive). * p < 0.01 compared to PSC and CVH F1/2. c Co-localization of ER stress marker GRP78 with senescent markers p16INK4a and p21WAF1/Cip1 in the damaged small bile ducts in PBC. Nuclear and cytoplasmic expression of senescent marker p16INK4a (right) and nuclear expression of p21WAF1/Cip1 (left) were seen in a part of biliary epithelial cells showing cytoplasmic expression of GRP78 in the damaged bile ducts in PBC. PBC, stage 2. Double immunostaining for GRP78 (brown) and senescent markers p16INK4a or p21WAF1/Cip1 (blue). Original magnification, ×400
Double immunostaining for ER stress marker GRP78 and senescent markers p16INK4a and p21WAF1/Cip1
Double immunostaining revealed that the expression of GRP78 was frequently observed in the damaged bile ducts in which senescent markers p16INK4a and p21WAF1/Cip1 were expressed in PBC (Fig. 5c). Whereas, the expression of GRP78 was rare in small bile ducts in control livers in which p16INK4a and p21WAF1/Cip1 were not expressed.
Discussion
The data obtained in this study are summarized as follows: (1) the expression of ER stress markers was significantly increased in cultured BECs treated with TM, GCDC, or PA; (2) Pretreatment with TUDCA significantly suppressed ER stress in BECs treated with TM, GCDC, or PA; (3) Autophagy, deregulated autophagy with p62 accumulation, and cellular senescence were induced in cultured BECs treated with TM, GCDC, or PA; (4) Pretreatment with TUDCA further increased the degree of autophagy in BECs treated with PA; (5) Pretreatment with TUDCA suppressed the stress-induced deregulated autophagy and cellular senescence in cultured BECs; (6) An intense granular and vesicular expression of ER stress markers, PDI and GRP78, was seen in damaged SBDs in PBC. The expression of PDI and GRP78 was significantly more extensive in SBDs in PBC, compared with control livers. (7) Increased expression of ER stress marker GRP78 was co-localized with senescent markers p16INK4a and p21WAF1/Cip1 in bile duct lesions in PBC.
In this study, we demonstrated firstly that ER stress might be involved in deregulated autophagy and subsequent cellular senescence in BECs. We have previously reported a deregulated autophagy and its possible role in abnormal expression of mitochondrial antigens in biliary epithelial lesions in PBC [12, 20, 21]. Triggers of deregulated autophagy, however, remained unclear in our previous studies [12, 20, 21]. In this study, we found that TM, a well-known ER stressor, and also a hydrophobic bile acid, GCDC, and a saturated free fatty acid, PA, induce ER stress, deregulated autophagy and cellular senescence in cultured BECs. These findings clearly suggest a presence of ER stress-mediated autophagy in BECs, as reported in neuroblastoma cells and other types of cells [26, 29, 30]. In addition, ER stress was increased in case of autophagy induced by serum deprivation in cultured BECs. Autophagy is linked to the ER stress at many levels and these two systems are cross-regulated [26, 27]. This result suggests that impaired autophagy may induce ER stress in BECs, as reported in other types of cells [27, 31], although the relationship between ER stress and autophagy is not fully clarified so far. The mechanism underlying PA toxicity remains incompletely understood and the reason why ER stress was not induced by OA is not clear, but this result agrees with previous reports [33].
This study revealed for the first time an involvement of ER stress in BECs in bile duct lesions in PBC. Although there have been studies on the involvement of ER stress mainly in hepatocytes in various liver disease [40], there have been few studies regarding ER stress in BECs, to our knowledge. It is conceivable that ER stress in biliary epithelial lesions may trigger autophagy, deregulated autophagy, and following cellular senescence, as shown in cultured BECs in this study. In fact, we demonstrated in this study that increased expression of ER stress marker GRP78 was co-localized with senescent markers p16INK4a and p21WAF1/Cip1 in bile duct lesions in PBC. The co-localization of ER stress marker in cultured senescent BECs was also observed in this study. We have also shown the co-localization of autophagy marker LC3 and deregulated autophagy marker p62 with senescent markers p16INK4a and p21WAF1/Cip1 in bile duct lesions in PBC in our previous studies [12, 20]. On the other hand, it is reported that impaired autophagy induces ER stress [27, 31]. Therefore, another possibility is that ER stress detected in biliary epithelial lesions may be induced by impaired autophagy in PBC. The reason why the staining of PDI or GRP78 was rather low in PSC in this study is unknown. Small bile ducts are generally intact in PSC and portal inflammation was rather weak in PSC livers examined in this study. This may be related to the rather weak expression of ER stress markers in PSC.
