
Abstract
Neuroendocrine tumors (NETs) [carcinoids, pancreatic neuroendocrine tumors (pNETs)] are becoming an increasing clinical problem because not only are they increasing in frequency, but they can frequently present with advanced disease that requires diagnostic and treatment approaches different from those used in the neoplasms that most physicians are used to seeing and treating. In the past few years there have been numerous advances in all aspects of NETs including: an understanding of their unique pathogenesis; specific classification systems developed which have prognostic value; novel methods of tumor localization developed; and novel treatment approaches described. In patients with advanced metastatic disease these include the use of newer chemotherapeutic approaches, an increased understanding of the role of surgery and cytoreductive methods, the development of methods for targeted delivery of cytotoxic agents, and the development of targeted medical therapies (everolimus, sunitinib) based on an increased understanding of the disease biology. Although pNETs and gastrointestinal NETs share many features, recent studies show they differ in pathogenesis and in many aspects of diagnosis and treatment, including their responsiveness to different therapies. Because of limited space, this review will be limited to the advances made in the management and treatment of patients with advanced metastatic pNETs over the past 5 years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
This article will concentrate on recent advances in the treatment of patients with pancreatic neuroendocrine tumors (pNETS) with advanced metastatic disease. This brief review is undertaken because not only are pNETs as well as other gastrointestinal neuroendocrine tumors (GI-NETs) (carcinoids), receiving increased attention recently, but also because there have been a number of advances in their understanding and treatment. pNETs are receiving increased attention because it is increasingly appreciated they are more frequent than previously believed; also, they are frequently more malignant than previously generally thought and can be a source of considerable morbidity, because many patients have a protracted course [1]. Other important advances include an increasing understanding that they have a different pathogenesis from pancreatic adenocarcinomas and GI-NETs [1–5]; the development of standardized classification systems that have prognostic significance [6–11]; the increased standardization of pathological reporting/classifications [12, 13]; the development of highly sensitive imaging modalities (primarily using somatostatin receptor imaging) [14, 15]; and the development of new target medical treatments (the mammalian target of rapamycin [mTOR] inhibitor, everolimus; the tyrosine kinase inhibitor, sunitinib) [16–21]; as well as other novel treatments such as peptide receptor radionuclide therapy (PRRT) using radiolabeled somatostatin analogues [22–24]. These changes have led to the recent publication of numerous general consensus guidelines for pNET management/treatment, including those from the European Neuroendocrine Tumor Society (ENETS) [25]; the North American Neuroendocrine Tumor Society (NANETS) [26]; the European Society for Medical Oncology (ESMO) [27], and those from the Nordic countries [28], as well as more specific consensus guidelines for the treatment of metastatic disease to the liver [29, 30] or more distant sites [29, 31–33]. In this article, advances in the past 5 years for pNETs will be briefly reviewed, concentrating on the evolving treatment of advanced metastatic disease.
Pancreatic neuroendocrine tumors (pNETs) are neuroendocrine tumors of the duodenal-pancreatic region that present many challenging problems in diagnosis and treatment [3, 7, 25, 26]. These problems occur both because pNETs can be hormonally active, ectopically secreting various biologically active substances that can cause specific syndromes requiring treatment (Table 1), and because 50–80 % are malignant (except for insulinomas) and require treatment approaches that differ from those used for most common adenocarcinomas [3, 7, 20, 25, 34, 35].
Most physicians consider pNETs to be very uncommon neoplasms that generally pursue a benign course. Both of these commonly held beliefs are only partially true. pNETs are not uncommon in autopsy studies, occurring in 0.5–1.5 % in various Western studies [3] and in 2.5 % in Japan [36]. However, pNETs are uncommon clinically because fewer than 1/1,000 of those that are found in autopsy studies cause clinical symptoms, with the result that they have a clinical incidence of 1–5 new cases/100,000 population per year with a prevalence of 10/100,000 population, resulting in their causing 1–4 % of all clinically apparent pancreatic tumors [3, 7]. In 2010 a study [37] reported the incidence of pNETs in Japan as 2.23/100,000 population/year, with a prevalence of 1.27/100,000 population. pNETs, similar to other NETs (carcinoids), are increasing in frequency in recent studies, particularly nonfunctional pNETs (NF-pNETs) detected in imaging studies performed for nondescript symptoms, screening, or other suspected diseases [38–40]. In terms of the relative frequency of the pNET syndromes (Table 1), in early series NF-pNETs comprised 1/3 of all pNETs reported, with a frequency approximately equal to that of insulinomas and slightly greater than that of gastrinomas [3, 41, 42]. In more recent series, NF-pNETs comprise a larger proportion of pNETs, in some cases reaching 75 % of the series [43], with up to 50 % of patients asymptomatic and 38 % with NF-pNETs discovered incidentally [25, 39, 40, 43]. The remaining pNETs are less common than NF-pNETs, insulinomas, and gastrinomas and are only infrequently seen (Table 1). In Japan, functional pNETs (F-pNETs) are reported to occur 30 % more frequently than NF-pNETs (prevalence-1.27 vs. 0.97/100,000) [37], with the most frequently occurring F-pNET being insulinoma (prevalence-0.37/100,000 population), which was fivefold more frequent than gastrinomas, 15-fold more frequent than glucagonomas, and >53-fold more frequent than somatostatinomas and vasoactive intestinal peptide-secreting pNETs (VIPomas) [37].
Also, in contrast to what is commonly believed, pNETs are frequently malignant (>50 %, except for insulinomas) (Table 1) and not infrequently pursue an aggressive course with metastases to the liver as well as more distant sites [3, 7, 42]. In one large population study from England of 4104 cases of malignant digestive endocrine tumors (pNETs, carcinoids), the 5-year survival was 56 % [44]. The extent, development, and growth of liver metastases are all associated with a poor prognosis [3, 42, 45–50] and their presence at some point occurs in up to 80 % of pNET patients in some series (Table 1). This is in contrast to lymph node metastases alone, which, in most series, have no or a minimal prognostic effect [34, 47, 51]. Median survivals for pNET patients with localized, regional, or distant disease for the 1310 patients in the United States Surveillance, Epidemiology, and End Results (SEER) database [45] were 124, 70, and 23 months, respectively. This resulted in 5- and 10-year survivals for pNET patients with localized disease of 71 and 52 %, respectively; for patients with regional disease 55 and 38 %, and for patients with distant metastases 23 and 9 % [45]. At initial presentation, the different pNETs vary in the percentage of patients with liver metastases, with a relative order of: insulinomas (5–15 %) < gastrinomas (20–35 %) < somatostatinomas < NF-pNETs, glucagonomas, adrenocorticotropic hormone (ACTH)-secreting pNETs (ACTHomas), and VIPomas (60–90 %). The marked effect of the presence of liver metastases is shown well by studies in patients with gastrinomas, where the 10-year disease-related survival in patients without liver metastases with or without lymph node metastases was 96 %, whereas for patients who developed liver metastases during follow up it was 85 % and for those with any liver metastases originally it was reduced to 26 % [47]. Furthermore, these studies also show the marked effect of the extent of the liver metastases on survival in patients with gastrinomas, with a 10-year survival of 96 % in patients without any metastases, 78 % in patients with liver metastases limited to one hepatic lobe, 80 % in those with limited metastatic disease in both lobes (<5 lesions), and 16 % in patients with diffuse metastases in both lobes [47, 48]. Unfortunately, only 3–15 % of patients in various series have limited liver metastases that might be surgically completely resectable and therefore specific treatments need to be directed at either the liver metastases or more distant disease in a significant proportion of pNET patients [52–54].
Staging, classification, and identification of prognostic factors prior to treatment
Recently a number of classification systems, which have prognostic value, have been proposed for pNETs as well as other NETs [6, 8, 9, 11, 13, 55, 56]. These include a WHO classification in 2004 [12]/2010 [57], a European Neuroendocrine Tumor Network (ENETN) classification in 2006 [58], and an International Union for Cancer Control/American Joint Cancer Committee/World Health Organization (UICC/AJCC/WHO) classification in 2010 [57, 59]. These classification systems propose TNM staging, which is based on primary pNET size and location, histological differentiation (well- or poorly differentiated), extent of the tumor (including local and distant metastases, and invasion), and the presence/absence of a hormonal syndrome, as well as tumor grading using a measure of proliferative activity (such as the Ki-67 index or mitotic index) [6, 8, 9, 11, 13, 55, 56]. The WHO classification distinguishes between well-differentiated and poorly differentiated pNETs, with the well-differentiated pNETs called neuroendocrine tumors (79–100 %) [44, 60–62], graded as G1 (Ki-67 ≤2 %) or G2 (Ki-67- 2–20 %), and poorly differentiated pNETs called neuroendocrine carcinomas (0–21 %) [44, 60–62] and graded as G3 (Ki67 >20 %) [57, 63]. In a number of studies [8, 11, 13, 62, 64], including one in Japan [65] the TNM staging has been shown to have important prognostic value for pNETs. Similarly, the tumor grade has been shown to have important prognostic value [64, 66] and is particularly important for treatment decisions, as discussed below, with well-differentiated pNETs treated differently from poorly differentiated tumors [7, 13, 60, 67, 68]. It is therefore important that, during the pretreatment evaluation, all patients with pNETs with advanced disease have a tumor biopsy and proper histological assessment of the pNET, allowing its proper classification [7, 9, 10, 63, 69].
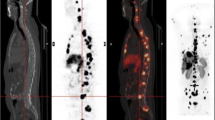
At present, as in the past, patients with pNETs with advanced disease, similar to all patients with pNETs, require assessment of the tumor extent and location for their proper management, and therefore imaging studies are required to choose the proper treatment [3, 14, 25, 26, 70]. Surgery remains the only potentially curative treatment modality and only thorough imaging studies can suggest whether this may be a treatment option in any pNET patient, including those with advanced disease [3, 14, 25, 26]. Initially in most centers a conventional imaging study [ultrasound, computed tomographic (CT) scan, magnetic resonance imaging (MRI) study, the latter two with contrast] is used to assess tumor extent and the possible location of the primary lesion [3, 14, 25, 26]. Increasingly the use of somatostatin receptor scintigraphy (SRS), which is based on the over/ectopic expression of somatostatin receptors by pNETs, is also routinely used, because numerous studies have demonstrated that SRS is more sensitive than conventional imaging studies for localizing pNETs, particularly for evaluating the extent of metastatic disease present [3, 14, 25, 26, 71, 72] (Fig. 1). An example of the increased sensitivity of SRS is shown in Fig. 1 in a patient with a previously resected pNET in whom, during a follow -up evaluation the CT scan did not show any recurrent pNET; however, SRS demonstrated metastatic disease both in the liver and in lymph nodes. Initially SRS included the widespread use of 111In-pentetreotide (Octreoscan; Mallinckrodt, Maryland Heights, MO, USA) [3, 14, 23, 25, 26, 71], and more recently there has been an increasing use of positron emission tomographic scanning using primarily 68Ga-radiolabeled somatostatin analogues [3, 15, 23, 25, 73–75]. The increased value of SRS has been shown in recent studies in which the use of SRS after conventional imaging studies changed the clinical management in 12–53 % (mean 30 %) of patients with pNETS [41, 73, 76, 77]. The SRS imaging result primarily affected the management of patients with advanced pNETs by providing information about the presence and density of somatostatin receptors in the pNET, which can affect the results/use of PRRT with radiolabeled somatostatin analogues (see section below on PRRT); the identification of distant metastases, especially to bone; and the identification of additional liver metastases or the identification of progression of metastatic disease either prior to or while the patient is receiving anti-tumor therapy [73–77]. The identification of bone metastases may have a particular effect on the management course, because these metastases are not infrequent in patients with pNETs with liver metastases (occurring in 33 % of pNET patients in one study) [78]; the presence of bone metastases is associated with a poor prognosis [47, 51] and is a contraindication to surgery generally; and their presence in certain sites many require specific directed therapies [32, 75]. Functional localization studies measuring hormonal gradients, which are used to localize functional pNET primaries not identified by other modalities, particularly after intra-arterial calcium stimulation for insulinomas or after secretin stimulation for gastrinomas, are only infrequently used for localizing metastatic disease in the liver [79–82].

