
Abstract
The paper presents a neural network based multi-classifier system for the identification of Escherichia coli promoter sequences in strings of DNA. As each gene in DNA is preceded by a promoter sequence, the successful location of an E. coli promoter leads to the identification of the corresponding E. coli gene in the DNA sequence. A set of 324 known E. coli promoters and a set of 429 known non-promoter sequences were encoded using four different encoding methods. The encoded sequences were then used to train four different neural networks. The classification results of the four individual neural networks were then combined through an aggregation function, which used a variation of the logarithmic opinion pool method. The weights of this function were determined by a genetic algorithm. The multi-classifier system was then tested on 159 known promoter sequences and 171 non-promoter sequences not contained in the training set. The results obtained through this study proved that the same data set, when presented to neural networks in different forms, can provide slightly varying results. It also proves that when different opinions of more classifiers on the same input data are integrated within a multi-classifier system, we can obtain results that are better than the individual performances of the neural networks. The performances of our multi-classifier system outperform the results of other prediction systems for E. coli promoters developed so far.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Finding genes in DNA strings is a problem of critical importance to genomic research. The large quantities of new DNA sequences being continually produced all over the world demand fast, accurate methods for automatic analysis. One of the most important challenges in the analysis of new DNA sequences is whether or not they contain any genes, and if so, determining exactly where they are. A number of methodologies have been put forward for solving this problem. These include the use of neural networks [12, 13, 27, 29, 31], decision trees [25], genetic programming [9], hidden Markov models [2, 8, 10, 11], fuzzy logic [16, 30], etc. The main goal of learning in classification problems is generalization; that is, for the purpose of this paper, how to induce a concept that accurately classifies genes not included in the training set. The difficulty with achieving a good generalization capability is that a learner cannot directly measure the generalization ability; instead, the learner relies on its inductive bias to hopefully produce an accurate classifier.
Neural networks have extensively been used in bioinformatics. It originated with the use of the Perceptron model to find ribosome binding sites in amino acid sequences [28]. Recent examples include the PredictProtein program Footnote 1 and the GRAIL genefinder Footnote 2. Snyder and Stormo [27] successfully used the Perceptron algorithm to find Escherichia coli translational initiation sites. Multi-layer neural networks trained using the back-propagation algorithm have also extensively been used in bioinformatics. Recent examples include the systems developed by Ma et al. [12] for DNA-sequence classification and data mining.
E. coli promoters are located immediately before an E. coli gene. Thus, the successful localization of the E. coli promoter leads to the identification of the E. coli gene. We propose a neural network based multi-classifier system, multiple neural network based system for promoter recognition (MultiNNProm, Fig. 1), for the identification of these promoter sites. The proposed system contains four neural networks, to which the same DNA sequence is presented using four different encoding methods. The outputs of the individual neural networks are then passed through a probability function and finally combined by an aggregation function. This aggregation function aggregates the results, in order to provide an answer as to whether the presented sequence is an E. coli promoter or not.

General scheme of the neural network based multi-classifier for promoter recognition
Genetic algorithms are able to search a large space of candidate hypotheses to identify the best hypothesis. They have been proven to be useful in situations where an evaluation method to calculate the fitness of a candidate hypothesis is available. The combination (aggregation) function used in MultiNNProm combines the classification results of the individual classifiers by calculating the entropy of each output and then aggregating them through a weighted sum. The values of the aggregating weights were obtained by initializing a random population of weights and then running a genetic algorithm, to obtain an optimal set of weights, by using the classification accuracy of the combined classifier as fitness value.
2 E. coli promoter recognition
DNA, deoxyribonucleic acid, is found in long chains, with each link called a nucleotide. Each nucleotide in DNA consists of a sugar molecule called deoxyribose that bonds to a phosphate molecule and to a nitrogen containing compound, called a base. DNA uses four bases in its structure: adenine(A), cytosine(C), guanine(G) and thymine(T). The order of the bases in a DNA molecule—the genetic code—determines the amino acid sequence of a protein. Genes are actually sections of a cell’s DNA. While every gene is made up of DNA bases, the entire DNA of an individual is not made up of genes only. Ironically, only two to three percent of human DNA is made up of genes (protein coding regions).
