
Abstract
Biliary atresia (BA) is a cholestatic disease with extrahepatic bile duct obstruction that requires early surgical intervention and occasionally liver transplantation (LT). Accumulation of toxic bile acids induces oxidative stress that results in cell damage, such as cell senescence, mitochondrial dysfunction and others. However, details of their reciprocal association and clinical significance are unexplored. Therefore, we used immuno-localization of markers for cell senescence (p16 and p21), nuclear double-strand DNA damage (γH2AX), autophagy (p62), and mtDNA damage (mtDNA copy number) in patients with BA who underwent Kasai portoenterostomy (KP) and LT. We studied liver biopsy specimens from 54 patients with BA, 14 who underwent LT and 11 from the livers of neonates and infants obtained at autopsy. In hepatocytes, p21 expression was significantly increased in KP. In cholangiocytes, p16 expression was significantly increased in LT, and p21 expression was significantly increased in KP. p62 expression was significantly increased in the KP hepatocytes and LT cholangiocytes. Furthermore, mtDNA copy number significantly decreased in KP and LT compared with the control. Cell senescence and mitochondrial DNA damage progression were dependent on the BA clinical stages and could possibly serve as the markers of indication of LT.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biliary atresia (BA) occurs from neonatal stage through infancy period, and is histologically characterized by cholestasis associated with sclerosing inflammation of the extrahepatic bile duct [1]. The possible etiology of biliary atresia has been proposed, including congenital bile duct dysplasia, viral infection, and immune disorders; however, none of these disorders have been established as direct causes of the disease [1, 2]. Patients with BA are usually treated with portoenterostomy, also termed Kasai portoenterostomy (KP) [3]. The clinical prognosis of patients with BA is generally evaluated in the presence or absence of jaundice disappearance and native liver survival [4]. The patient’s age at surgery and jaundice clearance rate are well known to be correlated with each other [3]. The jaundice disappearance rate was reported to exceed 70% in neonatal surgery cases but reached approximately 60% in surgical cases until 80 days after birth [3]. However, this rate gradually decreased with the delay in the surgical period [3]. In addition, even though jaundice clinically disappears, the clinical condition gradually deteriorates and liver transplantation is eventually required in many patients with BA [3]. Results from the Japanese Biliary Atresia Registry revealed that 40% of patients with BA eventually required liver transplantation (LT) [3]. Therefore, liver transplantation (LT) has been performed in BA patients with persistent or recurrent jaundice following Kasai portoenterostomy (KP), or those with clinically severe BA conditions [4].
Accumulation of toxic bile acids due to extrahepatic biliary obstruction has been reported to induce oxidative stress, which leads to cellular damage or dysfunctions, such as cell senescence, mitochondrial dysfunction, and DNA damage [5, 6]. Both mitochondrial dysfunction and cellular senescence are reported to be closely correlated with each other [7, 8]. Mitochondrial dysfunction is also known to generally promote and maintain cell senescence and directly contribute to aging-related mitochondrial dysfunction (SAMD) [9,10,11,12]. Recently, mitochondrial DNA (mtDNA) damage has been studied in various diseases associated with biliary obstruction and it possibly has a role in the pathophysiology of these diseases [13]. The number of copies of mtDNA is reported to be an indicator of mtDNA quality [14,15,16]. Therefore, maintaining copies of mitochondrial DNA is required for maintaining mitochondrial function, and reduced mitochondrial DNA copy numbers could reflect mitochondrial dysfunction [17, 18]. Among these factors, mitochondrial DNA copy number was reported to be reduced in hepatocytes in patients with BA and is of particular interest [19].
In the early stages of cell senescence, p53/p21 WAF1/Cip1 was reported to be activated by DNA damage as results of oxidative stress, expressional stress, and others. In addition, relatively high levels of p21 were also known to inhibit the kinase activity of cyclin D and CDK4 / 6 and to cause cell cycle arrest [20]. On the other hand, during the maintenance phase of the cell senescence, p16 was reported to be activated and inhibit the inhibition of the kinase activity of cyclin D and CDK4/6 [20]. The expression of senescence-associated cell cycle regulators including p16 INK4a and p21 WAF1/Cip1, is induced in the advanced stages of several chronic liver diseases, such as primary biliary cholangitis (PBC) and non-alcoholic steatohepatitis (NASH) [21]. p16 INK4a and p21 WAF1/Cip1 have also been reported to be induced in cholangiocytes in biliary atresia [22]. However, the association between the status of cell senescence/DNA damage and the clinical course of patients with BA remains virtually unknown. Therefore, we explored both mitochondrial and nuclear DNA damage in situ in pathological specimens of BA and their possible association with cellular senescent phenotypes, as well as their clinical significance in disease progression of BA patients who underwent KP and/or LT. BA liver samples obtained at the time of KP represented the relatively early stages and early changes in BA. In contrast, the BA liver samples obtained at the time of LT represented advanced stages of BA. In the present study, we used the livers of the same patients at KP and LT. Therefore, the changes associated with the disease course of BA in the same patients were evaluated by comparing the findings of BA liver samples taken at both KP and LT. Immuno-histochemical analysis of p16, p21, and Ki-67 was used to evaluate the status of cell senescence, and γH2AX for double-strand breaks of nuclear DNA damage [23]. In addition, mtDNAcn in hepatocytes was studied to further explore changes in mitochondrial damage during KP and LT. In addition, p62 was also examined as an index of autophagy, which was reported to contribute to the clearance of damaged cells in the local tissue microenvironment [24, 25]. These markers were also studied in the common bile duct at KP.