Several candidates of susceptible genes/loci in PBC have been identified in recent GWAS studies [5, 6]. It is of interest that ER-stress-related genes, such as ORM1-like protein 3 (ORMDL3), are included in the candidates associated with the susceptibility of PBC [5, 6]. ORMDL3 has also been linked to the pathogenesis of asthma, inflammatory bowel disease, and insulin-dependent diabetes mellitus [41, 42]. Recent studies have shown that ORMDL3 altered ER-mediated calcium homeostasis and facilitated the UPR, which is an endogenous inducer of inflammation [42]. The expression of ORMDL3 in PBC and other liver diseases is unknown so far. Cross-regulation between ER stress and autophagy and its role in pathogenesis of cell injuries and inflammation based on genetic susceptibility have been reported in intestinal epithelial cells in inflammatory bowel disease [41, 43]. Similarly to intestinal epithelial cells in inflammatory bowel disease, deregulated ER stress due to genetic susceptibility may be related to deregulated autophagy and pathogenesis of biliary epithelial lesions in PBC. This hypothesis remains to be elucidated.
This study clearly showed that pretreatment with TUDCA suppressed ER stress, deregulated autophagy, and cellular senescence in BECs. TUDCA and UDCA are chemical chaperones enhancing the adaptive capacity of the ER [44] and the effect of TUDCA on the reduction of ER stress has been shown in hepatocytes and other types of cells [45, 46]. On the other hand, UDCA is today regarded as the standard treatment of primary biliary cirrhosis (PBC) [3]. UDCA exerts anti-cholestatic, anti-fibrotic, and anti-proliferative effects in primary biliary cirrhosis (PBC) via mechanisms not yet fully understood [3]. This study provided evidence for the first time that TUDCA may be effective for the treatment of PBC by reducing ER stress in BECs. It is plausible that TUDCA reduces ER stress in BECs in PBC, which result in a reduction of deregulated autophagy and cellular senescence in BECs. The reason why the autophagy level was further increased by pretreatment with TUDCA in BECs treated with PA, but not in BECs treated with TM and GCDC, remains unclear. One possibility is that PA may activate another pathway related to autophagy in addition to ER stress-induced autophagy and TUDCA may affect the alternative pathway. However, the distinct molecular pathway remains to be clarified. Since PA induced deregulated autophagy and cellular senescence in this study, it is plausible that PA may play a role in the pathogenesis of biliary injuries. TUDCA may be effective in relieving lipotoxicity caused by PA via reduction of ER stress.
In conclusion, ER stress is involved in BECs in biliary epithelial lesions in PBC. ER stress may play a role in the pathogenesis of deregulated autophagy and cellular senescence in biliary epithelial lesions in PBC.
References
Portmann B, Nakanuma Y. Diseases of the bile ducts. In: Burt A, Portmann B, Ferrell L, editors. Pathology of the liver. London: Churchill Livingstone; 2011. p. 491–562.
Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73.
Lindor KD, Gershwin ME, Poupon R, et al. Primary biliary cirrhosis. Hepatology. 2009;50:291–308.
Nakanuma Y, Ohta G. Histometric and serial section observations of the intrahepatic bile ducts in primary biliary cirrhosis. Gastroenterology. 1979;76:1326–32.
Nakamura M, Nishida N, Kawashima M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet. 2012;91:721–8.
Mells GF, Floyd JA, Morley KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–32.