The Bioinformatics Initiative Footnote 3 lists a set of reasons as to why E. coli is currently the organism of choice for such studies, both as a reference organism for many studies on prokaryotic systems and as a source of information on proteins and metabolic pathways that are shared by eukaryotes as well.
A promoter is a region in the DNA where the transcription initiation takes place. In prokaryotes, the sequence of a promoter is recognized by the sigma (σ) factor of the RNA polymerase. E. coli promoters consist of two sites, known as binding sites. These are the locations to which E. coli polymerase, a kind of protein, binds onto in order to begin the transcription of the protein. The two binding sites are always located at the points known as the −35 hexamer box and the −10 hexamer box. These two names are derived from the statistical fact that these sites usually occur 35 positions and 10 positions, respectively, from the end of the promoter (Fig. 2) [6, 18].

The E. coli promoter, showing the locations of the −35 and −10 hexamer boxes
The spacer between the −10 hexamer box and the transcriptional initiation site has a variable length, the most probable length being 7. The spacer between the −10 site and the −35 site is also of variable length, and can vary between 15 and 21 bases. It is this variation that can make the recognition of these promoters difficult using traditional methodologies.
The E. coli promoter can have one of four σ factors. These are listed out in Table 1. The table also lists out the consensus sequences Footnote 4 for the recognition of these sites. A promoter should contain an element which is very close to, or identical to the consensus sequence. In E. coli, the RNA polymerase holoenzyme (with σ factor) contains all the information needed to accurately initiate transcription, given proper promoter signals (−35 recognition sequence plus −10 Pribnow box).
Ma et al. [12] used the software available at http://www-lecb.ncifcrf.gov/~toms/delia.html to display the sequence logos of 438 E. coli promoters that were aligned according to their transcriptional start sites (Fig. 3). This software used the Shannon Entropy to independently measure the non-randomness of each position. From Fig. 3, it can be seen that strong motifs exist at positions −35 and −10, and weak motifs exist at positions +1, −22, −29 and −44.

The sequence logos of 438 E. coli promoters
The location of an E. coli promoter is immediately before the E. coli gene. We are then interested in successfully localizing the E. coli promoter, to identify the position of the E. coli gene. Though the uncertainty of E. coli promoter characteristics adds to the difficulty of recognition, the above mentioned obvious and unobvious features can be employed to make the recognition possible.
3 Multiple classifiers
3.1 Why use multiple classifiers?
Individual classifier models are recently being challenged by combined pattern recognition systems, which show better performances than individual classifiers. The individual classifiers within a multi-classifier system complement each other and, finally, a better result is provided. Additionally, when a system incorporates more classifiers based on different methodologies, the inadequacies of one methodology may be nullified by the advantages offered by another methodology.
Thus, genes not recognized by one classifier will be recognized by another, and DNA falsely classified as being genes by one classifier will be rejected by others (Fig. 4). In Fig. 4, the shaded region represents the true positives, and the ellipses represent the space classified by each classifier as being true positives.

A classification problem with different classifiers being used for the same data set
The challenge in building such a system is the combination of the results provided by each classifier, in order to come up with the optimal result. The performance of the entire system can be proven to be never worse than that of the best individual expert. Dietterich [5] recently indicated that using an ensemble of classifiers we can achieve a better recognition than using a single classifier when (1) the recognition rate of each individual classifier of the ensemble is greater than 0.5, and (2) errors made by each individual classifier are uncorrelated.
In a situation where more classifiers are available, the simplest approach would be to select the best performing classifier and use it for the classification task [17]. This approach, although simple and easy to implement, does not guarantee a good performance [22]. It is highly probable that a combination of classifiers would outperform a singular classifier [24]. On the other hand, different and worse performing classifiers might only add to the complexity of the problem and provide even worse results than the worst classifier. Thus, it is a well-known fact that, if a multi-classifier system is to be successful, the different classifiers should have good individual performances and be sufficiently different from each other [26]. But, neither individual performances [21, 34], nor the diversity [24], provide an ideal measure of how successful the combination of the classifiers will be.