Materials and methods
Patients
Fifty-four liver biopsy specimens from patients with BA who underwent Kasai portoenterostomy (KP) and extracted liver specimens from 14 patients who underwent liver transplantation (LT) after Kasai portoenterostomy (KP) at Tohoku University Hospital from 2001 to 2017 were examined. Among the LT cases, 13 patients also underwent their first Kasai portoenterostomy surgical intervention from 2001 to 2017, during which biopsy specimens were taken and tentatively categorized as KP cases in this study. In 40 patients with BA who received KP, LT was not performed during their entire clinical course. In addition, among the cases above, 11 BA cases with residual common bile duct epithelium who underwent KP were also studied. For the control tissues of non-pathological liver (NL) in newborns and infants, 11 autopsy cases without any history of liver or metabolic diseases were retrieved from the autopsy files at Tohoku University Hospital between 2005 and 2017. The clinico-pathological characteristics of the KP, LT, and NL cases examined in this study are summarized in Supplemental Table 1 The present study protocol was approved by the Institutional Review Board of the Tohoku University School of Medicine (2019-1-996).
Definition of native liver survival and jaundice disappearance in patients with BA
The native liver survival period was defined as the number of days from the date of the initial KP operation to that of death from liver failure or LT. The clinical definition of jaundice disappearance was defined as total serum bilirubin levels less than 2.0 mg/dl, as described previously [26].
Histological and morphological evaluation of BA liver samples
Serial tissue sections were prepared from 10% formalin-fixed paraffin-embedded (FFPE) blocks cut at 3-μm thickness. Hematoxylin and eosin (H&E) staining, Elastica Masson (EM) stain, and CK19 immunohistochemistry (IHC) were performed for histopathological assessment of liver damage and disease progression (Supplemental Table 2).
The degree of ductular reaction (DR) was semi-quantitatively scored as 0 (negative), 1 (< 30% circumference in portal tracts), 2 (≥ 30% to < 60% circumference in portal tracts), or 3 (≥ 60% circumference in portal tracts), as described previously [27]. The degree of cholestasis was scored as 0 (negative), 1 (mild), 2 (moderate), or 3 (extensive), and that of giant cells was scored as 0 (negative), 1 (mild) or 2 (extensive) [22]. The degree of hepatic fibrosis was evaluated based on the criteria of the revised Inuyama classification system [28].
The status of cell senescence, DNA damage, and autophagy in BA liver samples
Serial tissue sections were analyzed using the streptavidin/biotin method. Images were captured using an Aperio AT2 microscope (Leica Microsystems, Wetzlar, Germany). Immunoreactivity was quantitatively evaluated in hepatocytes and cholangiocytes. The expression in hepatocytes was analyzed using Halo imaging analysis software (Indica Labs, Corrales, NM, USA) [29] and that in cholangiocytes was manually evaluated by two of the authors (Y. N. and Y. Y.) who were blinded to the clinico-pathological variables of the patients.
p16 and p21 were immuno-localized to the nuclei and evaluated using a labeling index (LI) ranging from 0 to 100% [29]. Cell senescence was evaluated with p16 and p21 immunoreactivity, as well as the absence of Ki-67 positivity. p62 was immuno-localized in the cytoplasm; therefore, the results were evaluated using the H-score, which was determined by the percentage of immuno-positive cells and their relative immuno-intensity. Immuno-intensity was scored according to the following criteria: 0, negative; 1, weak; 2, moderate; and 3, marked. The H-score was then calculated by multiplying the percentage of immuno-positive cells by the immuno-intensity on a scale from 0 to 300 [30].
mtDNA copy number analysis
Subsequent sets of serial tissue sections were prepared from FFPE tissue blocks of 10-μm thickness. Genomic DNA was extracted using the AllPrep DNA FFPE Kit (QIAGEN) following the manufacturer’s protocol and as previously reported [31]. DNA extraction was performed for 24 KP cases, 12 LT cases, and 11 NL cases in which sufficient amounts of the tissues were available for evaluation. mtDNAcn was also analyzed by qRT-PCR to study the relative mtDNAcn. Primers were designed to detect ND2 in mtDNA with infrequent deletion (Fw 5′-CCATCTTTGCAGGCACACTCATC-3′ and Rev 5′-ATCCACCTCAAC-TGCCTGCTATG-3′) [32, 33]. The product of the RPL13A house-keeping gene was used as a nuclear-coded gene of the diploid genome (Fw 5′-CCTGGAGGAGAAGAGGAAAG-′3 and Rev 5′-TTGAGGACCTCTGTGTATTT-′3). mtDNAcn was calculated using the following formula: relative mtDNAcn/diploid genome=2 POWER[-(Ct RPL13A-Ct ND2)]*2, as previously described [34].
Evaluation of immunoreactivity
Statistical analysis was performed using JMP Pro Version 15 (SAS Institute Inc., Cary, NC, USA). Multivariate analysis among individual pairs of groups was performed using the Steel–Dwass test. Paired-match analysis between the values obtained at the KP and LT periods in the same patients and paired-match analysis between cholangiocytes and common bile ducts were analyzed using the Wilcoxon signed-rank test. Correlations among clinico-pathological parameters and correlations among cholangiocytes and common bile ducts immunoreactivity were analyzed using the Spearman's rank correlation coefficient test. Native liver survival curves were tentatively drawn by Kaplan–Meier analysis and compared between subgroups based on their median (high and low) values [35]. Differences were considered statistically significant at a p value of < 0.05.
Results
Comparison of cell senescence, DNA damage, and autophagy markers among KP, LT, and NL cases
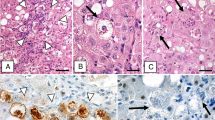
Representative IHC images are shown in Fig. 1. In hepatocytes, p21 expression was significantly higher in KP and LT than in NL samples (KP vs. NL, p = 0.0007; LT vs. NL, p = 0.0482; Fig. 2b). p62 expression was also significantly higher in KP and LT than in NL samples (KP vs. NL, p < 0.0001; LT vs. NL, p = 0.0056) (Fig. 2d), and higher in KP than LT (KP vs. LT, p = 0.0156; Fig. 2d).