Computed tomographic (CT) scan (top) and somatostatin receptor scintigraphy (SRS) (bottom) results in a patient with previously resected pancreatic neuroendocrine tumor (pNET). This patient previously (2 years prior) had a pNET resected and during follow up the CT scan was negative, as was the magnetic resonance imaging (MRI) scan and abdominal ultrasound (not shown); however, SRS performed with 111In-penetreotide (Octreoscan) demonstrated metastases both in the liver and in lymph nodes. These results illustrate the greater sensitivity of SRS over conventional imaging studies (CT, ultrasound, MRI) for detecting metastases in patients with malignant pNETs [3, 14, 25, 26, 71, 72]
Prior to and during the treatment of patients with advanced metastatic pNETs it is important to remember that the tumor behavior may differ markedly in different patients, which may affect the treatment approach. A number of clinical, laboratory, and histological factors, in addition to the tumor TNM classification and grading, are reported to have important prognostic value in patients with advanced disease and can be useful for planning treatment in different patients (Table 2). Poor prognosis is associated not only with the presence of liver metastases, but also with several features of liver metastases, including their extent, number, and rate of growth prior to treatment. A poor prognosis is also associated with the presence or development of bone or extrahepatic metastases; a new ectopic hormonal syndrome, particularly Cushing’s syndrome; various histological features that affect the grading/TNM classification, including high proliferative indices (Ki-67, mitotic count, poor differentiation) and the elevation of various tumor markers including alkaline phosphatase and chromogranin A (Table 2). Because the presence or the development of these prognostic factors in patients with pNETs with advanced disease can have an effect on clinical management they should be assessed in all patients both prior to and throughout their treatment protocols.
Specific treatments of patients with well-differentiated pNETs with advanced disease
Patients with advanced metastatic, hormonally active pNETs have two treatment problems that must be dealt with. Unfortunately in most cases no treatment, except for surgical cure, which is rarely possible, controls both of these problems and therefore they must both be considered. First, the hormone-excess state must be controlled because if inadequately treated, it can lead rapidly to complications and death. Next, therapy has to be directed at the metastatic pNET itself. In the case of NF-pNETs, which are not associated with a hormonal syndrome, the treatment can immediately be directed at the metastatic pNET itself.
It is now generally agreed that patients with well-differentiated pNETs (grades 1 or 2, Ki-67 ≤20) (termed neuroendocrine tumors) [63] should be treated differently from patients with poorly differentiated pNETs (termed neuroendocrine carcinomas) [63] (grade 3, Ki-67 >20) [7, 68, 83, 84]. Therefore, as discussed above, it is essential that a complete histological assessment of the tumor is available from a biopsy prior to treatment [9, 10, 69]. The current terminology of poorly differentiated pNETs includes tumors in the older literature described as high-grade neuroendocrine carcinomas, small cell carcinomas, undifferentiated carcinomas, anaplastic carcinomas, and large cell neuroendocrine carcinomas [63, 68, 83]. Poorly differentiated pNETs, similar to the behavior of poorly differentiated NETs in other sites, have a high occurrence of metastatic spread even in patients that appear to have localized disease, and therefore surgical resection is rarely curable [68, 83]. Furthermore, the medical treatment of poorly differentiated pNETs differs from that of advanced well-differentiated pNETs, in that the poorly differentiated pNETs are treated with cisplatin- and etoposide-based protocols as the initial therapy, which will be briefly discussed below [68, 83, 84].
Control of the hormone excess-state in the patient with advanced pNET
In a patient with a functional pNET it is essential to first control the hormone-excess state (VIPoma, insulinoma, Zollinger-Ellison syndrome, etc.) and to maintain control throughout all other anti-tumor treatments, because numerous studies demonstrate that if untreated or inadequately treated, the hormone-excess states are frequent causes of death [3, 25, 26, 41]. The acid hypersecretion in Zollinger-Ellison syndrome can be controlled by the use of parenteral or oral proton pump inhibitors, and if these are not available, high doses of histamine H2-receptor antagonists [3, 25, 85, 86]; the hypoglycemia in insulinoma can be controlled by frequent small feedings and the use of diazoxide, which controls the insulinoma hypersecretion by an effect on ATP-sensitive K+ channels [25, 26, 87]; and the hypersecretion of other hormones can be controlled by the use of short- and long-acting somatostatin analogues (Octreotide-LAR, Lanreotide-Autogel, Ipsen Pharma Biotech, Signes, France) [3, 25, 26]. Somatostatin analogues are also effective in some patients with insulinomas, although in others the hypoglycemia may be exacerbated because of the inhibition of counter-regulatory hormones [25, 88, 89]. Some newer methods have been recently described in refractory cases, especially in patients with malignant insulinomas and patients with insulinomas who cannot undergo surgical removal because of increased risk. These methods include ethanol ablation of the functional pNET [90–93]; the use of the mTOR inhibitor, everolimus, which has been effective at controlling the hypersecretory state as well as the tumor growth (see later section on Targeted therapy) [16, 17, 94–96]; and, recently, PRRT with radiolabeled somatostatin analogues has helped control the hypoglycemia in patients with malignant insulinomas in whom it was difficult to control the hypoglycemia [87, 97].
Surgical treatment of advanced pNETs
Specific tumoral resection
Surgical resection remains the only curative treatment for patients with pNETs and therefore it is generally recommended that it be carried out if all of the imaged disease or >90 % can be removed [25, 26, 52, 53, 96, 98]. Unfortunately, in the patients with liver metastases, <15 % of patients have limited disease in the liver which might be surgically completely resectable, and therefore other specific treatments need to be directed at either the liver metastases or more distant disease in a significant proportion of pNET patients [25, 52–54, 99, 100]. In general, debulking surgery is not recommended where resection cannot be complete or cannot result in the removal of >90 % of the metastatic tumor, because studies show this does not result in improved survival compared with that in patients who do not undergo debulking surgery [21, 25, 26, 98, 101, 102]. Furthermore, removal of the primary pNET in patients with unresectable liver metastases is not routinely recommended, because in patients with NF-pNETs, surgical resection of the primary tumor did not extend survival in patients with unresectable liver metastases [103]. Besides the small group of pNET patients with limited liver metastases, surgery is also playing an increasing role in patients with pNETs that were thought unresectable because of vascular involvement, not because of the presence of liver metastases. In one recent study [104], 17 % of 273 patients (46 patients) with pNETs had evidence on preoperative imaging studies of major vessel involvement (portal vein > superior mesenteric vein or artery > inferior vena cava > splenic vein) and were originally thought to be not resectable. At surgical exploration, in 91 % of the 46 patients the pNET could be surgically removed, with only 19 % requiring vascular reconstruction, resulting in 30 % remaining cured at 5-year follow up [104]. The authors of that study concluded that patients with pNETs with vascular abutment/invasion and even those with associated nodal or limited hepatic metastases should undergo surgical exploration [104]. This proposal is supported by a number of case reports and smaller series, including studies from Japan [105–109]. Particularly important in this group of patients are those in whom the pNET invades the splenic or mesenteric vein causing occlusion, which can result in portal hypertension, gastric/esophageal varices, and abdominal symptoms and/or severe upper gastrointestinal bleeding secondary to the varices [104]. In a number of studies the bleeding was completely resolved by resection of the pNET and the spleen [104, 110, 111].
Liver transplantation for advanced metastatic pNETs
In contrast to its non-use in most metastatic neoplasms, liver transplantation continues to be used in selected patients with metastatic pNETs confined to the liver [3, 29, 112–115]. However, its use for metastatic pNETs or other NETs remains controversial [21, 29, 115]. In a review in 2008 [116] of 85 patients with metastatic NETs (both pNETs [47 %] and carcinoids) who underwent hepatic transplantation in France, the overall 5-year survival rate was 47 %, the disease-free survival was 20 %, and the postoperative in-hospital mortality was 14 %. Independent factors for poor prognosis identified in this review [114] were an accompanying upper abdominal exenteration (relative risk [RR]; 3.7); primary NET in the duodenum or pancreas (RR = 2.93), and hepatomegaly (RR = 2.63). An analysis of the United Network for Organ Sharing database [21] between November 1988 and March 2011 identified 185 liver transplants for NETs in the United States; their overall 5-year survival was 58 %, which was less than the 74 % seen for all other patients transplanted. In a review [115] of reports from 24 monocentric series of liver transplantations for NETs involving 4–24 patients per center and 5 multicentric studies involving 30–103 patients per center, a number of risk factors were identified. The risk factors for a poor prognosis included, in a number of studies, the presence of extrahepatic metastatic disease at the time of the transplant; the performance of abdominal exenteration or multivisceral transplantation at the time of the liver transplant; the presence of a metastatic pNET rather than a metastatic GI carcinoid; age >50 years; the presence of extensive liver involvement (>50 %) compared with less involvement; and the presence of various histological features of the NET including a Ki-67 index of >10 % and abnormal E-cadherin staining. Because of the small percentage of patients who are disease-free after 5 years, the ENETS 2012 consensus guidelines for pNETs [29] conclude that liver transplantation should be viewed as providing palliative care and that cure remains the exception. It was therefore recommended [29] that liver transplantation be reserved for patients suffering life-threatening hormonal disturbances refractory to other treatments or patients with NF-pNETs with diffuse metastases refractory to all other treatments. Important selection criteria include a low Ki-67 (<10 %), normal E-cadherin in the tumor, no extrahepatic disease, the presence of a well-differentiated NET (grade G1 or G2), preferably age <50 years, and the transplantation performed without other concomitant large tumor resections [3, 21, 29, 30, 115].
Liver-directed strategies for treatment of advanced metastatic pNETs
Liver-directed strategies for hepatic metastases include various locoregional therapies including radiofrequency ablation (RFA), cryotherapy, hepatic arterial embolization, hepatic arterial chemoembolization, and hepatic arterial radioembolization [3, 29, 93, 117–119]. Similar to palliative liver surgery, there are no randomized trials of the comparative effectiveness of various hepatic locoregional therapies [29]. Therefore, the choice of which approach is used depends to a large degree on local expertise, but also on the location and extent of the liver metastases. Hepatic locoregional approaches are primarily considered in patients with metastatic pNETs with metastases limited to the liver or those patients having hepatic-predominant metastatic disease, especially in patients with functional pNETs in whom the hormone-excess state is not well controlled by other modalities [29, 119].
Liver-directed strategies: radiofrequency ablation and other locally ablative therapies
Locally ablative therapies (RFA, cryotherapy, ethanol injections) of metastatic liver tumor foci can be performed either using radiological techniques or at the time of surgery (laparoscopic/general surgery) [21, 29, 118, 120–123]. Of these therapies, RFA is the most commonly used and is increasingly used either alone or in combination with other treatments, particularly surgery [3, 21, 120–122, 124]. Various studies use different selection criteria, but factors that limit its effectiveness or are considered to be relative contraindications include large metastatic tumor deposits (>3.5–5 cm), large numbers of lesions (>5–15 lesions), and metastatic deposits near vital structures [3, 21, 29, 120–122, 124]. In one of the largest recent reports, involving 127 patients with metastatic NETs [122], of whom 69 were treated with RFA and the others by surgical resection (n = 29) or embolization (n = 29), RFA had the lowest complication rate, resulted in shorter hospital stays, and provided effective symptom relief in 91 % of the patients for a mean of 20 months. In other studies, response rates of 80–95 % are reported, with responses lasting up to 3 years [3, 21, 120, 123, 124]. RFA generally has low morbidity (<15 %), although, rarely, more serious complications can occur (bleeding, abscess formation) [3, 21, 120, 123]. In the NANETS 2010 guidelines and the ENETS 2012 guidelines [26, 29], it was stated that RFA’s effectiveness had not been established by any controlled study; however, it was stated that RFA could be an effective antitumor therapy and could be used for relieving symptoms in patients with pNET liver metastases as well as other metastatic NETs to the liver. Ablative therapies such as RFA [26, 29] were recommended for palliative therapy either in order to avoid a major surgical procedure or to effectively supplement a surgical procedure.