As explained in [24], the core element of classifier selection is the selection criteria. The most natural choice is the combined performance, which will also be the criterion for the selection of the combiner. The only drawback of this methodology is the exponential complexity of testing out all possible combinations of a given set of classifiers.
It has been proven that the usage of an ensemble of neural networks, for certain classification problems, can improve the classification performance when compared to the use of singular neural networks. Rost and Sander [23], and Riis and Krogh [20], provide results tested on protein secondary structure prediction. Baldi and Brunak [1] also list out an overview of applications in molecular biology.
Among different methodologies used for combining multiple neural network classifiers, the majority voting, neural networks, Bayesian inference and the Dempster–Shafer theory have proven to be the most popular ones [15, 32, 33]. The Dempster–Schafer method has proven to be successful, but has a considerable dependency on the function used for the alignment of probability. For the type of output produced by neural networks (numerical values), posterior class-conditional probabilities can be calculated. The calculation of these probabilities becomes relatively simple, especially when the number of output classes is small.
In the next section of the paper, we present two methods that can be used for the combination of results obtained through multiple neural network classifications. These two methods, the linear average predictor (LAP) and logarithmic opinion pool (LOP) methods, were introduced in [7]. We propose a modification of the LOP method—LOP2—which proved to be more adequate and produced better results when using genetic algorithms.
3.2 The LAP and LOP methods for combining classifiers
The method that we used for the combination of our four classifiers is a variation of the LOP method [7], called LOP2. We compare this method with the more commonly used LAP method. The LOP method is a general ensemble method which shows how to calculate the error of an ensemble using the individual errors of different classifiers (called the ensemble ambiguity). Hansen and Krogh [7] claimed that ensembles always improve the average performance.
An ensemble consists of M predictors f i , which are combined into the combined predictor F. Let each predictor produce an output in the form of a probability vector {f 1 i ,...,f N i }, where f j i (i=1,...,M) is the estimated probability that the input \(\vec x\) belongs to the class c j , j=1,...,N (N is the number of classes). There is also a vector of coefficients of the form {α 1,...,α M }, associated with each ensemble, where ∑Mi=1 α i =1, α i ≥ 0.
3.2.1 The LAP method
The combined predictor for the LAP method is defined as:
3.2.2 The LOP method
The combined predictor for the LOP method is defined by (2), where Z is the normalization factor defined by (3). This method is non-linear and asymmetric in comparison to the LAP method. A comparison of the results obtained using the two methods is given in Sect. 5.
3.2.3 The LOP2 method
The LOP combination method is changed by allowing the sum of coefficients α 1,...,α M to be different than 1, with α i also being allowed to take on negative values. We found that this small modification allowed the system to nullify small errors created by the more successful classifiers.
Genetic algorithms are used to search for the right coefficients α i for aggregating the classification results of the individual classifiers, as described in Sects. 4 and 5. Using the LOP method, where the sum of coefficients is always one, would pose very difficult problems in implementing the genetic algorithms, e.g. crossover, and would result in searching only a small part of the search space (the surface where the sum of coefficients is one).
4 Description of the MultiNNProm system
4.1 Overview of the system
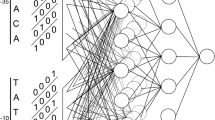
As shown in Fig. 5, the system is a neural network based multi-classifier system. We developed four neural networks, called NNE1, NNE2, NNC2 and NNC4, into which the same DNA sequence is inputted using four different encoding methods: E1, E2, C2 and C4 (see Table 2), respectively. The outputs of the individual neural networks are then passed onto a probability builder function, which assigns probabilities as to whether the presented sequence is an E. coli promoter or not. Finally, the outputs of the probability functions are aggregated by a result combiner, which combines the results and produces a final result as to whether the given sequence is an E. coli promoter or not. The final output is of the ‘yes’ or ‘no’ form.