Representative IHC images of the liver tissues from patients with BA. The positive control tissue for p16 IHC was that of cervical cancer (a), p16-positive hepatocytes (b), and cholangiocytes (c). The positive control tissue for p21 IHC was breast cancer (d), p21-positive hepatocytes (e), and hepatocytes (f). The positive control of Ki-67 was lymph node tissue (g). Ki67-positive hepatocytes (h) and cholangiocytes (i). The positive control tissue for γH2AX was breast cancer (j). γH2AX-positive hepatocytes (k) and cholangiocytes (l). The positive control tissue for p62 IHC was lung cancer (m). Hepatocytes with weak (n) and marked (o) p62 immunoreactivity. Cholangiocytes with weak (p) and marked (q) p62 immunoreactivity

Comparison of the markers examined in hepatocytes and cholangiocytes among KP, LT, and NL samples. ※ Indicates p < 0.05. Comparison of p16 (a) and p21 (b) in hepatocytes. KP and LT had significantly higher immunoreactivity than NL. Comparison of γH2AX (c) and p62 (d) in hepatocytes. KP and LT had significantly higher immunoreactivity than NL, while KP had significantly higher immunoreactivity than LT. Comparison of p16 in cholangiocytes (e). KP and LT had significantly higher expression than NL, while LT had significantly higher immunoreactivity than KP. Comparison of p21 in cholangiocytes (f). KP and LT had significantly higher immunoreactivity than NL. Comparison of γH2AX in cholangiocytes (g). LT had significantly higher immunoreactivity than KP and NL(h). Comparison of p16 examined in hepatocytes and cholangiocytes (i). Comparison of p21 examined in hepatocytes and cholangiocytes showed that p21 was significantly positively correlated between hepatocytes and cholangiocytes in the same patients (j). Comparison of γH2AX examined in hepatocytes and cholangiocytes, showing γH2AX was significantly positively correlated between hepatocytes and cholangiocytes in the same patient cases (k). Comparison of p62 examined in hepatocytes and cholangiocytes showing p62 was significantly positively correlated between hepatocytes and cholangiocytes in the same cases (l).
In cholangiocytes, p16 expression was significantly higher in KP and LT than in NL (KP vs. NL, p = 0.0022; LT vs. NL, p < 0.0001; Fig. 2e), and higher in LT than KP (LT vs. KP, p < 0.0001; Fig. 2e). p21 was significantly higher in KP and LT than in NL (KP vs. NL, p < 0.0001; LT vs. NL, p = 0.0004; Fig. 2f); and p62 expression was significantly higher in the LT group than in the KP and NL groups (LT vs. NL, p = 0.0001; LT vs. KP, p < 0.0001; Fig. 2h).
We also found that p21, γH2AX, and p62 were all significantly positively correlated between hepatocytes and cholangiocytes in the same BA patients (p21, p < 0.0001; γH2AX, p = 0.0293; p62, p = 0.0036; Fig. 2j-l). In addition, p16 tended to be positively correlated between hepatocytes and cholangiocytes in the same cases of BA, but the correlation was not statistically significant (p16; p = 0.0762; Fig. 2i).
Association of cell senescence, DNA damage, and autophagy markers in hepatocytes and cholangiocytes with histological features of BA
With relation to intrahepatic fibrosis, p16 immunoreactivity in hepatocytes was significantly higher in group 4 than in group 2 (group 4 vs. group 2, p = 0.0241; Fig. 3a). In cholangiocytes, p16 expression was significantly higher in group 4 than in groups 2 and 3 (group 4 vs. group 2, p < 0.0001; group 4 vs. group 3, p < 0.0001; Fig. 3e), while p21 expression was significantly higher in group 3 than in group 4 (group 3 vs. group 4, p = 0.0450; Fig. 3f). We found that p62 expression in hepatocytes was significantly higher in groups 2 and 3 than in group 4 (group 2 vs. group 4, p = 0.0490; group 3 vs. group 4, p = 0.0185; Fig. 3b); while in cholangiocytes, p62 expression was significantly higher in group 4 than in groups 2 and 3 (group 2 vs. group 4, p < 0.0001; group 3 vs. group 4, p = 0.0040; Fig. 3h).


Comparison of immunoreactivity of each marker examined in hepatocytes and cholangiocytes among the pathological factors examined in BA liver samples. ※ Indicates p < 0.05. Comparison of p16 in hepatocytes and fibrosis, DR, giant cell change, and cholestasis, indicating group 4 had significantly higher immunoreactivity than group 2 for fibrosis (a). Comparison of p21 in hepatocytes and fibrosis, DR, giant cell change and cholestasis, indicating group 2 had significantly higher immunoreactivity than group 1 for giant cell change (b). Comparison of γH2AX in hepatocytes and fibrosis, DR, giant cell change and cholestasis (c). Comparison of p62 in hepatocytes and fibrosis, DR, giant cell change and cholestasis, showing that group 2 and group 3 had significantly higher immunoreactivity than group 4 for fibrosis (d). Comparison of p16 in cholangiocytes and fibrosis, DR, giant cell change, and cholestasis indicating group 4 had significantly higher immunoreactivity for fibrosis than groups 2 and 3, while in the DR group, group 3 showed significantly higher immunoreactivity than group 2; and in cholangiocytes, group 3 showed significantly higher expression than group 1 (e). Comparison of p21 in cholangiocytes and fibrosis, DR, giant cell change, and cholestasis showing group 4 had significantly higher immunoreactivity for fibrosis than group 3 (f). Comparison of γH2AX in cholangiocytes and fibrosis, DR, giant cell change, and cholestasis (g). Comparison of p62 in cholangiocytes and fibrosis, DR, giant cell change, and cholestasis, which shows that group 4 had significantly higher immunoreactivity for fibrosis than groups 2 and 3; while in the DR, group 3 had significantly higher immunoreactivity than groups 1 and 2 (h).
We next asked which of these markers correlated with DR, and found that p16 immunoreactivity in cholangiocytes was significantly higher in group 3 than in group 2 (group 3 vs. group 2, p = 0.0020; Fig. 3e), and p62 expression in cholangiocytes was significantly higher in group 3 than in groups 1 and 2 (group 3 vs. group 1, p = 0.0135; group 3 vs. group 2, p = 0.0006; Fig. 3h).
