
Abstract
Purpose
This randomised, placebo-controlled single-blind trial investigated the safety and efficacy of SAMITAL®, a formulation of highly standardised botanical extracts, in the treatment of chemo/radiotherapy-induced oral mucositis (OM) in patients with head and neck cancer.
Methods
Patients received SAMITAL® or placebo four times daily for up to 50 days during scheduled chemo/radiotherapy. Severity of OM was monitored according to a modified WHO severity scale, and pain and quality-of-life assessments were based on the effect of symptoms of OM on relevant daily activities, according to a visual analogue scale.
Results
Mean scores for the severity of OM were significantly (p < 0.05 versus baseline) reduced from day 31 until the end of treatment in patients treated with SAMITAL® (n = 20). No significant improvement was observed in the placebo group (n = 10). Pain reduction was significant from day 4 till end of treatment with SAMITAL® and from days 7 to 21 in placebo patients. SAMITAL® also significantly improved quality of life, as shown by improvements in scores for relevant daily activities including eating, drinking and sleeping. All SAMITAL® patients completed the treatment period, but no placebo recipients completed treatment. No severe adverse events were observed with SAMITAL®, and systemic absorption of relevant active ingredients was undetectable.
Conclusions
SAMITAL® significantly decreased the severity of chemo/radiotherapy-induced OM in patients with head and neck cancer, with no treatment-related adverse events. Pain relief lasted through the treatment period, and improvements in quality of life were reflected by the significant benefits of SAMITAL® on activities like drinking, eating and speaking.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Almost all patients with head and neck cancer receiving chemo/radiotherapy treatment develop oral mucositis (OM), and OM is one of the most common dose-limiting toxic effects of chemo/radiotherapy in these patients [1–3]. The severity of OM varies from patient to patient, and also depends on treatment type, duration and intensity [4, 5]. Characterised by inflammatory lesions and ulcers of the mucosa, OM is associated with considerable pain, dysphagia and interference with nutrition (often requiring parenteral nutrition supplementation), interference with speech, and an increased susceptibility to infections, particularly bacterial and fungal infections [1]. However, despite the predictability of mucositis, its frequency, debilitating symptoms, and negative impact on the treatment programme and health economic costs of patients with head and neck cancer, an effective therapy for OM has long remained elusive.
SAMITAL® was developed with the aim of addressing the challenges of treating OM. It is a combination of three highly standardised botanical drug extracts. These extracts from Vaccinium myrtillus (bilberry), Macleaya cordata fruits and Echinacea angustifolia roots contribute to improving key stages of OM. SAMITAL® was produced to allow the formation of a gel-like suspension and subsequently prolonged exposure of the oral mucosa to the active ingredients. It is noteworthy that SAMITAL® composition is standardised and reproducible (Indena S.p.A.). A study conducted in Chile using SAMITAL® (study code LLCT06-SmT-06A) showed the formulation to be active in the treatment of gastrointestinal mucositis of varying aetiologies, prompting the development of SAMITAL® as a pharmaceutical product under the USA FDA’s Guidance for Industry: Botanical Drug Products released by the Center for Drug Evaluation and Research (June 2004). There is now extensive preclinical and clinical documentation [6–15] on the therapeutic efficacy of each active ingredient of SAMITAL® in the management of OM based on the biologically defined five-stage process of this disease [16]. V. myrtillus provides mucus protection and regeneration activity through improving fibroblast proliferation and stimulating glycosaminoglycan synthesis [6]. M. cordata affords antibacterial, antifungal and antiviral activity [7–11]. In addition, M. cordata is able to block the cascade of pro-inflammatory cytokines through inhibition of NF-κB activation [12]. E. angustifolia possesses anti-inflammatory and analgesic properties [13, 14]. In particular, the high affinity of alkylamides in E. angustifolia for the human CB2 receptor as well as evidence of agonism with TRPV1 receptors suggests that E. angustifolia may reduce peripheral pain [14, 15].
The aim of this Phase II single-blind study was to investigate the activity of SAMITAL® compared with placebo in patients with head and neck cancer with chemo/radiotherapy-induced OM. The safety profile of SAMITAL® and systemic exposure to the most relevant active ingredients were also investigated.