MultiNNProm architecture
4.2 Data set
The entire data set consisted of 483 E. coli promoter sequences and 600 non-promoter sequences. The positive data sequences were obtained from the PromEC Footnote 5 Web site. Each of the obtained E. coli sequences were 101 nucleotides in length and were aligned at the transcriptional start site. Each of the transcriptional start sites were positioned such that they each appeared at position 76 of the string. Thus, each of the positive sequences consisted of 101 nucleotides, starting from the −75 position, more exactly 75 nucleotides upstream of the transcriptional start site, and ending at the +25 position, which is 25 nucleotides downstream of the transcriptional start site. The negative data set was obtained from the database compiled by the E. coli GenomeProject, at the University of Wisconsin-Madison Footnote 6. The negative dataset consists of E. coli genes with the preceding promoter region deleted.
Using the given set of available data, we constructed three data sets: a training sequence, a positive test sequence and a negative test sequence. The training set consisted of 324 positive sequences and 429 negative sequences. The remaining positive and negative data were divided into a positive test set of 159 sequences and a negative test set of 171 sequences.
4.3 The encoding methods
As mentioned in Sect. 4.1, four encoding methods (E1, E2, C2 and C4) were used. A description of these encoding methods is outlined in Table 2. The networks were trained to produce an output of ‘−1’ if the DNA sequence was not deemed to be a promoter, and an output of ‘1’ if the DNA sequence was deemed to be a promoter.
By using different encoding methods, we have obtained slightly varying results with respect to the genes correctly classified, as shown in Sect. 5. The underlying conclusion was that each encoding method helped the network specialize in different types of promoters present within the training data set.
4.4 The individual neural network classifiers
We developed four neural networks, each formatted to accept one of the encoding methods mentioned above. Each neural network was trained on the training set using batch training with resilient Backpropagation [19]. Different configurations of hidden layers were considered. It was observed that for each configuration (number of layers), the accuracy of the system on the testing set had not increased with the number of neurons on the hidden layer.
It was also observed that the average accuracy improved as the number of hidden layers increased. Thus, it was concluded that, while the accuracy of the system does not increase beyond a certain threshold with an increase in the number of neurons in the hidden layer, it does improve with an increase in the number of hidden layers of the neural network. We accredited this increase in performance, along with the increase in the number of hidden layers, to the complexity of the DNA strings. We concluded that neural networks with a higher number of hidden layers were able to recognize relationships embedded in the DNA string of a higher order than networks with one or two hidden layers. That is, they were able to correctly learn and classify second and third order relationships among the bases of the DNA string, which in turn led to better generalization and more genes being correctly classified.
Taking a compromise between the size of the neural network and the time required to train the neural network, it was finally concluded that the most suited configuration in terms of accuracy would be a 3-hidden layer neural network. The number of neurons in the input layer is equal to the number of parameters presented to the system; in this case, the number of nucleotides that were presented to the system in one sequence. This technique was used in each of the four neural networks implementing the encoding methods given in Table 2. The configurations of the four neural networks are listed out in Table 3. Each network was trained until the mean squared error (MSE) reached a value below 1e-7. Starting with random weights, ten runs were performed on each network , and the network with the best performance was selected. Each trained neural network exhibited perfect performance (100% accuracy) on both the positive and negative data on the training set.
Another remarkable observation encountered with the trained system was that the values of the weights on the input layer. Figure 6 displays the distribution of the weights between the input layer and the first hidden layer of the neural network trained on the E1 encoding scheme. The distribution shows remarkable equivalence to the sequence logo graph generated by Ma et al. [12], from Fig. 3, in terms of where the emphasis on the inputs is the largest. This means the network automatically learns the consensus sequences and we don’t need to manually embed the motif information within the system.

The weights between the first and the second hidden layer in a neural network trained on the E1 encoding scheme
4.5 The probability function
Due to the fact that the LOP method requires the input to be in a probability vector form (see Sect. 3.2), we need to convert the output of each neural network into such a form. The probability function assigns a probability as to whether the given sequence is an E. coli promoter or not; in our case N=2, as we have two classes: ‘promoter’ and ‘non-promoter’. The probability functions used to calculate the probabilities for positive and negative data are defined in (4 and 5), where y is the value of the network output.
If the output of the neural network is 1 or more than 1, we deemed the given sequence to be an E. coli promoter. If the output of the neural network is −1 or less than −1, we concluded the given sequence as not being an E. coli promoter. We assigned a probability of 0.999 to either the ‘Yes’ value or the ‘No’ value in such a case, due to the use of the logarithm within the result combiner.