Regarding the correlation of each markers with giant cell changes in the liver, p21 immunoreactivity in hepatocytes was significantly higher in group 2 than in group 1 (group 2 vs. group 1, p = 0.0326; Fig. 3b). With respect to cholestasis, p16 expression in cholangiocytes was significantly higher in group 3 than in group 1 (group 3 vs. group 1, p = 0.0447; Fig. 3e).
Comparison of cell senescence, DNA damage, and autophagy markers among cholangiocytes and common bile ducts
Markers of cell senescence, DNA damage, and autophagy markers between cholangiocytes and common bile ducts in the same patients were also examined. p16 immunoreactivity in common bile ducts was significantly higher than cholangiocytes (p = 0.0166; Table 1). However, no significant correlation was detected between cholangiocytes and common bile ducts in the same cases (Fig. 4a–d).

Comparison of the markers examined in cholangiocytes and common bile ducts. ※ Indicates p < 0.05. Comparison of p16 examined in cholangiocytes and common bile ducts (a). Comparison of p21 examined in cholangiocytes and common bile ducts (b). Comparison of γH2AX examined in cholangiocytes and common bile ducts (c). Comparison of p62 examined in cholangiocytes and common bile ducts (d)
Matched analysis of individual markers in the same BA patients with BA between their first KP and subsequent LT
Cell senescence, DNA damage, and autophagy markers were compared between the time of the first surgical intervention with KP and that of the subsequent LT. We found that p16 immunoreactivity in both hepatocytes and cholangiocytes was significantly higher at the time of LT than KP (LT vs. KP, hepatocytes: p = 0.0164, cholangiocytes: p = 0.0001; Table 2), while p62 expression in cholangiocytes was significantly higher in LT than in KP (LT vs. KP, p = 0.0001; Table2).
Comparison of mtDNAcn among KP, LP and NL cases
mtDNAcn was significantly lower in KP and LT than in NL (KP vs. NL, p = 0.0244; LT vs. NL, p = 0.0141; Fig. 5); however, mtDNAcn in the LT group tended to be lower than that in the KP group, although this difference was not statistically significant.

Comparison of mtDNAcn among KP, LT, and NL. ※ Indicates p < 0.05. KP and LT were significantly lower than NL
Correlation between clinical characteristics and cell senescence, DNA damage, and autophagy marker expression in patients with BA
The expression of markers for cell senescence, DNA damage, and autophagy and mtDNAcn was compared using clinical data from patients with BA to evaluate their potential relationships (Table 3). Total bilirubin (T.Bil) levels were significantly and positively correlated with mtDNAcn (p = 0.0005), while direct bilirubin (D.Bil) levels were significantly positively correlated with p21 (p = 0.0142) in hepatocytes. In addition, p21 expression in cholangiocytes (p = 0.0394), and mtDNAcn (p = 0.0083) was also significantly correlated to D.Bil. Alanine transaminase (ALT) levels were also significantly positively correlated with p21 (p = 0.0020) and p62 (p = 0.0211) expression in hepatocytes, as well as with p21 expression in cholangiocytes (p = 0.0033). Cholinesterase (ChE) was significantly inversely correlated with the expression of p16 (p = 0.0248) and γH2AX (p = 0.0073) in hepatocytes, and p16 (p = 0.0001) and p62 (p = 0.0033) expression in cholangiocytes. Platelet (Plt) was also significantly inversely correlated with p16 expression in hepatocytes (p = 0.0406) and p16 (p = 0.0028) and p62 (p = 0.0200) expression in cholangiocytes; as well as with mtDNAcn (p = 0.0049).
Association between post-operative prognosis and native liver survival and cell senescence, DNA damage, and autophagy marker expression in patients with BA
Figure 6 shows the Kaplan–Meier curves of the individual factors examined in this study. No significant correlation was detected among the individual markers in terms of cell senescence, DNA damage, and autophagy in either hepatocytes or cholangiocytes and the date of native liver survival (Fig. 6a–h). However, native liver survival was significantly prolonged in BA cases with lower mtDNAcn (Fig. 6i), although no significant differences were detected when examining only those associated with jaundice disappearance after KP (Fig. 6j).

Native liver survival date analysis using Kaplan–Meier curve in patients with BA. ※ Indicates p < 0.05. p16 (a), p21 (b), γH2AX (c), and p62 (d) status in hepatocytes. p16 (e), p21 (f), γH2AX (g), and p62 (h) status in cholangiocytes. mtDNAcn status in all patient cases indicated that native liver survival date was significantly prolonged when mtDNAcn was low (i). mtDNAcn status in cases where jaundice disappeared (j)
Discussion
This is one of the first studies to simultaneously evaluate markers of cell senescence, DNA damage, and autophagy in both hepatocytes and cholangiocytes, as well as mtDNA copy number in patients with BA who underwent KP and/or LT during their clinical course. The results indicated a clinical significance of the relationship between the expression of these markers and mtDNA damage with BA disease progression. In addition, although few studies have examined liver tissues at KP and LT in the same patients, this is the first report to show that mitochondrial DNA damage and change occur during the clinical course of patients with BA.
The majority of senescence-inducing stressors have been reported to activate either the p16Ink4a and/or p53/p21 intracellular signaling, which have emerged as biomarkers of cellular senescence [36,37,38]. Bile salt was taken up by transporter Ntcp and accumulated in hepatocytes. Elevated levels of bile salt in hepatocytes caused endoplasmic reticulum stress and mitochondrial damage. This resulted in the release of inflammatory chemokines through Tlr9-dependent signaling pathways. Then reactive oxygen species (ROS) were released from the induced inflammatory cells, resulting in cytotoxicity [39]. During cell senescence, p21 can contribute to the early stages of the process that are associated with cell stress, making it a marker for the initiation of cell senescence; while p16 plays an important role in maintaining cell senescence [20, 40,41,42,43]. Therefore, increased p16 expression at the time of LT in the same BA case could reflect the progression of cell senescence during the clinical development of BA. With respect to hepatocytes, p21 expression increased in livers that received KP, while in cholangiocytes, p16 expression was high in LT and p21 was high in KP, which was consistent with previously reported findings [22]. The progression of cellular senescence in BA hepatocytes and cholangiocytes was similar following this period in the clinical course of BA; however, the profiles of the phenotypes were partially different. These results will need further investigation to understand how these cells senesce during BA progression.