Liver-directed strategies: embolization and chemoembolization
Embolization and chemoembolization are based on the finding that pNET metastases in the liver are highly vascular and they derive their blood supply primarily from the hepatic artery (70–80 %), whereas normal liver tissue derives most of its blood supply from the portal vein [3, 29, 117, 125, 126]. Therefore, occlusion of the hepatic arterial supply to the tumor affects the tumoral metastases much more than the normal liver. Although this occlusion can be performed at surgery, it is primarily performed using interventional radiology via sequential intra-arterial catheterization with either embolization of hepatic transarterial branches (transarterial embolization; TAE) alone or with the co-administration of chemotherapeutic agents (transarterial chemoembolization; TACE, with the agents doxorubicin, 5-fluorouracil, cisplatin, mitomycin C, and streptozotocin being used [3, 93, 117, 125, 127]. Contra-indications to the use of TAE/TACE include hepatic involvement of >50–75 % by the tumor, portal venous occlusion, liver failure, post-surgical biliary reconstruction, and poor performance status [3, 21, 125, 128]. In various studies, 50–100 % of patients with malignant pNETs had a symptomatic response and 25–86 % had an objective tumor response, the mean duration of which was 6–45 months [93, 120, 125, 127, 129, 130]. Although improved survival was reported in some studies after TAE or TACE in patients who were not surgical candidates, there are no randomized studies that have demonstrated this [3, 93, 119, 128]. Both TAE and TACE can be associated with side-effects with a mortality rate of <6 % and complication rates of 10–80 %, including a post-embolization syndrome of abdominal pain, nausea/vomiting, and fever [21, 93, 117, 125, 131]. Serious complications rarely occur, but include abscess of the liver, gallbladder necrosis, hepatic failure, and renal failure [21, 93, 117, 125, 131]. In both the recent NANETS 2010 and ENETS 2012 guidelines [26, 29] it was concluded that either TAE or TACE should be considered for palliative treatment in patients with hepatic-predominant pNETs that are not surgically resectable, especially if they are symptomatic despite therapy; it was also concluded that TAE or TACE are effective at controlling symptoms in most patients and result in an objective tumor response in 50 %; and they should be performed only in experienced centers.
Liver-directed strategies: radio-embolization or selective internal radiation therapy (SIRT)
Radio-embolization or SIRT with 90Yttrium (90Y) microspheres is a relatively new treatment, and because of the limited number of patients treated, it was considered still investigational in the recent ENETS 2012 guidelines [29]. Whereas 90Y microspheres have been used for some time to treat patients with unresectable liver metastases from colorectal cancer and hepatocellular carcinoma [132, 133], increasingly SIRT is now being used in patients with unresectable pNETs and other neuroendocrine tumors. To date, more than 500 patients have been treated with 90Y microspheres in 16 different studies [133–144]. Two types of 90Y microspheres are currently used for the treatment of unresectable liver metastases: 90Y resin microspheres (SIR-spheres, Sirtex Medical, Inc., North Ryde, NSW, Australia), which have a 20- to 60-μm diameter and a load of approximately 50 Bq/sphere, and glass microspheres (TheraSpheres, Nordion (Canada), Ottawa, Canada), which have a 20- to 30-μm diameter with a radioactive content of 2500 Bq/sphere [132, 133, 135]. 90Y is a beta emitter with a half-life of 64.2 h and an average energy of 0.94 MeV, resulting in a tissue penetration of 2.5 mm and a maximum tissue range of 1.1 cm; therefore, the radiation administered in the liver is completely absorbed by the liver. Prior to the intra-arterial administration of the 90Y microspheres a pretherapeutic angiography is performed with the injection of 99mTc-labeled macroaggregates of albumin, to determine that the catheter tip is in the appropriate location and to avoid injection of the 90Y microspheres into the gastroduodenal or cystic arteries, which can result in gastrointestinal ulceration or cholecystitis, respectively, if the catheter is in the wrong location when the 90Y microspheres are injected [132, 133]. A second potentially serious complication that can occur is radiation pneumonitis due to the 90Y microspheres collecting in the lung because of hepatic-pulmonary shunting, which can occur in patients with advanced metastatic disease [132, 133]. The amount of shunting can be calculated from the results of the 99mTc-albumin pretherapeutic angiographic studies, and the dose of the administered 90Y microspheres can be appropriately adjusted to avoid radiation pneumonitis [132, 133]. The mean overall objective response rate (complete and partial responses) with 90Y microspheres reported from 12 studies including more than 400 patients with unresectable hepatic metastases from various NETs (including pNETs) was 55 % (range 12.5–89 %) and stable disease was seen in 32 % (range 10–60 %) [135]. In four studies in which symptomatic response was assessed, there was improved quality of life or amelioration of symptoms post- 90Y microspheres treatment, and the mean survival was 30 months [135, 137, 138, 140]. Contraindications to the use of SIRT include inadequate liver reserve, the presence of excess shunting (due to vascular abnormalities) to the gastrointestinal tract or to the lung, the inability to isolate the liver arterial tree from the gastric and small bowel branches, and the presence of a compromised portal vein [133, 142, 145]. In general, the side effects of SIRT are reported to be less severe than those of chemoembolization or embolization [133]. Grade 2 and 3 constitutional side-effects (weight loss, fatigue, fever) occur in 43 and 1 % of patients; gastrointestinal side-effects (nausea, vomiting, pain, ulceration) in 25 and 5 %; radiation pancreatitis in <1 %, radiation-induced liver disease in <1 %, and radiation pneumonitis in <1 % [133, 142, 145].
Medical treatment of advanced metastatic pNETs
Chemotherapy of advanced metastatic pNETs
In contrast to the treatment of patients with metastatic carcinoid tumors, chemotherapy continues to play an important role in the treatment of pNET patients with advanced metastatic disease [3, 29, 146, 147]. The recommended chemotherapeutic regimens differ for patients with metastatic pNETs with well-differentiated tumors and those with poorly differentiated tumors [3, 29, 68, 125]. In this section, the chemotherapy of well-differentiated pNETs will be briefly discussed and in a later section the treatment of poorly differentiated pNETs is covered. There is no complete agreement on when chemotherapy, in the realm of all pNET anti-tumor treatments, should be used. In almost all studies chemotherapy is reserved for patients with inoperable disease, usually those with diffuse liver metastases, and it has not been shown to be beneficial as adjuvant therapy post-resection of liver metastases [3, 29, 125, 147, 148]. Because of its frequent side-effects, increasingly chemotherapy is recommended if biotherapy or targeted therapy fails, if the tumor is rapidly growing, if the metastatic pNET is symptomatic, or if markers of poor prognosis (such as bone or distant extrahepatic metastases) are present [3, 29, 32, 47, 49, 149]. Chemotherapy in patients with G1–G2 well-differentiated metastatic pNETs generally involves combinations of streptozotocin and 5-fluorouracil, with or without doxorubicin, and has an objective response rate of 20–45 %; complete responses are rare and the median responses are generally short (6–20 months) [3, 29, 119, 125, 150]. Poor responses are reported in patients with extensive liver involvement (>75 % tumor replacement) and in those who have received prior chemotherapy [150]. Streptozotocin-based treatments have considerable morbidity, with 70–100 % of patients developing some side-effect, including nausea/vomiting (70–100 %); also, 15–40 % may develop some degree of renal toxicity with long-term treatment [3, 125, 150]. Recently, temozolomide combined with capecitabine has been reported to show promise in the treatment of advanced pNETs [151–154]. In a recent retrospective study involving 30 patients [151], a partial response rate of 70 % was reported when temozolomide and capecitabine was used as first-line treatment for metastatic well-differentiated pNETs; the median progression-free survival rate was 18 months, the 2-year survival rate was 92 %, and only 4/30 (13 %) patients experienced a grade 3 or 4 adverse event. The efficacy of temozolomide-based treated for pNETs is supported by the results of other studies of this treatment [152–154], as well as by a study of cellular mechanisms determining NET responsiveness to alkylating agents such as temozolomide [66]. In the study by Kulke et al. [154], low tumoral levels of the DNA repair enzyme O6-methylguanine DNA methyltransferase (MGMT) occurred in pNETs and were associated with a high response to alkylating agents in these tumors, similar to that reported in other tumors, whereas carcinoid tumors possessed high levels of MGMT and these high levels were associated with a low response rate (2 %). Both the ENETS 2012 [29] and the NANETS 2010 [26] guidelines recommend the use of chemotherapy in selected patients with advanced metastatic inoperable well-differentiated (G1 or G2) pNETs and especially in patients with advanced unresectable progressive tumors, particularly if rapidly growing, symptomatic, or large-volume disease is present.
Biotherapy of advanced metastatic pNETs
Biotherapy of advanced metastatic pNETs: treatment with somatostatin analogues
Increasing evidence supports the conclusion that somatostatin analogues are not only effective for controlling the hormone-excess state in functional pNETs, but that these analogues also have important anti-tumor growth effects [3, 26, 29, 155–159]. The basis for the use of somatostatin analogues in patients with pNETs, similar to their use in other NETs, is that these tumors overexpress at least one of the five subtypes of somatostatin receptors (sst1–5) in 70–100 % of patients; also, in numerous experimental models of various tumors, including NETs, somatostatin analogues have anti-growth effects [3, 155–159]. Both in patients with pNETs, as well as in those with other NETs, there have been numerous studies reporting the effects of somatostatin analogues on tumor growth; however, to date, in only one study [i.e., the PROMID (Placebo controlled, double-blind, prospective, Randomized study of the effect of Octreotide LAR in the control of tumor growth in patients with metastatic Midgut tumors) study], in patients with GI-NETs (metastatic midgut carcinoid tumors), has a randomized, placebo-controlled trial been completed [160], although such a trial is also being carried out in patients with pNETs using Lanreotide-Autogel (120 mg/month) [CLARINET (Controlled study of Lanreotide Antiproliferative Response in NETs) study], but the results are not yet reported [29, 161]. In the PROMID study [160], octreotide LAR significantly extended the time to tumor progression (14.3 vs. 6 months, p < 0.000072) resulting in 67 % of the octreotide-treated patients having stable disease at 6 months compared with 37 % of the controls (p = 0.0079). In this study [160], patients with functional (carcinoid syndrome) and nonfunctional metastatic midgut carcinoid tumors responded equally; however, there was no effect on overall survival, perhaps because of the low numbers of deaths in both groups. Detailed analysis of factors associated with response showed the tumor response was significant only in patients with low hepatic tumor load (<10 %) and was more favorable in patients with resected primary tumors [160]. In various studies reporting the effect of somatostatin analogues on pNET tumor extent, objective tumor responses with a decrease in pNET tumor size are uncommon, occurring in <10 % of all patients; however, tumor stabilization is frequent, occurring in 40–80 % of patients [3, 26, 29, 155, 157–159]. Some studies report the tumoristatic effect is more frequently seen in slow-growing pNETs with low proliferative rates; therefore, some recommend that patients with rapidly growing pNETs or those with higher proliferative indices be initially treated with other modalities [3, 29, 149, 157, 162, 163]. In some cases the tumoristatic effect (growth stabilization) can be long-lasting (>2 years) [149, 164, 165]. At present it is unclear by which exact mechanism(s) the antigrowth effects of somatostatin analogues are mediated in vivo, although different studies suggest these mechanisms could include the ability of these analogues to stimulate apoptosis, activate phosphatases, suppress the release of various stimulatory growth factors, inhibit the signaling of various growth factor receptors such as that through the insulin-like growth factor (IGF) 1 receptor, have immunomodulatory effects, and inhibit angiogenesis [166]. In general, with long-term treatment with somatostatin analogues, side-effects occur in 50 % of patients, but these effects are mild and uncommonly lead to cessation of therapy [41]. The most frequent side-effects are pain at the injection site, and gastrointestinal symptoms [15–20 %] that frequently resolve with prolonged treatment, with the latter effect perhaps being due to the motility effects of somatostatin analogues [41, 155, 157, 167]. Potentially more important long-term side-effects are the development of glucose intolerance/diabetes; steatorrhea, which is usually mild; and cholelithiases (with 10–80 % [mean 29 %] of patients developing biliary/gallbladder sludge, although only 1 % develop symptomatic gallbladder disease) [41, 167]. At present the exact role of somatostatin analogues for anti-growth effects in patients with pNETs is unclear because of the lack of data from a controlled trial. The ENETS 2012 guidelines conclude that somatostatin analogues may be of use in pNET patients with slowly proliferative metastatic disease, and therefore should be considered as a therapeutic option if tumors are G1 [29]. Similarly the NANETS 2010 guidelines conclude that somatostatin analogues are often first tried for their antiproliferative effects because of their low side-effect profile. Lastly, in the recent National Comprehensive Cancer Network (NCCN) guidelines, somatostatin analogues are included as one of the therapies to be considered (level 2B evidence) for patients with locoregional, unresectable, and/or metastatic pNETs [168, 169].