4.6 Result aggregation
The outputs of the four neural networks were combined using the LOP2 method described in Sect. 3.2. The system was tested on three combination methods, namely, classic majority voting, LAP and LOP2. The resulting observations showed us that the results obtained through the LOP2 method provided a much better recognition rate for the test data, both positive and negative. The comparison between these three aggregation methods is listed out in Sect. 5.
The LOP2 method was implemented as follows. Let the outputs of the four neural networks be symbolized by O i , where 1≤ i ≤4. We also defined four coefficients α i , where 1≤ i ≤4. Then, the combined predictor is defined by (6—8).
O.Positive is the probability of the given sequence being a promoter, and is calculated by the aggregation function of the combined system, whereas O.Negative is the probability of the given sequence being a non-promoter. It is obvious that O.Positive+O.Negative=1. The final conclusion on whether the given sequence is an E. coli promoter or not is reached using (9).
So, if O.Positive is greater than O.Negative, the given sequence is classified as a promoter, otherwise as a non-promoter. The methodology used to determine the values of the aggregating coefficients is described in the next section.
4.7 Determination of the aggregating coefficients using genetic algorithms
Genetic algorithms (GAs) are especially effective in searching large and difficult solution spaces, where other searching methods, such as gradient based methods, fail. The search space associated with the coefficients for combining more individual classifiers in our problem, though not an extremely large space, can be a good problem for GAs.
We initialized a set of ten populations, each containing 60 random chromosomes with real values ranging from −10 to 10. Each random chromosome corresponded to a vector representing the four weights utilized within the aggregating function. Each of these populations were then exercised through a genetic algorithm, iterating through 60 generations. The six best performing chromosomes of each population were then extracted and a new population was created. This new population was further exercised through a genetic algorithm for a further 60 generations. The best performing chromosomes of the resulting population were then selected to implement the aggregating function.
The fitness function utilized for the selection of chromosomes is the classification error exhibited by the multi-classifier system on the testing dataset. This function calculated the percentage of erroneous identifications Footnote 7 with respect to the total number of testing data presented. Thus, the genetic algorithm attempted to minimize the total error displayed by the system with respect to the test data. The genetic algorithm was implemented using a precision of 0.9, which meant that the six best performing chromosomes were included in their whole within the next generation. Also, a crossover probability of 0.7 and a mutation probability of 0.1 were used along with roulette-wheel selection for its implementation.
5 Experimental results
5.1 Evaluation of results
In order to evaluate the predictive accuracy of a gene-finding program, we need to compare the promoters (exons) predicted by the program with the actual coding promoters. From this comparison, we calculate the nucleotide level and exon (promoter) level accuracy measures.
Let us denote the following values for true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN):
- NTP::
-
the number of coding nucleotides predicted as coding,
- NTN::
-
the number of non-coding nucleotides predicted as non-coding,
- NFP::
-
the number of coding nucleotides predicted as non-coding,
- NFN::
-
the number of non-coding nucleotides predicted as coding.
Then, we define the sensitivity (10) as the proportion of coding nucleotides that are correctly predicted as coding, and the specificity (11) as the proportion of nucleotides that are actually coding and are predicted as coding.
Here, NNG , NPO signify the total number of negative and positive sequences, respectively. Both of these are widely used measures for the evaluation of the accuracy of gene prediction programs. Both Sp and Sn range independently over [0, 1], with perfect prediction occurring only when both measures are equal to 1. Another measure that is used for the evaluation of the performance of neural networks is precision (12), where C and N denote the number of test sequences correctly classified and the total number of sequences presented, respectively.
5.2 Performance evaluation of individual classifiers
Each neural network performed perfectly on the training data set and displayed a recognition rate of 100% when presented with them after training. The test sequences were exercised through the four neural networks and the results obtained are listed out in Table 4. These performances are summarized in Fig. 7. The results obtained were then used to calculate the specificity, sensitivity and precision of the four neural network classifiers. These results are tabulated in Table 5, and show us that the best performance is exhibited by NNC4, whereas the worst performance is exhibited by NNE2.