The correlation of these findings with clinical data from patients with BA also revealed that p16 expression in both hepatocytes and cholangiocytes was significantly inversely correlated with that of ChE and Plt, which could represent viability or biological synthetic activities of hepatocytes. In addition, p16 expression in hepatocytes was significantly positively correlated with the stages of fibrosis, while that in cholangiocytes correlated with the stages of fibrosis and the degree of DR and cholestasis. We found that p21 expression was also significantly positively correlated with giant cell changes in hepatocytes, while decreased expression in cholangiocytes correlated with the progression of fibrosis. These results indicated the synchronization of the progressive status between the disease period and cellular senescence in both hepatocytes and cholangiocytes. Giant cell changes in the liver were reported to be closely associated with cellular senescence [44], which is consistent with the results of this study.
We also used γH2AX as a marker of DNA double-strand breaks caused by genotoxic substances, such as UV and radiation [45], to further explore the association between the status of DNA damage and senescence status of BA liver samples. DNA damage and altered replication are integral components of the aging phenotype [38, 46]. Our results demonstrated that the number of γH2AX-positive cells was very small at every stage of the disease, suggesting that double-stranded DNA damage did not frequently occur in BA liver cells, despite abundant noxious stimuli. This is possibly because of more stable DNA in BA cells in contrast to those of various diseases that present with genomic instability, such as neoplastic diseases. 8-hydroxydeoxyguanosine (8-OHdG), a marker of DNA damage due to oxidative stress [47], was reported to be present in hepatocytes of BA liver [48]. 8-OHdG was reported to be produced by reactive oxygen species and induce point mutations during DNA replication [47, 49], whereas γH2AX was a DNA double-strand breaks marker generally reported as the most deleteriously cytotoxic event caused by irradiation and other noxious stimuli. [45]. Therefore, biliary obstruction could promote ROS oxidative stress in BA liver, subsequently inflicting genotoxicity on the hepatocytes as previously reported but could not reach to the double-stranded DNA cleavage. However, further investigation is required to test this interesting hypothesis.
Autophagy represents genetically regulated mechanisms that involve intracellular turnover of cellular proteins and damaged organelles. p62 is an adaptor protein involved in the delivery of ubiquitin-bound cargo to the auto-phagosome and helps to regulate the formation of protein aggregates [25, 50,51,52]. Accumulation of p62 was reported to reflect dysfunctional autophagy [53,54,55]. In this study, BA livers that received KP had significantly higher levels of p62 expression in hepatocytes, while those that received LT showed a significant increase in cholangiocytes. In addition, hepatocyte p62 expression demonstrated a significant inverse correlation with fibrosis progression. Therefore, in hepatocytes, induction of autophagy occurs in the early stages of BA in the liver, while the relative dysfunction of autophagy could result in cytosolic p62 accumulation. In advanced stages of BA, liver damage is considered irreversible, possibly beyond the autophagy-regulating clearance. In contrast, the induction of autophagy in cholangiocytes was considered to be maintained even at the stage of LT.
Mitochondrial dysfunction due to mtDNA damage has been reported in various disorders, including neurodegenerative diseases, myopathy, and diabetes [56,57,58,59]. Mitochondrial dysfunction usually results in cellular dysfunction and senescence [9,10,11]. Of particular interest, mtDNA was found to be more susceptible to stress-induced damage, including oxidative DNA damage, than nuclear DNA because of its proximity to the potential intracellular sites of oxidative phosphorylation and its intrinsic lack of histones [60, 61]. The TCA cycle is highly active in hepatocytes and mitochondrial function is closely related to hepatocyte function [62, 63]. Therefore, mtDNA damage due to cholestasis may represent a very early stage of hepatocyte dysfunction and cell senescence. In this study, a significant decrease in mtDNAcn was detected in BA livers that had received KP and LT compared to normal livers. Although no significant difference was detected, mtDNAcn tended to decrease in BA livers that had received LT compared to KP livers. These results indicate that mtDNA degradation could occur during cell aging and stages of BA progression in the liver. In addition, when we assessed the correlation of the results with native liver survival in patients with BA, the liver survival rate tended to be higher in those that had a lower mtDNAcn. However, no significant correlation was detected between the mtDNAcn and liver survival time when examined in samples from patients whose jaundice disappeared after KP. Therefore, the effects of other potential factors, such as patient age at time of surgery, could also contribute to the clinical progression of BA in the liver.
BA is a disease caused by obstruction of the extrahepatic bile duct due to sclerosing inflammation of unknown cause [1]. In this study, there were no significant correlation between cholangiocytes and common bile ducts in the same patients in cell senescence, DNA damages, and autophagy markers. Those findings indicated that the degree of sclerosing inflammation of the extrahepatic bile duct, which could represent the major pathophysiology of BA, was not necessarily correlated with that of liver injury. Only p16 immunoreactivity was higher in the common bile duct (p = 0.0166; Table 1). These results therefore suggested that cell senescence in the common bile duct could be more advanced compared to the intrahepatic bile duct and reached the maintenance phase [20, 40,41,42,43]. However, this hypothesis needs further investigation.
Figure 7 presents a hypothetical scheme that depicts the association between the senescence, DNA damage, and autophagy markers we tested and the progression of the BA disease period based on the findings of our present study. As demonstrated in Fig. 7, cell senescence might be induced in both hepatocytes and cholangiocytes at relatively early stages of the disease and senescence phenotypes become pronounced as the disease progresses into the advanced stages of BA in the liver. In addition, mtDNA degradation can become more pronounced with disease progression, but can also be detected at earlier stages of the disease, which possibly reflects damage inflicted on hepatocytes. Moreover, cytosolic p62 accumulation becomes pronounced as the BA progresses; however, we detected a discrepancy between hepatocytes and cholangiocytes in the BA liver samples. This finding could reflect a potential discrepancy in the phase of autophagy impairment between hepatocytes and cholangiocytes in the BA liver, but further investigation is required for clarification of this interpretation.