Biotherapy of advanced metastatic pNETs: treatment with interferon
Similar to somatostatin analogues, interferon is reported to be effective at controlling symptoms caused by the hormone-excess state of various functional PNETs, as well as having an anti-growth effect, which is primarily tumoristatic, resulting in tumor stabilization (30–80 %), rather than resulting in a decrease in tumor size (<15 % of cases) [3, 7, 157]. The anti-tumor effect of interferon is thought to be partially mediated by stimulating an increase in bcl-2, resulting in decreased proliferation, by blocking the cell-cycle progression in the G1 phase; by inhibiting angiogenesis by decreasing the expression of vascular endothelial growth factor (VEGF) and VEGF receptor (R); by up-regulating somatostatin receptors (sst2 subtype); and by stimulating the immune system [3, 7, 157]. Side-effects develop in almost all patients with interferon treatment, with the most frequent being a flu-like syndrome (80–97 %); followed by anorexia, weight loss (60 %), and fatigue (51 %). These side-effects frequently resolve with continued treatment. Other side-effects include bone-marrow toxicity (leucopenia, anemia, thrombocytopenia); hepatotoxicity (31 %); hyperlipidemia (31 %); autoimmune disorders, particularly thyroid disease (19 %); and, rarely, central nervous system (CNS) side-effects, including depression, mental disorders, and visual problems [3, 157]. The ENETS 2012 guidelines concluded that patients with low proliferating (G1), slowly progressive pNETs and those with somatostatin-negative tumors should be considered as candidates for interferon treatment; that interferon treatment should be avoided in patients with large hepatic burdens; that somatostatin analogue treatment has less severe side-effects and thus should be considered first; and that if interferon treatment is used it should be titrated individually so that the leukocyte count is reduced to approximately 3,000/µl [29, 167].
Targeted medical therapy of advanced metastatic pNETs
Targeted medical therapy: mTOR inhibitors (everolimus)
mTOR (Fig. 2) is a serine-threonine kinase that plays an important role in mediating proliferation, cell growth, and apoptosis in both normal and neoplastic tissues [2, 4, 170, 171]. Numerous in vitro and in vivo studies provide evidence that this molecule plays an important role in the growth of NETs, particularly pNETs [4, 16, 17, 146, 170–174].

Mammalian target of rapamycin (mTOR) pathway and everolimus. Activation of the PI3 K (phosphoinositde 3-kinase)-Akt pathway is observed in many types of cancers. This pathway is involved in cell growth and proliferation, through the serine-threonine kinase mTOR. mTOR acts as a central regulator of growth, proliferation, cellular metabolism, and angiogenesis. Everolimus is a targeted oral inhibitor of mTOR and demonstrates anti-tumor activity in pancreatic neuroendocrine tumors (pNETs), as shown in the figure. IGF-1 Insulin-like growth factor-1
Everolimus [RAD001, Affinitor, Novaritis AG, Basel Switzerland (Fig. 2)] is an orally active mTOR inhibitor that has been shown to have anti-growth effects in a number of studies involving pNETs [170, 175, 176], including a recent large, placebo-controlled, double-blind trial [RADIANT-3 (RAD001 In Advanced Neuroendocrine Tumors, Novaritis AG, Basel, Switzerland) trial] [17]. In this latter study [17], 410 patients with advanced, progressive pNETs were treated with everolimus (10 mg/day) or placebo, and everolimus demonstrated a significant improvement in progression-free survival (11 vs. 4.6 months, p < 0.0001) and increased by a factor of 3.7 the proportion of patients with progression-free survival at 18 months (37 vs. 9 % with placebo). A subgroup analysis [17] demonstrated that everolimus treatment was of benefit in different subgroups of patients, including those with or without previous anti-tumor treatments. Overall survival was not different between the two groups of patients; however, patients randomly assigned to placebo were able to cross-over to everolimus if the disease progressed, therefore limiting the ability to calculate the effect of everolimus alone on overall survival [16, 17]. A subsequent analysis [177] of 40 Japanese patients (23 treated with everolimus, 17 with placebo) included in the everolimus RADIANT-3 study described above [17], showed that everolimus treatment resulted in a significant 17-month improvement in progression-free survival, (19.45 vs. 2.8 months), and an 81 % reduction of progression/death (hazard ratio [HR] = 0.19, 95 % confidence interval [CI] 0.08–0.48, p < 0.001). In the RADIANT-3 study [17] everolimus caused a twofold increase in adverse events, with most events being grade 1 or 2, although grade 3 or 4 adverse events did occur. The most common grade 3 or 4 side-effects were hematological, diarrhea, stomatitis, or hyperglycemia (ranging from 3 to 7 %) [17]. The side-effects were generally manageable with dose reduction or drug interruption, or both [17].
This study [17] resulted in the approval of everolimus for use in both Europe and the United States in patients with pNETs that were unresectable or metastatic and well-differentiated. However, there is no agreement on the exact place or sequence of use of everolimus in the treatment of patients with advanced, unresectable pNETs. The ENETS 2012 [29] guidelines conclude that everolimus represents a novel therapeutic option in patients with surgically non-resectable pNETs after progression following chemotherapy. They conclude that, at present, everolimus, similarly to sunitinib (discussed below) should be considered as first-line therapy only in selected cases [29]. In contrast, the United States NCCN [168] and a recent review of treatment of patients with metastatic pNETs to the liver [21] listed the use of everolimus as a possible first-line treatment for unresectable well-differentiated pNETs.
Targeted medical therapy: tyrosine kinase inhibitor (sunitinib)
The tyrosine kinase receptors are a family of receptors consisting of more than 20 members which include the receptors for epidermal growth factor and related peptides (EGFR), IGFRs, platelet-derived growth factor receptors (PDGFRs), hepatocyte growth factor (c-MET), stem cell factor (receptor = c-KIT), VEGFRs, and a number of others. These receptors function as tyrosine kinases when activated, which results in the phosphorylation of numerous tyrosine kinase receptor residues which activate or function as docking sites for numerous intracellular molecules that are particularly important in mediating growth-related cascades, angiogenesis, apoptosis, and differentiation [172, 178].
Numerous studies have shown that pNETs, similar to other NETs, frequently possess a number of tyrosine kinase receptors and that these receptors can have important growth effects in these tumors [130, 178–184]. Because of this feature, a number of inhibitors of growth factor cascades have been developed, and these inhibitors, including monoclonal antibodies to growth factors or their receptors, as well as small molecule inhibitors of the receptor’s tyrosine kinase activity, show promise in the treatment of pNETs and other NETs [19, 178, 182]. In this section, results with the best-studied member of this class of inhibitors for pNETs (sunitinib) will be briefly reviewed and in a later section on future treatments, a number of others will be briefly discussed.
Sunitinib (SU11248; Sutent, Pfizer, New York, NY) is an orally active, small molecule inhibitor of the tyrosine kinase activity of PDGFRs, VEGFR-1, VEGFR-2, c-KIT, and FLT3 [178]. Numerous in vitro and in vivo studies, as well as Phase 2 studies [182, 185] and a recently completed international double-blind, randomized Phase 3 study [18], have demonstrated that sunitinib has anti-growth activity in pNETs. In a Phase 2 study [186] in Japanese patients, 12 patients with pNETs with unresectable/metastatic disease were enrolled and received 37.5 mg/day of sunitinib. The clinical benefit ratio was 75 %, including 42 % with a partial tumor response and 33 % with stable disease lasting for ≥24 weeks [186]. In the international double-blind, multicenter Phase 3 study [18], 171 patients with progressive malignant pNETs were randomly assigned to either sunitinib (37.5 mg/day) (n = 86) or placebo (n = 85). Sunitinib treatment resulted in a doubling of the progression-free survival (11.4 vs. 5.5 months for placebo, p < <0.001), an increase in the rate of objective tumor response (9 vs. 0 %, p = 0.007), and an increase in the overall survival [18]. Similar to everolimus, sunitinib was effective in various subgroups, including patients who had or had not previously received other anti-tumor treatments [16, 18]. Sunitinib treatment was associated with a threefold increased occurrence of side-effects compared with the control, although most side-effects were grade 1 or grade 2. However, some grade 3 or 4 adverse events did occur, with the most common being neutropenia (12 %), and hypertension (9.6 %) [18]. Despite these side-effects no difference was noted in a quality of life index with sunitinib treatment, and the side-effects were generally manageable with either dose reduction, cessation of treatment, or both [16, 18].
This Phase 3 study [18] resulted in the approval of sunitinib for use in both Europe and the United States in patients with well-differentiated pNETs that were unresectable or metastatic. However, there is no agreement on the exact place or sequence of use of sunitinib in the treatment of patients with advanced, unresectable pNETs. The ENETS 2012 [29] guidelines conclude that sunitinib, similar to everolimus, represents a novel therapeutic option in patients with surgically non-resectable pNETs after progression following chemotherapy. They conclude that, at present, everolimus/sunitinib should be considered as first-line therapy only in selected cases [29]. In contrast, the United States NCCN [168] and a recent review of treatment of patients with metastatic pNETs to the liver [21] listed the use of everolimus/sunitinib as a possible first-line treatment for unresectable well-differentiated pNETs.
Peptide receptor radionuclide therapy (PRRT) with radiolabeled somatostatin analogues
Peptide receptor radionuclide therapy (PRRT) with radiolabeled somatostatin analogues is based on the over/ectopic expression of somatostatin receptors by 60–100 % of pNETs, which allows targeting of a cytotoxic radiolabeled compound to the tumor [3, 22, 187–189]. Two different radiolabels are most frequently used: analogues labeled with 90Yttrium(90Y), which strongly emit β-particles, have a maximum energy of 2.27 MeV, maximal tissue penetration of 12 mm, and a half-life of 2.7 days; or somatostatin analogues labeled with 177Lutetium (177Lu), which emit β-particles and gamma rays, have a maximum energy of 0.5 MeV, maximal tissue penetration of 2 mm, and a half-life of 6.7 days [3, 22, 189]. A number of different somatostatin analogues and attached chelators to allow binding of the radioisotope have been used in various studies, with the most frequent chelators being DTPA (diethylene triamine penta-acetic acid) and DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetra-acetic acid) and with the most used peptide-chelator combinations being [DOTA0,Tyr3]octreotate (DOTATATE) or [DOTA0,Tyr3]octreotide(DOTATOC)[22, 189]. 90Y-[DOTA0,Tyr3]octreotide, 90Y-[DOTA0,Tyr3]octreotate, or 90Y-[DOTA0]lanreotide were examined in 10 studies involving more than 440 patients with various malignant NETs (pNETs, carcinoids), and these treatments resulted in complete tumor response occurring in 0–6 % of the patients, partial tumor regression in 7–37 %, and tumor stabilization (i.e., no additional growth) in 42–86 % [3, 22, 187, 189]. Results with 177Lu [DOTA0,Tyr3]octreotate were reported from 510 patients with various malignant NETs (40 % pNETs), with a complete response being found in 2 %, partial tumor regression in 28 %, minor tumor response in 16 %, and tumor stabilization in 35 % [190–192]. In one of these studies [190], in which tumor response was assessed in 310 patients with NETs (40 % pNETs), prognostic factors for any response with 177Lu-[DOTA0,Tyr3]octreotate were the presence of high uptake of the radioisotope by the tumor on the Octreoscan and a Karnofsky performance score of >70 [22, 190]. The median duration of an objective response was 46 months and the median disease-related survival was not reached (>48 months). Factors associated with decreased disease-specific survival after 177Lu -[DOTA0,Tyr3]octreotate treatment [190] included failure to achieve a tumor response and the presence of progressive disease (p < 0.001), extensive liver involvement compared with moderate/none (p < 0.001), Karnofsky index of ≤70 (p = 0.001), baseline weight loss (p = 0.001), the presence of bone metastases (p = 0.004), and the presence of a gastrinoma/insulinoma/VIPoma (p = 0.04). Side-effects did occur with PRRT but were usually mild [3, 22, 189, 190, 192]. Acute side-effects with such treatment (pain, vomiting, nausea) occurred in approximately 30 % of patients, but were usually mild and could be treated symptomatically [3, 22, 189, 190, 192]. More severe side-effects included hematological toxicity (15 % transient, 0.8 % developed a myelodysplastic disorder), liver toxicity (0.6 %), and renal toxicity, primarily with patients receiving 90Y-labeled somatostatin analogues (these side-effects could be limited by co-administration of an amino acid solution with the treatment) [3, 22, 189, 190, 192]. Although a number of analyses comparing the results of PRRT with those of other anti-tumor therapies for patients with unresectable metastatic NETs (pNETs and carcinoids) have suggested that PRRT is a promising therapy [190, 193, 194], no prospective, controlled study supports these conclusions at present, although one such study is now underway. Hence, in the NANETS 2010 [26], the ENETS 2012 [25], the Nordic 2010 [28], and the ESMO [27] guidelines, PRRT is listed as an experimental or investigational treatment.