True positives and true negatives recognized by the individual neural networks
5.3 Performance evaluation of the combined system
The classification performances, including the specificity, sensitivity and precision of the combined system using the LOP2 method, are listed out in Tables 6 and 7. These results are compared, in Fig. 8, with the results obtained for the individual neural networks. It is clear that the combination of these four neural networks provides us with a better recognition rate in terms of precision, specificity and sensitivity. Thus, the performance of the system has been improved by a considerable margin.

Comparison of the specificity, sensitivity and precision of the four neural networks and the combined system
The outputs of the four neural networks were also combined by using majority voting and the LAP method. Table 8 lists out a comparison on the specificity, sensitivity and precision between these two methods and the LOP2 method. We did not conduct any simulations for the implementation of the LOP method due to the complexity involved in maintaining the condition where the sum of the coefficients is always equal to one. The obtained results point out the fact that, although the combination of the four neural networks, using either method, provides us with better results than the individual neural networks, the results obtained through the LOP2 method are better than those using the other two aggregating methods.
Table 9 compares the results we obtained, using the approach described in this paper, with previous work done on the same problem. Thus, our system can be seen as a considerable improvement compared with recent research on the recognition of E. coli promoters.
6 Conclusion
In this paper, we presented a novel approach for the recognition of E. coli promoters in strings of DNA. The proposed system showed a substantial improvement in the recognition rate of these promoters, specifically in the recognition of true positives, i.e. rejection of non-promoters. This resulted in our system displaying a far higher specificity than all other systems developed thus far.
We consider the reason for this improvement to be twofold. Firstly, it is the use of larger neural networks than those that have been used thus far. This led to a better rate of recognition and generalization. Secondly, it is the use of multiple neural networks, each accepting the same set of inputs in different forms. We observed the fact that the false positives and false negatives of one network could be wiped out by true positives and true positives in other networks. Thus, the combination of the opinions of more classifiers led us to a system that performed much better than the individual components. Our results substantiate Dietterich’s conclusion [5].
One of the major obstacles encountered during the design and implementation of the neural network was the difficulty in obtaining optimal configurations for the neural networks. Future work for the improvement of this system would involve the development of a framework that can be used for the optimal design of neural networks geared towards gene and promoter recognition. This would involve the optimization of both the neural network configuration and the encoding methods used.
Notes
http://www.embl-heidenberg.de/ predictprotein / predictprotein.html
The consensus sequence is an ideal sequence for the interaction with its regulatory protein.
false positives and false negatives
References
Baldi P, Brunak S (1998) Bioinformatics–the machine learning approach. MIT Press, Cambridge
Birney E (2001) Hidden Markov Models in biological sequence analysis. IBM J Res Dev 45(3/4):449–454
Brunak S, Engelbrecht J, Knudsen S (1991) Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 220(1):49–65
Demeler B, Zhou GW (1991) Neural network optimization for E. coli promoter prediction. Nucleic Acids Res 19(7):1593–1599
Dietterich TG (1997) Machine learning research: four current directions. AI Mag 18(4):97–136
Galas DJ, Eggert M, Waterman MS (1985) Rigorous pattern-recognition methods for DNA sequences: analysis of promoter sequences from E. coli. J Mol Biol 186(1):117–128
Hansen JV, Krogh A (1995) A general method for combining in predictors tested on protein secondary structure prediction. http://www.citeseer.ist.psu.edu/324992.html
Henderson, J, Salzberg S, Fasman K (1997) Finding genes in DNA with a hidden markov model. J Comput Biol 4(2):127–141