Methods
Study setting and design
This Phase II, monocentric, placebo-controlled, single-blind (patients were blinded to the treatment) clinical study (registration code SAM 01/10) was conducted at the Gokhale Hospital in Pune, India, between January and September 2010. The study was conducted in accordance with the Declaration of Helsinki, and in accordance with WHO guidelines for Good Clinical Practice and the current Indian Council of Medical Research (2000) Ethical Guidelines for Biomedical Research on human subjects. The protocol for this trial was approved by the League Health Independent Ethics Committee of Mumbai (approval date 1 April 2010).
Primary objective
The primary objective was the efficacy of SAMITAL® compared with placebo in terms of the severity of OM in patients undergoing chemo/radiotherapy for head and neck cancer. OM grade was evaluated according to an investigator-modified WHO scale (see details below).
Secondary objectives
-
Evaluation on a daily basis of the intensity of OM-associated pain according to scores on a patient-assessed visual analogue scale (VAS),
-
Evaluation of quality-of-life using a self-administered daily questionnaire on OM [17],
-
Monitoring of adverse events and compliance with drug treatment protocol,
-
Evaluation of systemic exposure to sanguinarine and its main metabolite, dihydrosanguinarine (DHSA).
Patient consent
Prior to screening, patients were given adequate verbal and written information regarding study objectives and procedures and the possible risks involved. All patients gave signed informed consent before inclusion in the study.
Patients
Participating patients were adult patients with head and neck cancer with measurable chemo/radiotherapy-induced OM (grade ≥3). Patients with grade ≥3 OM were included in the study because the incidence of World Health Organization (WHO) grade 3 or 4 OM in patients receiving high-dose head and neck radiation approaches 100 % and recent data show that around 40 % of patients with OM—caused by chemo-radiotherapy and/or molecular targeted therapy for the treatment of advanced squamous cell carcinoma of the head and neck—had grade 3 OM despite the fact that all the patients receive OM prophylaxis [18]. Exclusion criteria were psychological or logistical conditions that precluded administration of SAMITAL® or placebo, previous hypersensitivity to the active ingredients of SAMITAL® refusal or inability to provide consent, and known inability to complete the study.
Intervention and procedures
As this was a monocentric study, we were constrained by patient numbers and to optimise the number receiving active treatment patients were randomised to SAMITAL® or placebo at a ratio of 2:1. Following baseline assessment, patients received four oral administrations of one SAMITAL® or placebo sachet (at approximately 08.00, 13.00, 18.00 and 24.00 hours) daily for 7 days a week and for a total of 7 weeks/50 days, corresponding to the length of time of a cycle of radiotherapy/chemotherapy. The contents of each sachet were mixed with 20 mL of drinkable water at room temperature, and stirred for approximately 1 min. The solution was left to stand for at least 15 min until thickened. The solution was then split into three to four aliquots. The patient was instructed to take one aliquot (about one teaspoonful) of the liquid orally and swish around for at least 1 min, then to either spit it out or swallow it. The four aliquots were administered over a 30-min period. Following administration of all aliquots, patients were instructed not to drink or rinse the mouth for a further 10 min. SAMITAL® treatment (four sachets per day) was continued until the end of the chemo/radiotherapy program (up to a maximum of 50 days).
As the study was single-blind, a matching placebo identical to SAMITAL® (except for the exclusion of the study drug) was formulated. In order to maintain blinding of the placebo formulation, two colouring agents FD&C Red No.40 (Allura Red, E129) and FD&C Blue No 1 (E133) were added. The inactive ingredients used in the SAMITAL® formulation were compliant with Ph.Eur. Monographs, and the colouring agents used in the placebo were food grade (as proved by the EU codes). Like the active granules, the placebo granules were packaged in sachets, each containing 1.5 g granules, and the sachets including the patient information leaflet were presented in similar boxes to that of SAMITAL®.
During SAMITAL® treatment, patients continued to receive routine cancer treatment—induction chemotherapy followed by radical chemo-radiation. All patients were treated with two cycles of induction chemotherapy followed by definitive chemo-radiation for squamous cell carcinoma of the head and neck region. Cisplatin (40 mg/m2) weekly for 5 weeks was the standard chemotherapeutic agent used in all patients during radiotherapy. Radiation was delivered using conformal technique, and tissues containing macroscopic and microscopic disease were treated to doses of 60 Gy in 30 fractions.