Possible future treatments of patients with well-differentiated pNETs with advanced disease
Other strategies targeting the mTOR pathway
With basic science studies, as well as results showing the effectiveness of the mTOR inhibitor, everolimus, supporting the importance of the mTOR pathway in pNET pathogenesis and growth, a number of other newer therapies are also being aimed at this pathway. These include the possibility of overcoming resistance to the mTOR inhibitors that frequently emerges over time with continued treatment, in part due to the up-regulation of AKT via the IGF-1R/PI3K pathway, by down-regulating this effect with the simultaneous use of somatostatin analogues or the inhibition of IGF-1 signaling by administering the monoclonal antibody, cixutumumab. In turn, the PI3 K/AKT/mTOR pathway can be inhibited at different points by the use of kinase inhibitors such as BEZ235 or INC128 [19, 195]. In addition, another mTOR inhibitor (temsirolimus) shows promise and is being evaluated [172, 196], as well as atiprimod, a pro-apoptotic and anti-angiogenic compound which inhibits both the STAT3 and mTOR/AKT pathways [172].
Strategies targeting angiogenesis
pNETs, similar to other NETs, are highly vascular tumors [172, 179]. A number of studies report the effect of bevacizumab, a monoclonal antibody directed against VEGF, combined with other therapies (octreotide, temozolomide, capecitabine, everolimus) having some benefit in patients with various NETs (pNETs and carcinoids)[19, 119, 172, 178, 179, 197–199]. Thalidomide is an orally active immunomodulatory agent that causes inhibition of tumor necrosis factor (TNF)-alpha, but that also has anti-angiogenic activity, principally by inhibiting the VEGF and basic fibroblast growth factor (FGF) pathways [172]. It is being further evaluated in well-differentiated metastatic NETs, because it showed activity in one study when combined with temozolomide in the treatment of advanced NETs (carcinoids and pNETs) [172, 200]. The IGF-1R1 can be inhibited by MK-0646, a monoclonal antibody that blocks IGF-1R, or by AMG479, which is a human monoclonal antibody that antagonizes IGF-1R by inhibiting its interaction with IGF-1 or IGF-2; these two monoclonal antibodies are being evaluated in NETs [19, 173, 201].
Strategies targeting growth factor receptors using other tyrosine kinase inhibitors
In addition to sunitinib, whose results with pNETs are reviewed above, numerous other tyrosine kinase inhibitors show some activity in various NETs, including pNETs, and are undergoing additional studies. These include imatinib, with activity against bcr-abl, PDGFR, and c-KIT; sorafenib, an inhibitor of VEGFR-2, VEGFR-3, RAF, PDGFR, FLT-3, and c-KIT; vatalanib, an inhibitor of VEGF-1 (Flt-1), VEGFR-2 (FLK-1/KDR), and at higher concentrations c-KIT, PDGFR-beta, and c-FMS Tk; gefitinib (Iressa; Astra-Zeneca, Wilmington, DE) a small molecule inhibitor of the EGFR [172, 202]; and pazopanib, with activity against VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-alpha, PDGF-beta, and c-Kit [172, 178, 182, 198, 201];
Strategies targeting somatostatin receptors
Both octreotide and lanreotide, the currently approved somatostatin analogues used in the treatment of pNETs, have high affinity for only somatostatin receptor subtypes 2 and 5 (sst2, sst5). However, pNETs and other NETs frequently possess other somatostatin receptor subtypes [3, 156, 157, 188]. Pasireotide (SOM230) has high affinity for sst1, sst2, sst3, and sst5 [203] and it is being evaluated both for its possible enhanced anti-growth effects on NETs and for its antisecretory effects [19, 202].
Treatment of patients with poorly differentiated pNETs with advanced disease
Poorly differentiated pancreatic neuroendocrine tumors account for <1 % of all malignant pNETs and 2–3 % of all pNETs [68]. These high-grade malignancies are characterized by histological features characteristic of aggressive growth (Grade 3 with high Ki 67 index, >20 %, but usually 50–90 %; presence of necrosis; nuclear atypia), rapid growth, and poor clinical prognosis [8, 63, 66, 68, 83, 204]. Poorly differentiated pNETs differ from the well-differentiated pNETs discussed in the sections above not only in their biological behavior and prognosis, but also in a number of other features. These include: poorly differentiated pNETs frequently have low densities or the absence of somatostatin receptors, and therefore somatostatin receptor scintigraphy is rarely useful in these patients and somatostatin analogues are not used for their antiproliferative action in these tumors; on immunohistochemical staining, these poorly differentiated pNETs may lack chromogranin A; however, synaptophysin is usually present and is therefore is useful to help identify the tumor as a NET; and in up to 40 % of poorly differentiated pNETs, elements of non-neuroendocrine cancers may be present [63, 68, 83]. To image the extent of poorly differentiated pNETs, CT scanning, MRI scanning, and [18F]-fluorodeoxy-glucose positron emission tomographic scanning are usually used for both the initial staging and to monitor response to therapy [83]. Most patients with poorly differentiated pNETs have regional or distant metastases at diagnosis, and although surgery is rarely curative it should be considered in patients with limited disease at initial presentation [66, 68]. Systemic chemotherapy should be used in patients with inoperable disease if the patients have adequate performance status and no contra-indications [68, 83]. The recommended treatment is cisplatin-based drugs combined with etoposide either alone or in combination with other agents (vincristine, paclitaxel); such treatment induces remission in 14–80 % of patients, with a mean duration of response of <12 months [27, 68, 83, 84, 147, 205, 206]. This chemotherapy can be associated with major toxicity, especially myelosuppression and gastrointestinal toxicities (nausea/vomiting) [84, 206, 207].
Abbreviations
- ACTHomas:
-
ACTH-secreting pancreatic neuroendocrine tumors
- CNS:
-
Central nervous system
- ENETS:
-
European Neuroendocrine Tumor Society
- ESMO:
-
European Society for Medical Oncology
- GI:
-
Gastrointestinal
- GI-NETs:
-
Gastrointestinal neuroendocrine tumors (carcinoids of gastrointestinal tract)
- TAE/TACE:
-
Transarterial embolization/chemoembolization
- IGF:
-
Insulin-like growth factor
- UICC/AJCC/WHO:
-
International Union for Cancer Control/American Joint Cancer Committee/World Health Organization
- MRI:
-
Magnetic resonance imaging
- NANETS:
-
North American Neuroendocrine Tumor Society
- NCCN:
-
National Comprehensive Cancer Network
- NETs:
-
Neuroendocrine tumors (carcinoids, pancreatic neuroendocrine tumors)
- NF-pNETs:
-
Nonfunctional pancreatic neuroendocrine tumors
- pNETs:
-
Pancreatic neuroendocrine tumors
- PDGFRs:
-
Platelet-derived growth factor receptors
- PRRT:
-
Peptide receptor radionuclide therapy
- RFA:
-
Radiofrequency ablation
- SIRT:
-
Selective internal radiation
- SRS:
-
Somatostatin receptor scintigraphy
- VEGFR:
-
Vascular endothelial growth factor receptor
- VIPomas:
-
Vasoactive intestinal peptide-secreting pancreatic neuroendocrine tumors
References
Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61–72.
Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–203.
Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology. 2008;135:1469–92.
de Wilde RF, Edil BH, Hruban RH, Maitra A. Well-differentiated pancreatic neuroendocrine tumors: from genetics to therapy. Nat Rev Gastroenterol Hepatol. 2012;9:199–208.
Capurso G, Festa S, Valente R, et al. Molecular pathology and genetics of pancreatic endocrine tumours. J Mol Endocrinol. 2012;49:R37–50.
Oberg K. Neuroendocrine tumors of the digestive tract: impact of new classifications and new agents on therapeutic approaches. Curr Opin Oncol. 2012;24:433–40.
Oberg K. Pancreatic endocrine tumors. Semin Oncol. 2010;37:594–618.
Rindi G, Falconi M, Klersy C, et al. TNM staging of neoplasms of the endocrine pancreas: results from a large international cohort study. J Natl Cancer Inst. 2012;104:764–77.
Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39:707–12.
Klimstra DS, Modlin IR, Adsay NV, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am J Surg Pathol. 2010;34:300–13.
Pape UF, Jann H, Muller-Nordhorn J, et al. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer. 2008;113:256–65.
Kloppel G. Tumour biology and histopathology of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:15–31.
Bettini R, Boninsegna L, Mantovani W, et al. Prognostic factors at diagnosis and value of WHO classification in a mono-institutional series of 180 non-functioning pancreatic endocrine tumours. Ann Oncol. 2008;19:903–8.
Sundin A, Vullierme MP, Kaltsas G, Plockinger U. ENETS guidelines for the standards of care in patients with neuroendocrine tumours: radiological examinations in patients with neuroendocrine tumours. Neuroendocrinology. 2009;90:167–83.
Oberg K. Gallium-68 somatostatin receptor PET/CT: Is it time to replace (111)Indium DTPA octreotide for patients with neuroendocrine tumors? Endocrine. 2012;42:3–4.
Jensen RT, Delle Fave G. Promising advances in the treatment of malignant pancreatic endocrine tumors. N Engl J Med. 2011;364:564–5.
Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–23.
Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–13.
Pavel M. Translation of molecular pathways into clinical trials of neuroendocrine tumors. Neuroendocrinology. 2012 [Epub ahead of print].
Halperin DM, Kulke MH. Management of pancreatic neuroendocrine tumors. Gastroenterol Clin North Am. 2012;41:119–31.
Harring TR, Nguyen NT, Goss JA, O’Mahony CA. Treatment of liver metastases in patients with neuroendocrine tumors: a comprehensive review. Int J Hepatol. 2011;2011:154541.
van Vliet EI, Teunissen JJ, Kam BL, de Jong M, Krenning EP, Kwekkeboom DJ. Treatment of gastroenteropancreatic neuroendocrine tumors with peptide receptor radionuclide therapy. Neuroendocrinology. 2012 [Epub ahead of print].
Kwekkeboom DJ, Kam BL, Van Essen M, et al. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17:R53–73.
Kwekkeboom DJ, Krenning EP, Scheidhauer K, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: somatostatin receptor imaging with (111) In-pentetreotide. Neuroendocrinology. 2009;90:184–9.
Jensen RT, Cadiot G, Brandi ML, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology. 2012;95:98–119.
Kulke MH, Anthony LB, Bushnell DL, et al. NANETS treatment guidelines: well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas. 2010;39:735–52.
Oberg K, Jelic S. Neuroendocrine gastroenteropancreatic tumors: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20 Suppl 4:147–9.