Fussey S, Guest J, James O, et al. Identification and analysis of the major M2 autoantigens in primary biliary cirrhosis. Proc Natl Acad Sci USA. 1988;85:8654–8.
Kita H, Matsumura S, He XS, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Investig. 2002;109:1231–40.
Shimoda S, Van de Water J, Ansari A, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Investig. 1998;102:1831–40.
Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75.
Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6.
Sasaki M, Miyakoshi M, Sato Y, et al. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liv Int. 2012;32:487–99.
Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010;189:925–35.
Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35.
Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92.
Paludan C, Schmid D, Landthaler M, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–6.
Nedjic J, Aichinger M, Emmerich J, et al. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400.
Sasaki M, Miyakoshi M, Sato Y, et al. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest. 2010;90:835–43.
Sasaki M, Miyakoshi M, Sato Y, et al. Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis. Liver International: Official J Int Assoc Study Liver. 2013;33:312–20.
Sasaki M, Ikeda H, Haga H, et al. Frequent cellular senescence in small bile ducts in primary biliary cirrhosis: a possible role in bile duct loss. J Pathol. 2005;205:451–9.
Sasaki M, Ikeda H, Sato Y, et al. Decreased expression of Bmi1 is closely associated with cellular senescence in small bile ducts in primary biliary cirrhosis. Am J Pathol. 2006;169:831–45.
Sasaki M, Ikeda H, Yamaguchi J, et al. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology. 2008;48:186–95.
Sasaki M, Ikeda H, Nakanuma Y. Activation of ATM signaling pathway is involved in oxidative stress-induced expression of mito-inhibitory p21(WAF1/Cip1) in chronic non-suppurative destructive cholangitis in primary biliary cirrhosis: an immunohistochemical study. J Autoimmun. 2008;31:73–8.
Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–7.
Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78.
Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17.
Rouschop KM, van den Beucken T, Dubois L, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Investig. 2010;120:127–41.
Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31.
Gonzalez-Rodriguez A, Mayoral R, Agra N, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179.
Katayanagi K, Kono N, Nakanuma Y. Isolation, culture and characterization of biliary epithelial cells from different anatomical levels of the intrahepatic and extrahepatic biliary tree from a mouse. Liver. 1998;18:90–8.
Salvado L, Coll T, Gomez-Foix AM, et al. Oleate prevents saturated-fatty-acid-induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia. 2013;56:1372–82.
Martinez SC, Tanabe K, Cras-Meneur C, et al. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes. 2008;57:846–59.
Sasaki M, Van De Water J, Kenny TP et al. Immunoglobulin gene usage and immunohistochemical characteristics of human monoclonal antibodies to the mitochondrial autoantigens of primary biliary cirrhosis induced in the XenoMouse. Hepatology. 2001;34:631–7.
Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7.
Nakanuma Y, Sasaki M. Expression of blood-group-related antigens in the intrahepatic biliary tree and hepatocytes in normal livers and various hepatobiliary diseases. Hepatology. 1989;10:174–8.
Roskams TA, Theise ND, Balabaud C, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739–45.
Desmet V, Gerber M, Hoofnagle J, et al. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. 1994;19:1513–20.
Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809.
Kaser A, Blumberg RS. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology. 2011;140:1738–47.
Cantero-Recasens G, Fandos C, Rubio-Moscardo F, et al. The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum Mol Genet. 2010;19:111–21.
Adolph TE, Tomczak MF, Niederreiter L, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–6.
Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40.
Anderson CD, Upadhya G, Conzen KD, et al. Endoplasmic reticulum stress is a mediator of posttransplant injury in severely steatotic liver allografts. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2011;17:189–200.
Chen Y, Liu CP, Xu KF, et al. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am J Nephrol. 2008;28:1014–22.
Acknowledgments
This study was supported in part by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, and Science and Technology of Japan (21590366 and 24590409).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sasaki, M., Yoshimura-Miyakoshi, M., Sato, Y. et al. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J Gastroenterol 50, 984–995 (2015). https://doi.org/10.1007/s00535-014-1033-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-014-1033-0