Concomitant treatments were recorded in the case report form. Although the study did not allow any topical agents for the supportive care of mucositis, all patients received diclofenac, ibuprofen and/or paracetamol alone or in combination as well as hexetidine mouthwash and clotrimazole mouth paint as required. In addition, all patients were given advice and care on how best to manage their OM before and after enrolment into the study.
Efficacy evaluation
One of the problems with conducting research in patients with OM is the lack of a universally accepted, validated, and objective scoring system—over the years, a series of objective and subjective parameters have been used to measure its severity. Some scoring systems assess only the anatomic distribution of mucosal lesions, whereas others incorporate the patient’s ability to chew and swallow. Two of the most commonly used systems are the WHO Oral Toxicity score and the National Cancer Institute Common Toxicity Criteria for OM. The latter has separate scores for appearance (erythema and ulceration) and function (pain and ability to eat solids, liquids or nothing by mouth) while the WHO score grades the severity of the condition from 0 (no OM) to 4 (swallowing not possible such that patient needs supplementary nutrition). Our research group has developed an OM scoring system for use in clinical trials which is a combination of the International Mucositis Scale (score range) and the WHO Mucositis Scale (grade range). This investigator-modified WHO scale is essentially similar to the WHO scoring system, but it also incorporates an assessment of pain in the parameters of the score (Table 1). Furthermore the investigator-modified WHO scale is the hospital standard and is approved by the Ethics Committee.
The primary efficacy variable was the evaluation of the severity of mucositis of the oral cavity according to the investigator-modified WHO scale described above. The secondary efficacy variables of pain, and irritation and inflammation for the purposes of quality-of-life assessment, were assessed according to scores on a VAS. Oral pain was assessed on an 11-point (0 to 10) scale, where 0 = no pain and 10 = unbearable pain. Irritation in the throat and oral cavity was assessed on a five-point (0 to 4) scale where 0 = no irritation, 1 = slight irritation, 2 = moderate irritation, 3 = severe irritation and 4 = extreme irritation. Inflammation in the throat and oral cavity was assessed on a five-point (0 to 4) scale, where 0 = no inflammation, 1 = just a little inflammation, 2 = somewhat inflamed, 3 = quite inflamed and 4 = impossible to perform activity.
Quality of life was evaluated using a self-administered daily questionnaire assessing the effect of OM symptoms (based on the scale scores above) on the following individual activities: swallowing, drinking, eating, speaking and sleeping. Alimentation was assessed on the basis of ability to swallow liquid and solid food according to yes/no criteria. Finally, the need for analgesics/anti-inflammatories and the occurrence of fever were also recorded.
Safety evaluation
The nature and severity of adverse effects including serious adverse events, either spontaneously reported by the patient or noted by the physician, were recorded. Compliance with treatment was also monitored.
Bioanalysis of sanguinarine
The systemic absorption of sanguinarine in five SAMITAL®-treated patients was assessed using a bioanalytical method, developed and validated using high-performance liquid chromatography mass spectrometer to measure plasma levels [19]. In addition, DHSA, the main metabolite of sanguinarine, was measured in the same blood samples. These markers have been selected since of all the active constituents, these have the greatest potential toxicity [20].
Data analysis
Demographic and clinical data were analyzed descriptively as means, medians or proportions. Curves in figures were generated using the Kaplan–Meier method. Differences throughout the study in data collected within each study group were evaluated using the Friedman test. Comparisons between the SAMITAL® and the placebo groups were not performed.
Results
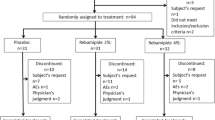
A total of 30 patients were enrolled to receive single-blind treatment with SAMITAL® (n = 20) or placebo (n = 10). Baseline demographic and clinical details (as assessed on day 3) are shown in Table 2. All patients had a diagnosis of stage III head and neck squamous cell carcinoma. The majority of patients (n = 24; 80 %) were male, and all had a performance status of 0–1. All patients completed the planned chemo/radiotherapy regimen.