Janson ET, Sorbye H, Welin S, et al. Nordic guidelines 2010 for diagnosis and treatment of gastroenteropancreatic neuroendocrine tumours. Acta Oncol. 2010;49:740–56.
Pavel M, Baudin E, Couvelard A, et al. ENETS consensus guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2012;95:157–76.
Steinmuller T, Kianmanesh R, Falconi M, et al. Consensus guidelines for the management of patients with liver metastases from digestive (neuro) endocrine tumors: foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2008;87:47–62.
Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378:2005–12.
Kos-Kudla B, O’Toole D, Falconi M, et al. ENETS consensus guidelines for the management of bone and lung metastases from neuroendocrine tumors. Neuroendocrinology. 2010;91:341–50.
Kianmanesh R, Ruszniewski P, Rindi G, et al. ENETS consensus guidelines for the management of peritoneal carcinomatosis from neuroendocrine tumors. Neuroendocrinology. 2010;91:333–40.
Krampitz GW, Norton JA, Poultsides GA, Visser B, Sun L, Jensen RT. Lymph nodes and survival in duodenal and pancreatic neuroendocrine tumors. Arch. Surg. 2012;(in press) (abstr).
Kulke MH, Bendell J, Kvols L, Picus J, Pommier R, Yao J. Evolving diagnostic and treatment strategies for pancreatic neuroendocrine tumors. J Hematol Oncol. 2011;4:29–37.
Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci. 1991;36:933–42.
Ito T, Sasano H, Tanaka M, et al. Epidemiological study of gastroenteropancreatic neuroendocrine tumors in Japan. J Gastroenterol. 2010;45:234–43.
Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–72.
Haynes AB, Deshpande V, Ingkakul T, et al. Implications of incidentally discovered, nonfunctioning pancreatic endocrine tumors: short-term and long-term patient outcomes. Arch Surg. 2011;146:534–8.
Gullo L, Migliori M, Falconi M, et al. Nonfunctioning pancreatic endocrine tumors: a multicenter clinical study. Am J Gastroenterol. 2003;98:2435–9.
Jensen RT. Endocrine neoplasms of the pancreas. In: Yamada T, Alpers DH, Kalloo AN, Kaplowitz N, Owyang C, editors. Textbook of gastroenterology. Oxford, England: Wiley-Blackwell; 2009. p. 1875–920.
Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;10:1727–33.
Zerbi A, Falconi M, Rindi G, et al. Clinicopathological features of pancreatic endocrine tumors: a prospective multicenter study in Italy of 297 sporadic cases. Am J Gastroenterol. 2010;105:1421–9.
Lepage C, Rachet B, Coleman MP. Survival from malignant digestive endocrine tumors in England and Wales: a population-based study. Gastroenterology. 2007;132:899–904.
Yao JC, Eisner MP, Leary C, et al. Population-based study of islet cell carcinoma. Ann Surg Oncol. 2007;14:3492–500.
Jensen RT. Natural history of digestive endocrine tumors. In: Mignon M, Colombel JF, editors. Recent advances in pathophysiology and management of inflammatory bowel diseases and digestive endocrine tumors. Paris, France: John Libbey Eurotext Publishing Co.; 1999. p. 192–219.
Yu F, Venzon DJ, Serrano J, et al. Prospective study of the clinical course, prognostic factors and survival in patients with longstanding Zollinger-Ellison syndrome. J Clin Oncol. 1999;17:615–30.
Weber HC, Venzon DJ, Lin JT, et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108:1637–49.
Sutliff VE, Doppman JL, Gibril F, et al. Growth of newly diagnosed, untreated metastatic gastrinomas and predictors of growth patterns. J Clin Oncol. 1997;15:2420–31.
Durante C, Boukheris H, Dromain C, et al. Prognostic factors influencing survival from metastatic (stage IV) gastroenteropancreatic well-differentiated endocrine carcinoma. Endocr Relat Cancer. 2009;16:585–97.
Panzuto F, Nasoni S, Falconi M, et al. Prognostic factors and survival in endocrine tumor patients:comparison between gastrointestinal and pancreatic localization. Endocr Relat Cancer. 2005;12:1083–92.
Sarmiento JM, Que FG. Hepatic surgery for metastases from neuroendocrine tumors. Surg Oncol Clin North Am. 2003;12:231–42.
Norton JA, Warren RS, Kelly MG, Zurek MB, Jensen RT. Aggressive surgery for metastatic liver neuroendocrine tumors. Surgery. 2003;134:1057–65.
Solorzano CC, Lee JE, Pisters PW, et al. Nonfunctioning islet cell carcinoma of the pancreas: survival results in a contemporary series of 163 patients. Surgery. 2001;130:1078–85.
Helle KB. Regulatory peptides from chromogranin A and secretogranin II. Cell Mol Neurobiol. 2010;30:1145–6.
Niederle MB, Hackl M, Kaserer K, Niederle B. Gastroenteropancreatic neuroendocrine tumours: the current incidence and staging based on the WHO and European Neuroendocrine Tumour Society classification: an analysis based on prospectively collected parameters. Endocr Relat Cancer. 2010;17:909–18.
Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO World Health Organization classification of tumors and genetics of the digestive system. Lyon, France: IARC press; 2010.
Rindi G, Kloppel G, Alhman H, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2006;449:395–401.
Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Manual. New York: Springer; 2010.
Panzuto F, Boninsegna L, Fazio N, et al. Metastatic and locally advanced pancreatic endocrine carcinomas: analysis of factors associated with disease progression. J Clin Oncol. 2011;29:2372–7.
Pomianowska E, Gladhaug IP, Grzyb K, et al. Survival following resection of pancreatic endocrine tumors: importance of R-status and the WHO and TNM classification systems. Scand J Gastroenterol. 2010;45:971–9.
Scarpa A, Mantovani W, Capelli P, et al. Pancreatic endocrine tumors: improved TNM staging and histopathological grading permit a clinically efficient prognostic stratification of patients. Mod Pathol. 2010;23:824–33.
Kloppel G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 2011;18 Suppl 1:S1–16.
Ekeblad S, Skogseid B, Dunder K, Oberg K, Eriksson B. Prognostic factors and survival in 324 patients with pancreatic endocrine tumor treated at a single institution. Clin Cancer Res. 2008;14:7798–803.
Tsuchiya A, Koizumi M, Ohtani H. World Health Organization Classification (2004)-based reevaluation of 95 nonfunctioning “malignant” pancreatic endocrine tumors reported in Japan. Surg Today. 2009;39:500–9.
Hentic O, Couvelard A, Rebours V, et al. Ki-67 index, tumor differentiation, and extent of liver involvement are independent prognostic factors in patients with liver metastases of digestive endocrine carcinomas. Endocr Relat Cancer. 2011;18:51–9.
Krausz Y, Freedman N, Rubinstein R, et al. (68)Ga-DOTA-NOC PET/CT imaging of neuroendocrine tumors: comparison with (111)In-DTPA-Octreotide (OctreoScan(R)). Mol Imaging Biol. 2011;13:583–93.
Nilsson O, Van Cutsem E, Delle Fave G, et al. Poorly differentiated carcinomas of the foregut (gastric, duodenal and pancreatic). Neuroendocrinology. 2006;84:212–5.
Kloppel G, Couvelard A, Perren A, et al. ENETS guidelines for the standards of care in patients with neuroendocrine tumors: towards a standardized approach to the diagnosis of gastroenteropancreatic neuroendocrine tumors and their prognostic stratification. Neuroendocrinology. 2009;90:166.
Krudy AG, Doppman JL, Jensen RT, et al. Localization of islet cell tumors by dynamic CT: comparison with plain CT, arteriography, sonography and venous sampling. Am J Roentgenol. 1984;143:585–9.
Gibril F, Reynolds JC, Doppman JL, et al. Somatostatin receptor scintigraphy: its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas: a prospective study. Ann Intern Med. 1996;125:26–34.
Gibril F, Jensen RT. Diagnostic uses of radiolabelled somatostatin-receptor analogues in gastroenteropancreatic endocrine tumors. Dig Liver Dis. 2004;36:S106–20.
Ruf J, Heuck F, Schiefer J, et al. Impact of Multiphase 68Ga-DOTATOC-PET/CT on therapy management in patients with neuroendocrine tumors. Neuroendocrinology. 2010;91:101–9.
Buchmann I, Henze M, Engelbrecht S, et al. Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2007;34:1617–26.
Putzer D, Gabriel M, Henninger B, et al. Bone metastases in patients with neuroendocrine tumor: 68Ga-DOTA-Tyr3-octreotide PET in comparison to CT and bone scintigraphy. J Nucl Med. 2009;50:1214–21.
Termanini B, Gibril F, Reynolds JC, et al. Value of somatostatin receptor scintigraphy: a prospective study in gastrinoma of its effect on clinical management. Gastroenterology. 1997;112:335–47.
Schillaci O, Spanu A, Scopinaro F, et al. Somatostatin receptor scintigraphy with 111In-pentetreotide in non-functioning gastroenteropancreatic neuroendocrine tumors. Int J Oncol. 2003;23:1687–95.
Gibril F, Doppman JL, Reynolds JC, et al. Bone metastases in patients with gastrinomas: a prospective study of bone scanning, somatostatin receptor scanning, and MRI in their detection, their frequency, location and effect of their detection on management. J Clin Oncol. 1998;16:1040–53.
Imamura M. Recent standardization of treatment strategy for pancreatic neuroendocrine tumors. World J Gastroenterol. 2010;16:4519–25.
Doppman JL, Miller DL, Chang R, et al. Gastrinomas: localization by means of selective intraarterial injection of secretin. Radiology. 1990;174:25–9.
Gibril F, Doppman JL, Chang R, Weber HC, Termanini B, Jensen RT. Metastatic gastrinomas: localization with selective arterial injection of secretin. Radiology. 1996;198:77–84.
Morganstein DL, Lewis DH, Jackson J, et al. The role of arterial stimulation and simultaneous venous sampling in addition to cross-sectional imaging for localisation of biochemically proven insulinoma. Eur Radiol. 2009;19:2467–73.
Strosberg JR, Coppola D, Klimstra DS, et al. The NANETS consensus guidelines for the diagnosis and management of poorly differentiated (high-grade) extrapulmonary neuroendocrine carcinomas. Pancreas. 2010;39:799–800.
Iwasa S, Morizane C, Okusaka T, et al. Cisplatin and etoposide as first-line chemotherapy for poorly differentiated neuroendocrine carcinoma of the hepatobiliary tract and pancreas. Jpn J Clin Oncol. 2010;40:313–8.
Osefo N, Ito T, Jensen RT. Gastric acid hypersecretory states: recent insights and advances. Curr Gastroenterol Rep. 2009;11:433–41.
Jensen RT, Niederle B, Mitry E, et al. Gastrinoma (duodenal and pancreatic). Neuroendocrinology. 2006;84:173–82.
de Herder WW, van SE, Kwekkeboom D, Feelders RA. New therapeutic options for metastatic malignant insulinomas. Clin Endocrinol (Oxf). 2011;75:277–84.
Vezzosi D, Bennet A, Rochaix P, et al. Octreotide in insulinoma patients: efficacy on hypoglycemia, relationships with Octreoscan scintigraphy and immunostaining with anti-sst2A and anti-sst5 antibodies. Eur J Endocrinol. 2005;152:757–67.
Vezzosi D, Bennet A, Courbon F, Caron P. Short- and long-term somatostatin analogue treatment in patients with hypoglycaemia related to endogenous hyperinsulinism. Clin Endocrinol (Oxf). 2008;68:904–11.
Vleggaar FP, Bij de Vaate EA, Valk GD, Leguit RJ, Siersema PD. Endoscopic ultrasound-guided ethanol ablation of a symptomatic sporadic insulinoma. Endoscopy. 2011;43 Suppl 2 UCTN:E328–9.
Levy MJ, Thompson GB, Topazian MD, Callstrom MR, Grant CS, Vella A. US-guided ethanol ablation of insulinomas: a new treatment option. Gastrointest Endosc. 2012;75:200–6.