Koza JR, Andre D (1995) Automatic discovery of protein motifs using genetic programming. http://citeseer.ist.psu.edu/2158.html
Krogh A (1997) Two methods for improving performance of a HMM and their application for gene finding. In: Proceedings of the 5th international conference on intelligent systems for molecular biology. AAAI Press, Menlo Park, pp 179–186
Kulp D, Haussler D, Reese MG, Eeckman FHÄ (1996) Generalized hidden markov model for the recognition of human genes in DNA. In: Proceedings of the 4th international conference on intelligent systems for molecular biology. AAAI Press, Menlo Park, pp 134–142
Ma Q, Wang JTL, Wu CH (2001) Application of Bayesian neural networks to biological data mining: a case study in DNA sequence classification. IEEE Trans Syst Man Cybern Part C 31(4):468–475
Ma Q, Wang JTL (1999) Recognizing promoters in DNA using Bayesian neural networks. In: Proceedings of the IASTED international conference, artificial intelligence and computing, 9–12 August. http://www.citeseer.ist.psu.edu/ma99recognizing.html
Mahadevan I, Ghosh I (1994) Analysis of E. coli promoter structures using neural networks. Nucleic Acids Res 22(11):2158–2165
Mandler EJ, Schurmann J (1988) Combining the classification results of independent classifiers based on the Dempster/Schafer theory of evidence. Pattern Recognit Artif Intell X:381–393
Ohno-Machado L, Vinterbo S, Weber G (2002) Classification of gene expression data using fuzzy logic. J Intell Fuzzy Syst 12(1):19–24
Partridge D, Yates WB (1996) Engineering multiversion neural-net systems. Neural Comput 8:869–893
Pedersen AG, Jensen LJ, Brunak S, Stærfeldt A, Ussery DW (2000) A DNA structural atlas for Escherichia coli. J Mol Biol 299:907–390
Reidmiller M, Braun H (1993) A direct adaptive method for faster Backpropagation learning: the RPROP algorithm. In: International conference on neural networks (ICNN-93, San Francisco, CA). IEEE Press, Piscataway, pp 586–591
Riis SK, Krogh A (1996) Improving prediction of protein secondary structure using neural networks and multiple sequence alignments. J Comput Biol 3:163–183
Rogova G (1994) Combining the results of several neural network classifiers. Neural Netw 7(5):777–781
Roli F, Giacinto G (2002) Hybrid methods in pattern recognition, chapter design of multiple classifier systems. Worldwide Scientific Publishing, pp 199–226
Rost B, Sander C (1993) Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol 232(2):584–599
Ruta D, Gabrys B (2001) Analysis of the correlation between majority voting error and the diversity measures in multiple classifier systems. In: Proceedings of the SOCO/ISFI’2001 conference, ISBN: 3-906454-27-4, Abstract p 50, Paper no.#1824-025, Paisley
Salzberg S, Delcher AL, Fasman KH, Henderson J (1998) A decision tree system for finding genes in DNA. J Comput Biol Winter 5(4):667–80
Sharkey ACJ, Sharkey NE (1997) Combining diverse neural networks. Knowl Eng Rev 12(3):231–247
Snyder EE, Stormo G (1995) Identification of protein coding regions in genomic DNA. J Mol Biol 248:1–18
Stormo GD, Schneider TD, Gold LM, Ehrenfeucht A (1982) Use of the Perceptron algorithm to distinguish translation initiation sites in E. coli. Nucleic Acids Res 10:2997–3011
Uberbacher EC, Mural RJ (1991) Locating protein-coding regions in human DNA sequences by a multiple sensor-neural network approach. Proc Natl Acad Sci USA 88:11261–11265
Woolf PJ, Wang Y (2000) A fuzzy logic approach to analysing gene expression data. Physiol Genomics 3:9–15
Wu CH (1997) Artificial neural networks for molecular sequence analysis. Comput Chem 21(4):237–256
Xu L, Krzyzak A, Suen CY (1991) Associative Switch for combining multiple classifiers. In: Proceedings of the international joint conference on neural networks, IEEE Press, Seattle, pp I-43–48
Xu L, Krzyzak A, Suen CY (1992) Methods for combining multiple classifiers and their applications to handwriting recognition. IEEE Trans Syst Man Cybern 22(3):418–435
Zenobi G, Cuningham P (2001) Using diversity in preparing ensembles of classifiers based on different feature subsets to minimize generalization error. In: Proceedings of the 12th European conference on machine learning, pp 576–587
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ranawana, R., Palade, V. A neural network based multi-classifier system for gene identification in DNA sequences. Neural Comput & Applic 14, 122–131 (2005). https://doi.org/10.1007/s00521-004-0447-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00521-004-0447-7