Overall, 17 out of 20 patients who received SAMITAL® completed the 50-day study period and had evaluable data. Three patients withdrew from the study for personal reasons within 3 days of enrolment and were lost to follow-up. None of the 10 placebo recipients completed the study: five completed 7 days, two 14 days and one patient each completed 21, 28 and 35 days of treatment.
Primary efficacy endpoint
SAMITAL® was associated with significant improvements from baseline in OM grade according to the investigator-modified WHO scale. From day 31, a significantly greater reduction from baseline (p < 0.05) in mean OM score was observed in patients who received SAMITAL® (from 2.94 ± 0.43 to 2.00 ± 0.35) but not in those receiving placebo (OM score = 3.00 in all patients, with no improvements throughout the follow-up; Fig. 1a). These improvements were reflected in the percentage change from baseline in OM score, which was significant with SAMITAL® from day 31 (−4 % at day 31, −32 % at the end of the follow-up; p < 0.05) but not with placebo (Fig. 1b). No episodes of grade 4 OM were reported.

a, b Mucositis severity score following administration of SAMITAL® or placebo four times daily for a maximum of 50 days in patients with chemo/radiotherapy-induced mucositis associated with treatment for head and neck cancer. a Improvement from baseline in mean score according to the WHO scale and b percent change from baseline. Of 30 patients randomised in a 2:1 ratio, data are available for 17 of 20 SAMITAL® and 3 of 10 placebo recipients. Changes from baseline from day 31 were significant (p < 0.05) for the SAMITAL® group but not for the placebo group
Secondary efficacy variables
SAMITAL® was associated with a significant reduction from baseline (p < 0.05) in mean pain score beginning on day 4, and lasting for the 50-day study period (Fig. 2a). However, in placebo patients, significant improvements from baseline (p < 0.05) were only observed from days 7–21 (Fig. 2b). Three patients required rescue analgesic treatments during the first 3 days of SAMITAL® treatment; analgesic treatments were then no longer needed.

a Improvement from baseline in mean pain score according to a visual analogue scale following administration of SAMITAL® or placebo four times daily for a maximum of 50 days in patients with chemo- and/or radiotherapy-induced mucositis associated with treatment for head and neck cancer. Of 30 patients randomised in a 2:1 ratio, data are available for 17 of 20 SAMITAL® and 3 of 10 placebo recipients. b Changes from baseline from day 4 onwards were significant for SAMITAL® and from days 7 until 21 for placebo (p < 0.05, compared with baseline for both comparisons)
Similarly, for the quality of life parameters evaluated according to the modified daily questionnaire, significant improvements from baseline scores occurred with SAMITAL® in swallowing (baseline, 2.94 ± 0.83; end of the follow-up, 2.41 ± 0.94; p < 0.05 versus baseline from day 5 onwards), drinking (baseline, 1.88 ± 1.05; end of the follow-up, 1.53 ± 0.80; p < 0.05 versus baseline from day 18 onwards), eating (baseline, 3.24 ± 1.03; end of the follow-up, 2.88 ± 0.93; p < 0.05 versus baseline from day 5 onwards), sleeping (baseline, 2.18 ± 1.01; end of the follow-up, 1.00 ± 0.61; p < 0.05 versus baseline from day 2 onwards) and speaking (baseline, 2.24 ± 0.90; end of the follow-up, 1.47 ± 0.62; p < 0.05 versus baseline from day 5 onwards). By contrast, for placebo-treated patients, scores for these parameters were either unchanged (swallowing, eating), showed slight nonsignificant improvements from baseline to day 8 (drinking) or showed variability in outcomes from improvements to worsening throughout the study period (sleeping, speaking).
This preliminary study was designed to assess the safety and efficacy of SAMITAL® and to explore potential outcome measures to help guide design of future controlled large-scale trials—in accordance with this premise, irritation and inflammation were also measured but these were not primary variables and as such are not reported in extenso here. In brief, a significant improvement in the irritation score from day 3 onwards was observed with SAMITAL® (p < 0.05 versus baseline), but this effect was not evident in the placebo group. A similar trend was observed for inflammation.
Adverse events
Of the 17 patients who completed 50 days of SAMITAL® treatment and the therapeutic program for head and neck cancer, five reported nausea and vomiting. In detail three experienced severe vomiting and two had moderate vomiting; however, these events were solved without requiring any modification of the administration scheme of SAMITAL®. In the placebo group, five patients reported adverse events including nausea (n = 3), diarrhoea (n = 1) and cough (n = 1) and withdrew from the study before day 14.