Jurgensen C, Schuppan D, Neser F, Ernstberger J, Junghans U, Stolzel U. EUS-guided alcohol ablation of an insulinoma. Gastrointest Endosc. 2006;63:1059–62.
Kvols LK, Turaga KK, Strosberg J, Choi J. Role of interventional radiology in the treatment of patients with neuroendocrine metastases in the liver. J Natl Compr Canc Netw. 2009;7:765–72.
Fiebrich HB, Siemerink EJ, Brouwers AH, et al. Everolimus induces rapid plasma glucose normalization in insulinoma patients by effects on tumor as well as normal tissues. Oncologist. 2011;16:783–7.
Kulke MH, Bergsland EK, Yao JC. Glycemic control in patients with insulinoma treated with everolimus. N Engl J Med. 2009;360:195–7.
Ong GS, Henley DE, Hurley D, Turner JH, Claringbold PG, Fegan PG. Therapies for the medical management of persistent hypoglycaemia in two cases of inoperable malignant insulinoma. Eur J Endocrinol. 2010;162:1001–8.
Chandra P, Yarandi SS, Khazai N, Jacobs S, Umpierrez GE. Management of intractable hypoglycemia with Yttirum-90 radioembolization in a patient with malignant insulinoma. Am J Med Sci. 2010;340:414–7.
Falconi M, Bartsch DK, Eriksson B, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology. 2012;95:120–34.
Falconi M, Bettini R, Boninsegna L, Crippa S, Butturini G, Pederzoli P. Surgical strategy in the treatment of pancreatic neuroendocrine tumors. JOP. 2006;7:150–6.
Carty SE, Jensen RT, Norton JA. Prospective study of aggressive resection of metastatic pancreatic endocrine tumors. Surgery. 1992;112:1024–31.
Falconi M, Bassi C, Bonora A, et al. Role of chemoembolization in synchronous liver metastases from pancreatic endocrine tumours. Dig Surg. 1999;16:32–8.
Chamberlain RS, Canes D, Brown KT, et al. Hepatic neuroendocrine metastases: does intervention alter outcomes? J Am Coll Surg. 2000;190:432–45.
Bettini R, Mantovani W, Boninsegna L, et al. Primary tumour resection in metastatic nonfunctioning pancreatic endocrine carcinomas. Dig Liver Dis. 2009;41:49–55.
Norton JA, Harris EJ, Chen Y, et al. Pancreatic endocrine tumors with major vascular abutment, involvement, or encasement and indication for resection. Arch Surg. 2011;146:724–32.
Tsuchikawa T, Kondo S, Hirano S, et al. Distal pancreatectomy and portal vein resection without vascular reconstruction for endocrine tumors with massive intraportal growth: report of a case. Hepatogastroenterology. 2011;58:1029–31.
Hellman P, Andersson M, Rastad J, et al. Surgical strategy for large or malignant endocrine pancreatic tumors. World J Surg. 2000;24:1353–60.
Handa M, Nakada T, Kajitsuka S, Hirose M, Sato Y. Portosystemic A-V fistula and portal hypertension associated with islet-cell tumor of the pancreas. Nippon Geka Gakkai Zasshi. 1985;86:953–8.
Kawakami H, Kuwatani M, Hirano S, et al. Pancreatic endocrine tumors with intraductal growth into the main pancreatic duct and tumor thrombus within the portal vein: a case report and review of the literature. Intern Med. 2007;46:273–7.
Ochiai T, Masuda T, Nishizawa M, et al. Curative resection of a huge malignant pancreatic endocrine tumor by pancreatoduodenectomy with portal and superior mesenteric vein resection and reconstruction using the right ovarian vein: report of a case. Surg Today. 2011;41:1260–5.
Okuno M, Sakaguchi S, Nagayama M, et al. Nonfunctioning islet cell carcinoma presenting bleeding gastric varices and splenomegaly. Jpn J Surg. 1984;14:244–7.
Yamaguchi T, Takahashi H, Kagawa R, et al. Nonfunctioning pancreatic endocrine tumor presenting with hemorrhage from isolated gastric varices. Am Surg. 2005;71:1027–30.
Pascher A, Klupp J, Neuhaus P. Transplantation in the management of metastatic endocrine tumours. Best Pract Res Clin Gastroenterol. 2005;19:637–48.
Olausson M, Friman S, Herlenius G, et al. Orthotopic liver or multivisceral transplantation as treatment of metastatic neuroendocrine tumors. Liver Transpl. 2007;13:327–33.
Le Treut YP, Gregoire E, Belghiti J, et al. Predictors of long-term survival after liver transplantation for metastatic endocrine tumors: an 85-case French multicentric report. Am J Transplant. 2008;8:1205–13.
Gregoire E, Le Treut YP. Liver transplantation for primary or secondary endocrine tumors. Transpl Int. 2010;23:704–11.
Murthy R, Kamat P, Nunez R, et al. Yttrium-90 microsphere radioembolotherapy of hepatic metastatic neuroendocrine carcinomas after hepatic arterial embolization. J Vasc Interv Radiol. 2008;19:145–51.
Vogl TJ, Naguib NN, Zangos S, Eichler K, Hedayati A, Nour-Eldin NE. Liver metastases of neuroendocrine carcinomas: interventional treatment via transarterial embolization, chemoembolization and thermal ablation. Eur J Radiol. 2009;72:517–28.
Eriksson J, Stalberg P, Nilsson A, et al. Surgery and radiofrequency ablation for treatment of liver metastases from midgut and foregut carcinoids and endocrine pancreatic tumors. World J Surg. 2008;32:930–8.
Basuroy R, Srirajaskanthan R, Ramage JK. A multimodal approach to the management of neuroendocrine tumour liver metastases. Int J Hepatol. 2012;2012:819193.
O’Toole D, Ruszniewski P. Chemoembolization and other ablative therapies for liver metastases of gastrointestinal endocrine tumours. Best Pract Res Clin Gastroenterol. 2005;19:585–94.
Elias D, Goere D, Leroux G, et al. Combined liver surgery and RFA for patients with gastroenteropancreatic endocrine tumors presenting with more than 15 metastases to the liver. Eur J Surg Oncol. 2009;35:1092–7.
Karabulut K, Akyildiz HY, Lance C, et al. Multimodality treatment of neuroendocrine liver metastases. Surgery. 2011;150:316–25.
Akyildiz HY, Mitchell J, Milas M, Siperstein A, Berber E. Laparoscopic radiofrequency thermal ablation of neuroendocrine hepatic metastases: long-term follow-up. Surgery. 2010;148:1288–93.
Mazzaglia PJ, Berber E, Milas M, Siperstein AE. Laparoscopic radiofrequency ablation of neuroendocrine liver metastases: a 10-year experience evaluating predictors of survival. Surgery. 2007;142:10–9.
Toumpanakis C, Meyer T, Caplin ME. Cytotoxic treatment including embolization/chemoembolization for neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:131–44.
Gu P, Wu J, Newman E, Muggia F. Treatment of liver metastases in patients with neuroendocrine tumors of gastroesophageal and pancreatic origin. Int J Hepatol. 2012;2012:131659.
Reddy SK, Clary BM. Neuroendocrine liver metastases. Surg Clin North Am. 2010;90:853–61.
Nazario J, Gupta S. Transarterial liver-directed therapies of neuroendocrine hepatic metastases. Semin Oncol. 2010;37:118–26.
Osborne DA, Zervos EE, Strosberg J, et al. Improved outcome with cytoreduction versus embolization for symptomatic hepatic metastases of carcinoid and neuroendocrine tumors. Ann Surg Oncol. 2006;13:572–81.
Srirajaskanthan R, Toumpanakis C, Meyer T, Caplin ME. Review article: future therapies for management of metastatic gastroenteropancreatic neuroendocrine tumours. Aliment Pharmacol Ther. 2009;29:1143–54.
Lewis MA, Jaramillo S, Roberts L, Fleming CJ, Rubin J, Grothey A. Hepatic artery embolization for neuroendocrine tumors: postprocedural management and complications. Oncologist. 2012;17:725–31.
Deleporte A, Flamen P, Hendlisz A. State of the art: radiolabeled microspheres treatment for liver malignancies. Expert Opin Pharmacother. 2010;11:579–86.
Kennedy AS, Salem R. Radioembolization (yttrium-90 microspheres) for primary and metastatic hepatic malignancies. Cancer J. 2010;16:163–75.
Memon K, Lewandowski RJ, Mulcahy MF, et al. Radioembolization for neuroendocrine liver metastases: safety, imaging, and long-term outcomes. Int J Radiat Oncol Biol Phys. 2012;83:887–94.
Vyleta M, Coldwell D. Radioembolization in the treatment of neuroendocrine tumor metastases to the liver. Int J Hepatol. 2011;2011:785315.
Kennedy A, Coldwell D, Sangro B, Wasan H, Salem R. Integrating radioembolization into the treatment paradigm for metastatic neuroendocrine tumors in the liver. Am J Clin Oncol. 2012;35:293–301.
Paprottka PM, Hoffmann RT, Haug A, et al. Radioembolization of symptomatic, unresectable neuroendocrine hepatic metastases using yttrium-90 microspheres. Cardiovasc Intervent Radiol. 2012;35:334–42.
Lacin S, Oz I, Ozkan E, Kucuk O, Bilgic S. Intra-arterial treatment with 90yttrium microspheres in treatment-refractory and unresectable liver metastases of neuroendocrine tumors and the use of 111in-octreotide scintigraphy in the evaluation of treatment response. Cancer Biother Radiopharm. 2011;26:631–7.
Shaheen M, Hassanain M, Aljiffry M, et al. Predictors of response to radio-embolization (TheraSphere(R)) treatment of neuroendocrine liver metastasis. HPB (Oxford). 2012;14:60–6.
King J, Quinn R, Glenn DM, et al. Radioembolization with selective internal radiation microspheres for neuroendocrine liver metastases. Cancer. 2008;113:921–9.
Whitney R, Valek V, Fages JF, et al. Transarterial chemoembolization and selective internal radiation for the treatment of patients with metastatic neuroendocrine tumors: a comparison of efficacy and cost. Oncologist. 2011;16:594–601.
Kennedy AS, Dezarn WA, McNeillie P, et al. Radioembolization for unresectable neuroendocrine hepatic metastases using resin 90Y-microspheres: early results in 148 patients. Am J Clin Oncol. 2008;31:271–9.
Saxena A, Chua TC, Bester L, Kokandi A, Morris DL. Factors predicting response and survival after yttrium-90 radioembolization of unresectable neuroendocrine tumor liver metastases: a critical appraisal of 48 cases. Ann Surg. 2010;251:910–6.
Rhee TK, Lewandowski RJ, Liu DM, et al. 90Y Radioembolization for metastatic neuroendocrine liver tumors: preliminary results from a multi-institutional experience. Ann Surg. 2008;247:1029–35.
Lee E, Leon Pachter H, Sarpel U. Hepatic arterial embolization for the treatment of metastatic neuroendocrine tumors. Int. J Hepatol. 2012;2012:471203.
Riccardi F, Rizzo M, Festino L, et al. Therapy innovation for the treatment of pancreatic neuroendocrine tumors. Expert Opin Ther Targets. 2012;16 Suppl 2:S91–102.
Eriksson B, Annibale B, Bajetta E, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: chemotherapy in patients with neuroendocrine tumors. Neuroendocrinology. 2009;90:214–9.
Maire F, Hammel P, Kianmanesh R, et al. Is adjuvant therapy with streptozotocin and 5-fluorouracil useful after resection of liver metastases from digestive endocrine tumors? Surgery. 2009;145:69–75.
Hoffmann KM, Gibril F, Entsuah LK, Serrano J, Jensen RT. Patients with multiple endocrine neoplasia type 1 with gastrinomas have an increased risk of severe esophageal disease including stricture and the premalignant condition, Barrett’s esophagus. J Clin Endocrinol Metab. 2006;91:204–12.
Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–71.
Strosberg JR, Fine RL, Choi J, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer. 2011;117:268–75.
Maire F, Hammel P, Faivre S, et al. Temozolomide: a safe and effective treatment for malignant digestive endocrine tumors. Neuroendocrinology. 2009;90:67–72.