Predicted cellular injury or damage processes in the liver during biliary atresia (BA). Bile acid accumulation and oxidative stress occur in hepatocytes and cholangiocytes due to cholestasis. In hepatocytes, cellular senescence and mitochondrial damage occur as liver damage progresses. At early stages of BA in livers that undergo KP, the cell clearance mechanisms can be initiated; however, when the liver damage is advanced such that LT was required, the clearance mechanism is decreased. Liver damage can be reversed at early stages of BA in livers that receive KP, but is considered irreversible at advanced stages of BA in livers that require LT. In cholangiocytes, cell senescence progresses similarly to hepatocytes, but the clearance mechanism can be maintained at advanced clinical stages of BA in livers that receive LT
The age matching in this study was not obtained between the control group and BA cases, especially in the LT cases. In addition, BA disease specificity was not evaluated in the present study; however, our results suggest that cell senescence, mitochondrial DNA damage, and autophagy dysfunction are all associated with the progression of liver cellular damage in patients with BA.
Abbreviations
- ALT:
-
Alanine transaminase
- BA:
-
Biliary atresia
- ChE:
-
Cholinesterase
- D.Bil:
-
Direct bilirubin
- DR:
-
Ductular reaction
- FFPE:
-
Formalin-fixed paraffin-embedded
- H&E:
-
Hematoxylin–eosin
- IHC:
-
Immunohistochemistry
- KP:
-
Kasai portoenterostomy
- LT:
-
Liver transplantation
- mtDNA:
-
Mitochondrial DNA
- mtDNAcn:
-
Mitochondrial DNA copy number
- NL:
-
Non-pathological liver
- Plt:
-
Platelet
- ROS:
-
Reactive oxygen species
- T.Bil:
-
Total bilirubin
- 8-OHdG:
-
8-Hydroxydeoxyguanosine
References
Hartley JL, Davenport M, Kelly DA (2009) Biliary atresia. Lancet 374:1704–1713. https://doi.org/10.1016/S0140-6736(09)60946-6
Zagory JA, Nguyen MV, Wang KS (2015) Recent advances in the pathogenesis and management of biliary atresia. Curr Opin Pediatr 27:389–394. https://doi.org/10.1097/MOP.0000000000000214
Nio M, Registry JBA (2017) Japanese biliary atresia registry. Pediatr Surg Int 33:1319–1325. https://doi.org/10.1007/s00383-017-4160-x
Sasaki H, Tanaka H, Wada M, Kazama T, Nishi K, Nakamura M, Kudo H, Kawagishi N, Nio M (2014) Liver transplantation following the Kasai procedure in treatment of biliary atresia: a single institution analysis. Pediatr Surg Int 30:871–875. https://doi.org/10.1007/s00383-014-3552-4 (Epub 2014 Jul 27)
Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, Abete P (2018) Oxidative stress, aging, and diseases. Clin Interv Aging 13:757–772. https://doi.org/10.2147/CIA.S158513
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL (2013) Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123:966–972. https://doi.org/10.1172/JCI64098 (Epub 2013 Mar 1)
Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, Zappavigna S, Sperlongano P, Irace C, Caraglia M, Misso G (2020) Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol 98:139–153. https://doi.org/10.1016/j.semcdb.2019.05.022 (Epub 2019 Jun 27)
Dabravolski SA, Bezsonov EE, Orekhov AN (2021) The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed Pharmacother 142:112041. https://doi.org/10.1016/j.biopha.2021.112041 (Epub 2021 Aug 16)
Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly-Y M, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, von Zglinicki T, Korolchuk VI, Passos JF (2016) Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35:724–742. https://doi.org/10.15252/embj.201592862 (Epub 2016 Feb 4)
Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, Birch-Machin MA, Kirkwood TB, von Zglinicki T (2007) Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLOS Biol 5:e110. https://doi.org/10.1371/journal.pbio.0050110
Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23:303–314. https://doi.org/10.1016/j.cmet.2015.11.011 (Epub 2015 Dec 10)
Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T (2010) Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 6:347. https://doi.org/10.1038/msb.2010.5 (Epub 2010 Feb 16)
Arduini A, Serviddio G, Tormos AM, Monsalve M, Sastre J (2012) Mitochondrial dysfunction in cholestatic liver diseases. Front Biosci (Elite Ed) 4:2233–2252. https://doi.org/10.2741/539
Clay Montier LL, Deng JJ, Bai Y (2009) Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics 36:125–131. https://doi.org/10.1016/S1673-8527(08)60099-5
Holmuhamedov E, Jahangir A, Bienengraeber M, Lewis LD, Terzic A (2003) Deletion of mtDNA disrupts mitochondrial function and structure, but not biogenesis. Mitochondrion 3:13–19. https://doi.org/10.1016/S1567-7249(03)00053-9
Malik AN, Czajka A (2013) Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 13:481–492. https://doi.org/10.1016/j.mito.2012.10.011 (Epub 2012 Oct 22)
Jeng JY, Yeh TS, Lee JW, Lin SH, Fong TH, Hsieh RH (2008) Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J Cell Biochem 103:347–357. https://doi.org/10.1002/jcb.21625
Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, Walter P, Yaffee M (1995) Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta 1271:67–74. https://doi.org/10.1016/0925-4439(95)00012-s