Ekeblad S, Sundin A, Janson ET, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986–91.
Kulke MH, Hornick JL, Frauenhoffer C, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res. 2009;15:338–45.
Sideris L, Dube P, Rinke A. Antitumor effects of somatostatin analogs in neuroendocrine tumors. Oncologist. 2012;17:747–55.
Appetecchia M, Baldelli R. Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumours, current aspects and new perspectives. J Exp Clin Cancer Res. 2010;29:19–31.
Plockinger U, Wiedenmann B. Biotherapy. Best Pract Res Clin Endocrinol Metab. 2007;21:145–62.
Strosberg J, Kvols L. Antiproliferative effect of somatostatin analogs in gastroenteropancreatic neuroendocrine tumors. World J Gastroenterol. 2010;16:2963–70.
Panzuto F, Di Francesco V, Iannicelli E, et al. Long-term clinical outcome of somatostatin analogues for treatment of progressive, metastatic, well-differentiated entero-pancreatic endocrine carcinoma. Ann Oncol. 2006;17:461–6.
Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–63.
Blumberg J, Liyanage N, Caplin M, UK and Ireland NET/ENET Society. The Clarinet study-assessing the effect of Lanreotide Autogel on tumor progression-free survival in patients with non-functioning gastroenteropancreatic neuroendocrine tumors (GEP-NETs). NANETS-2011-Neuroendocrine Tumor Symposium (abstr C4).
Oberg K, Eriksson B. Endocrine tumours of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753–81.
Butturini G, Bettini R, Missiaglia E, et al. Predictive factors of efficacy of the somatostatin analogue octreotide as first line therapy for advanced pancreatic endocrine carcinoma. Endocr Relat Cancer. 2006;13:1213–21.
Yamaguchi M, Yamada Y, Hosokawa Y, et al. Long-term suppressive effect of octreotide on progression of metastatic gastrinoma with multiple endocrine neoplasia type 1: seven-year follow up. Intern Med. 2010;49:1557–63.
Granberg D, Jacobsson H, Oberg K, Gustavsson J, Lehtihet M. Regression of a large malignant gastrinoma on treatment with Sandostatin LAR: a case report. Digestion. 2008;77:92–5.
Guillermet-Guibert J, Lahlou H, Pyronnet S, Bousquet C, Susini C. Somatostatin receptors as tools for diagnosis and therapy: Molecular aspects. Best Pract Res Clin Gastroenterol. 2005;19:535–51.
Oberg K, Ferone D, Kaltsas G, Knigge UP, Taal B, Plockinger U. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: biotherapy. Neuroendocrinology. 2009;90:209–13.
The NCCN clinical practice guidelines in oncology for neuroendocrine tumors version 1.2012. Version1.2012. 2012. online. go to www.nccn.org. Ref Type: Computer Program
Miljkovic MD, Girotra M, Abraham RR, Erlich RB. Novel medical therapies of recurrent and metastatic gastroenteropancreatic neuroendocrine tumors. Dig Dis Sci. 2012;57:9–18.
Yao JC. Molecular targeted therapy for carcinoid and islet-cell carcinoma. Best Pract Res Clin Endocrinol Metab. 2007;21:163–72.
Phan AT. Metastatic pancreatic neuroendocrine tumors (pNET): placing current findings into perspective. Cancer Treat Rev. 2012 [Epub ahead of print].
Capurso G, Fazio N, Festa S, Panzuto F, de Braud F, Delle Fave G. Molecular target therapy for gastroenteropancreatic endocrine tumours: biological rationale and clinical perspectives. Crit Rev Oncol Hematol. 2009;72:110–24.
Fazio N, Cinieri S, Lorizzo K, et al. Biological targeted therapies in patients with advanced enteropancreatic neuroendocrine carcinomas. Cancer Treat Rev. 2010;36 Suppl 3:S87–94.
Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245–55.
Varas M, Gornals J, Ponseti JM, et al. Pancreatic endocrine tumors or apudomas. Rev Esp Enferm Dig. 2011;103:184–90.
Yao JC, Phan AT, Chang DZ, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol. 2008;26:4311–8.
Ito T, Okusaka T, Ikeda M et al. Everolimus versus placebo in Japanese patients with advanced pancreatic neuroendocrine tumors (pNETs); Japanese subgroup analysis of RADIANT-3. J. Clin. Oncol. 2011;29:289 (abstr).
Raymond E, Hobday T, Castellano D, Reidy-Lagunes D, Garcia-Carbonero R, Carrato A. Therapy innovations: tyrosine kinase inhibitors for the treatment of pancreatic neuroendocrine tumors. Cancer Metastasis Rev. 2011;30 Suppl 1:19–26.
Faivre S, Sablin MP, Dreyer C, Raymond E. Novel anticancer agents in clinical trials for well-differentiated neuroendocrine tumors. Endocrinol Metab Clin North Am. 2010;39:811–26.
Furukawa M, Raffeld M, Mateo C, et al. Increased expression of insulin-like growth factor I (IGF-1) and/or its receptor (IGF-1R) in gastrinomas is associated with low curability, increased growth and development of metastases. Cancer Res. 2005;11:3233–42.
Peghini PL, Iwamoto M, Raffeld M, et al. Overexpression of epidermal growth factor and hepatocyte growth factor receptors in a proportion of gastrinomas correlates with aggressive growth and lower curability. Clin Cancer Res. 2002;8:2273–85.
Pavel ME, Wiedenmann B. Novel therapeutic agents for the treatment of gastroenteropancreatic neuroendocrine tumors. Horm Metab Res. 2011;43:844–53.
Capdevila J, Salazar R. Molecular targeted therapies in the treatment of gastroenteropancreatic neuroendocrine tumors. Target Oncol. 2009;4:287–96.
Fjallskog ML, Lejonklou MH, Oberg KE, Eriksson BK, Janson ET. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin Cancer Res. 2003;9:1469–73.
Kulke MH, Lenz HJ, Meropol NJ, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403–10.
Okusaka T, Ito T, Nishida T, et al. Phase 11 study of sunitinib (SU) in Japanese patients with unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors (NET). J. Clin. Oncol. 2012;30:381 (abstr).
Virgolini I, Traub T, Novotny C, et al. Experience with indium-111 and yttrium-90-labeled somatostatin analogs. Curr Pharm Des. 2002;8:1781–807.
Oberg K. Somatostatin analog octreotide LAR in gastro-entero-pancreatic tumors. Expert Rev Anticancer Ther. 2009;9:557–66.
Van Essen M, Krenning EP, Kam BL, de Jong M, Valkema R, Kwekkeboom DJ. Peptide-receptor radionuclide therapy for endocrine tumors. Nat Rev Endocrinol. 2009;5:382–93.
Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0, Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124–30.
Kwekkeboom DJ, Teunissen JJ, Bakker WH, et al. Radiolabeled somatostatin analog [177Lu-DOTA0, Tyr3]octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol. 2005;23:2754–62.
Forrer F, Valkema R, Kwekkeboom DJ, de Jong M, Krenning EP. Neuroendocrine tumors. Peptide receptor radionuclide therapy. Best Pract Res Clin Endocrinol Metab. 2007;21:111–29.
Kwekkeboom DJ, de Herder WW, Krenning EP. Somatostatin receptor-targeted radionuclide therapy in patients with gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:173–85.
Oberg KE, Reubi JC, Kwekkeboom DJ, Krenning EP. Role of somatostatins in gastroenteropancreatic neuroendocrine tumor development and therapy. Gastroenterology. 2010;139:742–53.
Dong M, Phan AT, Yao JC. New strategies for advanced neuroendocrine tumors in the era of targeted therapy. Clin Cancer Res. 2012;18:1830–6.
Duran I, Kortmansky J, Singh D, et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95:1148–54.
Yao JC, Phan A, Hoff PM, et al. Targeting vascular endothelial growth factor in advanced carcinoid tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26:1316–23.
Chan JA, Kulke MH. New treatment options for patients with advanced neuroendocrine tumors. Curr Treat Options Oncol. 2011;12:136–48.
Kulke MH, Siu LL, Tepper JE, et al. Future directions in the treatment of neuroendocrine tumors: consensus report of the national cancer institute neuroendocrine tumor clinical trials planning meeting. J Clin Oncol. 2011;29:934–43.
Kulke MH, Stuart K, Enzinger PC, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24:401–6.
Raut CP, Kulke MH. Targeted therapy in advanced well-differentiated neuroendocrine tumors. Oncologist. 2011;16:286–95.
Eriksson B. New drugs in neuroendocrine tumors: rising of new therapeutic philosophies? Curr Opin Oncol. 2010;22:381–6.
Schmid HA, Schoeffter P. Functional activity of the multiligand analog SOM230 at human recombinant somatostatin receptor subtypes supports its usefulness in neuroendocrine tumors. Neuroendocrinology. 2004;80 Suppl 1:47–50.
Schmitt AM, Anlauf M, Rousson V, et al. WHO 2004 criteria and CK19 are reliable prognostic markers in pancreatic endocrine tumors. Am J Surg Pathol. 2007;31:1677–82.
Ruf J, Schiefer J, Furth C, et al. 68Ga-DOTATOC PET/CT of neuroendocrine tumors: spotlight on the CT phases of a triple-phase protocol. J Nucl Med. 2011;52:697–704.
Hainsworth JD, Spigel DR, Litchy S, Greco FA. Phase II trial of paclitaxel, carboplatin, and etoposide in advanced poorly differentiated neuroendocrine carcinoma: a Minnie Pearl Cancer Research Network Study. J Clin Oncol. 2006;24:3548–54.
Olsen IH, Langer SW, Jepsen I, et al. First-line treatment of patients with disseminated poorly differentiated neuroendocrine carcinomas with carboplatin, etoposide, and vincristine: a single institution experience. Acta Oncol. 2012;51:97–100.
Clancy TE, Sengupta TP, Paulus J, Ahmed F, Duh MS, Kulke MH. Alkaline phosphatase predicts survival in patients with metastatic neuroendocrine tumors. Dig Dis Sci. 2006;51:877–84.
Lo CY, Van Heerden JA, Thompson GB, Grant CS, Soreide JA, Harmsen WS. Islet cell carcinoma of the pancreas. World J Surg. 1996;20:878–84.
Janson ET, Holmberg L, Stridsberg M, et al. Carcinoid tumors: analysis of prognostic factors and survival in 301 patients from a referral center. Ann Oncol. 1997;8:685–90.
Madeira I, Terris B, Voss M, et al. Prognostic factors in patients with endocrine tumours of the duodenopancreatic area. Gut. 1998;43:422–7.
Maton PN, Gardner JD, Jensen RT. Cushing’s syndrome in patients with Zollinger-Ellison syndrome. N Engl J Med. 1986;315:1–5.
Arnold R, Wilke A, Rinke A, et al. Plasma chromogranin a as marker for survival in patients with metastatic endocrine gastroenteropancreatic tumors. Clin Gastroenterol Hepatol. 2008;6:820–7.
Arnold R, Rinke A, Klose KJ, et al. Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: a randomized trial. Clin Gastroenterol Hepatol. 2005;3:761–71.
Strosberg J, Gardner N, Kvols L. Survival and prognostic factor analysis of 146 metastatic neuroendocrine tumors of the mid-gut. Neuroendocrinology. 2009;89:471–6.
Acknowledgments
This work was partially supported by intramural funds of NIDDK, NIH.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ito, T., Igarashi, H. & Jensen, R.T. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): recent insights and advances. J Gastroenterol 47, 941–960 (2012). https://doi.org/10.1007/s00535-012-0642-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-012-0642-8
Keywords
- Neuroendocrine tumor
- Pancreatic endocrine tumor
- Liver metastases
- Gastrinoma
- Insulinoma
- Surgery
- Chemotherapy
- Streptozotocin
- Everolimus
- Sunitinib
- Somatostatin
- Octreotide
- Lanreotide
- Hepatic transarterial embolization
- Liver transplantation
- Peptide receptor radionuclide therapy
- Somatostatin receptor
- SIRT
- Chemoembolization
- Unresectable liver metastases