Wang J, Xu J, Xia M, Yang Y, Shen Z, Chen G, Dong R, Zheng S (2019) Correlation between hepatic oxidative damage and clinical severity and mitochondrial gene sequencing results in biliary atresia. Hepatol Res 49:695–704. https://doi.org/10.1111/hepr.13324 (Epub 2019 Apr 1)
Kumari R, Jat P (2021) Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol 9:645593. https://doi.org/10.3389/fcell.2021.645593
Sasaki M, Ikeda H, Yamaguchi J, Miyakoshi M, Sato Y, Nakanuma Y (2010) Bile ductular cells undergoing cellular senescence increase in chronic liver diseases along with fibrous progression. Am J Clin Pathol 133:212–223. https://doi.org/10.1309/AJCPWMX47TREYWZG
Sasaki M, Kuo FY, Huang CC, Swanson PE, Chen CL, Chuang JH, Yeh MM (2018) Increased expression of senescence-associated cell cycle regulators in the progression of biliary atresia: an immunohistochemical study. Histopathology 72:1164–1171. https://doi.org/10.1111/his.13476 (Epub 2018 Mar 9)
Mah LJ, El-Osta A, Karagiannis TC (2010) gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24:679–686. https://doi.org/10.1038/leu.2010.6 (Epub 2010 Feb 4)
White E, Lowe SW (2009) Eating to exit: autophagy-enabled senescence revealed. Genes Dev 23:784–787. https://doi.org/10.1101/gad.1795309
Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614. https://doi.org/10.1083/jcb.200507002 (Epub 2005 Nov 14)
Nio M, Wada M, Sasaki H, Kazama T, Tanaka H, Kudo H (2016) Technical standardization of Kasai portoenterostomy for biliary atresia. J Pediatr Surg 51:2105–2108. https://doi.org/10.1016/j.jpedsurg.2016.09.047 (Epub 2016 Sep 16)
Kuo FY, Huang CC, Chen CL, Chuang JH, Riehle K, Swanson PE, Yeh MM (2015) Immunohistochemical characterization of the regenerative compartment in biliary atresia: a comparison between Kasai procedure and transplant cases. Hum Pathol 46:1633–1639. https://doi.org/10.1016/j.humpath.2015.07.003 (Epub 2015 Jul 15)
Ichida F, Tsuji T, Omata M, Ichida T, Inoue K, Kamimura T, Yamada G, Hino K, Yokosuka O, Suzuki H (1996) New Inuyama classification: new criteria for histological assessment of chronic hepatitis. Int Hepatol Commun 6:112–119. https://doi.org/10.1016/S0928-4346(96)00325-8
Yamazaki Y, Nakamura Y, Shibahara Y, Konosu-Fukaya S, Sato N, Kubota-Nakayama F, Oki Y, Baba S, Midorikawa S, Morimoto R, Satoh F, Sasano H (2016) Comparison of the methods for measuring the Ki-67 labeling index in adrenocortical carcinoma: manual versus digital image analysis. Hum Pathol 53:41–50. https://doi.org/10.1016/j.humpath.2015.10.017 (Epub 2016 Mar 7)
Ueki S, Fujishima F, Kumagai T, Ishida H, Okamoto H, Takaya K, Sato C, Taniyma Y, Kamei T, Sasano H (2020) GR, Sgk1, and NDRG1 in esophageal squamous cell carcinoma: their correlation with therapeutic outcome of neoadjuvant chemotherapy. BMC Cancer 20:161. https://doi.org/10.1186/s12885-020-6652-7
Omata K, Anand SK, Hovelson DH, Liu CJ, Yamazaki Y, Nakamura Y, Ito S, Satoh F, Sasano H, Rainey WE, Tomlins SA (2017) Aldosterone-producing cell clusters frequently harbor somatic mutations and accumulate with age in normal adrenals. J Endocr Soc 1:787–799. https://doi.org/10.1210/js.2017-00134
Duregon E, Volante M, Cappia S, Cuccurullo A, Bisceglia M, Wong DD, Spagnolo DV, Szpak-Ulczok S, Bollito E, Daffara F, Berruti A, Terzolo M, Papotti M (2011) Oncocytic adrenocortical tumors: diagnostic algorithm and mitochondrial DNA profile in 27 cases. Am J Surg Pathol 35:1882–1893. https://doi.org/10.1097/PAS.0b013e31822da401
Izzotti A, Saccà SC, Longobardi M, Cartiglia C (2010) Mitochondrial damage in the trabecular meshwork of patients with glaucoma. Arch Ophthalmol 128:724–730. https://doi.org/10.1001/archophthalmol.2010.87
Révész D, Verhoeven JE, Picard M, Lin J, Sidney S, Epel ES, Penninx BWJH, Puterman E (2018) Associations between cellular aging markers and metabolic syndrome: findings from the CARDIA study. J Clin Endocrinol Metab 103:148–157. https://doi.org/10.1210/jc.2017-01625
Gokon Y, Fujishima F, Taniyama Y, Ishida H, Yamagata T, Sawai T, Uzuki M, Ichikawa H, Itakura Y, Takahashi K, Yajima N, Hagiwara M, Nishida A, Ozawa Y, Sakuma T, Kanba R, Sakamoto K, Zuguchi M, Saito M, Kamei T, Sasano H (2020) Immune microenvironment in Barrett’s esophagus adjacent to esophageal adenocarcinoma: possible influence of adjacent mucosa on cancer development and progression. Virchows Arch 477:825–834. https://doi.org/10.1007/s00428-020-02854-0 (Epub 2020 Jun 12)
Ju Z, Choudhury AR, Rudolph KL (2007) A dual role of p21 in stem cell aging. Ann N Y Acad Sci 1100:333–344. https://doi.org/10.1196/annals.1395.036
Shay JW, Roninson IB (2004) Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23:2919–2933. https://doi.org/10.1038/sj.onc.1207518
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M (2019) Cellular senescence: defining a path forward. Cell 179:813–827. https://doi.org/10.1016/j.cell.2019.10.005
Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, Wang Y, Mehal WZ, Jain D, Boyer JL (2017) Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2(5):e90780. https://doi.org/10.1172/jci.insight.90780
Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:1501–1512. https://doi.org/10.1101/gad.13.12.1501
Stein GH, Drullinger LF, Soulard A, Dulić V (1999) Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 19:2109–2117. https://doi.org/10.1128/MCB.19.3.2109
Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M (2017) Unmasking transcriptional heterogeneity in senescent cells. Curr Biol 27:2652-2660.e4. https://doi.org/10.1016/j.cub.2017.07.033 (Epub 2017 Aug 30)
Petrova NV, Velichko AK, Razin SV, Kantidze OL (2016) Small molecule compounds that induce cellular senescence. Aging Cell 15:999–1017. https://doi.org/10.1111/acel.12518 (Epub 2016 Sep 14)
Ikeda H, Sasaki M, Sato Y, Harada K, Zen Y, Mitsui T, Nakanuma Y (2009) Large cell change of hepatocytes in chronic viral hepatitis represents a senescent-related lesion. Hum Pathol 40:1774–1782. https://doi.org/10.1016/j.humpath.2009.06.009 (Epub 2009 Sep 4)
Kopp B, Khoury L, Audebert M (2019) Validation of the γH2AX biomarker for genotoxicity assessment: a review. Arch Toxicol 93:2103–2114. https://doi.org/10.1007/s00204-019-02511-9 (Epub 2019 Jul 9)
González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D (2021) A guide to assessing cellular senescence in vitro and in vivo. FEBS J 288:56–80. https://doi.org/10.1111/febs.15570 (Epub 2020 Oct 10)
Kasai H (1997) Analysis of a form of oxidative DNA damage, 8-hydroxy-2’-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat Res 387(3):147–163. https://doi.org/10.1016/s1383-5742(97)00035-5
Tiao MM, Lin TK, Kuo FY, Huang CC, Du YY, Chen CL, Chuang JH (2007) Early stage of biliary atresia is associated with significant changes in 8-hydroxydeoxyguanosine and mitochondrial copy number. J Pediatr Gastroenterol Nutr 45(3):329–334. https://doi.org/10.1097/MPG.0b013e3180cc2c0f
Shibutani S, Takeshita M, Grollman AP (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349(6308):431–434. https://doi.org/10.1038/349431a0
Lamark T, Kirkin V, Dikic I, Johansen T (2009) NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8:1986–1990. https://doi.org/10.4161/cc.8.13.8892 (Epub 2009 Jul 30)
Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 283:22847–22857. https://doi.org/10.1074/jbc.M802182200 (Epub 2008 Jun 4)
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. https://doi.org/10.1074/jbc.M702824200 (Epub 2007 Jun 19)
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223. https://doi.org/10.1038/ncb2021 (Epub 2010 Feb 21)
Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, Hansdottir S, Hunninghake GW (2010) Identification of an autophagy defect in smokers’ alveolar macrophages. J Immunol 185:5425–5435. https://doi.org/10.4049/jimmunol.1001603 (Epub 2010 Oct 4)
Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y (2012) A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int 32:487–499. https://doi.org/10.1111/j.1478-3231.2011.02656.x (Epub 2011 Sep 27)
Coppedè F, Stoccoro A (2019) Mitoepigenetics and neurodegenerative diseases. Front Endocrinol (Lausanne) 10:86. https://doi.org/10.3389/fendo.2019.00086
Mehta AR, Gregory JM, Dando O, Carter RN, Burr K, Nanda J, Story D, McDade K, Smith C, Morton NM, Mahad DJ, Hardingham GE, Chandran S, Selvaraj BT (2021) Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol 141(2):257–279. https://doi.org/10.1007/s00401-020-02252-5 (Epub 2021 Jan 4)
Bychkov IO, Itkis YS, Tsygankova PG, Krylova TD, Mikhaylova SV, Klyushnikov SA, Pechatnikova NL, Degtyareva AV, Nikolaeva EA, Seliverstov YA, Kurbatov SA, Dadali EL, Rudenskaya GE, Illarioshkin SN, Zakharova EY (2021) Mitochondrial DNA maintenance disorders in 102 patients from different parts of Russia: mutational spectrum and phenotypes. Mitochondrion 57:205–212. https://doi.org/10.1016/j.mito.2021.01.004 (Epub 2021 Jan 21)
Fazzini F, Lamina C, Raftopoulou A, Koller A, Fuchsberger C, Pattaro C, Del Greco FM, Döttelmayer P, Fendt L, Fritz J, Meiselbach H, Schönherr S, Forer L, Weissensteiner H, Pramstaller PP, Eckardt KU, Hicks AA, Kronenberg F, Investigators GCKD (2021) Association of mitochondrial DNA copy number with metabolic syndrome and type 2 diabetes in 14 176 individuals. J Intern Med 290:190–202. https://doi.org/10.1111/joim.13242 (Epub 2021 Feb 20)
Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 94:514–519. https://doi.org/10.1073/pnas.94.2.514
Druzhyna NM, Wilson GL, LeDoux SP (2008) Mitochondrial DNA repair in aging and disease. Mech Ageing Dev 129:383–390. https://doi.org/10.1016/j.mad.2008.03.002 (Epub 2008 Mar 13)
Rui L (2014) Energy metabolism in the liver. Compr Physiol 4:177–197. https://doi.org/10.1002/cphy.c130024
Auger C, Alhasawi A, Contavadoo M, Appanna VD (2015) Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol 3:40. https://doi.org/10.3389/fcell.2015.00040
Acknowledgements
We appreciate the skillful technical assistance of Mr. Katsuhiko Ono, Mr. Yoshiaki Onodera (Department of Pathology, Tohoku University Graduate School of Medicine, Sendai, Japan) and Ms. Yayoi Aoyama (Department of Pathology, Tohoku University Hospital, Sendai, Japan). We would like to thank Editage (www.editage.com) for English language editing. Supported by Health and Labor Sciences Research Grants (19FC1008).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nakajima, Y., Yamazaki, Y., Gao, X. et al. Association between mitochondrial and nuclear DNA damages and cellular senescence in the patients with biliary atresia undergoing Kasai portoenterostomy and liver transplantation. Med Mol Morphol 55, 131–145 (2022). https://doi.org/10.1007/s00795-022-00314-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-022-00314-z