No adverse events were deemed to be related to treatment. These episodes of nausea and vomiting in both the SAMITAL® and placebo groups were considered by the investigators as due to chemo/radiation therapy.
Bioanalysis of sanguinarine and DHSA
No detectable concentrations of sanguinarine or DHSA were recorded. Plasma levels of sanguinarine and DHSA (in nanograms per millilitre) were below the low limit of quantification in all five plasma samples collected from SAMITAL®-treated patients on completion of study period.
Discussion
This single-blind, placebo-controlled study evaluated the efficacy and safety of SAMITAL® in OM caused by chemo- and/or radiotherapy in patients with head and neck cancer. SAMITAL® was associated with a significant reduction versus baseline in OM severity, and there were significant improvements in pain, irritation, swallowing, eating, drinking, speaking and sleeping and all quality of life parameters.
Local pain is one of the most troublesome patient-reported problems associated with OM, and analgesics used to treat this symptom are often inadequate, requiring rapid and substantial dose increases, particularly during the final weeks of therapy. The significant reduction from baseline in pain scores with SAMITAL®, observed from day 4 until the end of treatment (50 days), provides confirmation of the analgesic properties of the active components of SAMITAL®, probably related to the alkylamides in E. angustifolia. It was recently reported that selective activity of alkylamides on the cannabinoid type 2 receptor indirectly activates tumour necrosis factor-α mRNA expression ultimately providing anaesthetic-like activity [13]. Moreover, E. angustifolia has shown evidence of agonism with TRPV1 receptors suggesting the ability of the extract to reduce peripheral pain [15].
Interestingly, significant improvements from baseline in pain scores were observed with placebo administration, but this was observed only from days 7 through 21, at which time the effect was reduced. This is indicative of an initial placebo effect on pain and is in accordance with findings reported by Turner et al. [21], who showed a transient effect of placebo on pain in oncological patients.
Nausea and vomiting are common side effects of cancer treatment, resulting in reduced nutritional intake, and dysphagia associated with OM adds an extra health burden to the already debilitated cancer patient. Severe OM frequently results in the need for parenteral nutrition, which carries well-documented health risks [22]. After SAMITAL® therapy, the significant improvements from baseline in swallowing, drinking and eating scores, from as early as day 5 for swallowing and eating, not only confers a major benefit to this patient population, whereby the need for parenteral nutritional support can be avoided, but also in terms of overall health and well-being.
The tolerability results from this study confirm those from additional proof-of-concept studies in which SAMITAL® was found to be well tolerated and safe [23]. In the present study, 5 out of 20 patients in the SAMITAL® group reported nausea and vomiting after receiving SAMITAL® but were able to continue with treatment and completed the entire 7-week SAMITAL® treatment programme. Furthermore, 5 out of 10 patients randomised to placebo experienced nausea and diarrhoea and they withdrew from the study after 2 weeks. These adverse events were therefore considered not to be related to SAMITAL® treatment. All but three SAMITAL® recipients completed the 50-day study period, while none of the patients in the placebo group completed the 50-day treatment period. The three patients who withdrew did so within 3 days of enrolment due to personal reasons.
No detectable concentrations of sanguinarine or its principle metabolite DHSA were detected in plasma samples from patients completing the study supporting the hypothesis that SAMITAL® has a local rather than systemic effect.
Although results from this preliminary study provide positive clinical information to support the use of SAMITAL® in cancer treatment-induced OM, it has a number of limitations. These include the small sample size, the concomitant use of analgesics/anti-inflammatories (which does not allow drawing definite conclusions on the effects of SAMITAL® on pain), the lack of double blinding and the large number of dropouts in the placebo group. These limitations make it unwise to draw general conclusions on the safety and efficacy of SAMITAL®; however, we can nevertheless say that SAMITAL® provides some much needed relief in patients with OM for which currently available therapies are not adequate control of the debilitating symptoms of the disease. This was a preliminary study, and the results of which are encouraging and provide evidence that SAMITAL® can play an important role in the management of chemotherapy/radiation-induced OM.
Conclusion
This placebo-controlled, single-blind study indicates that SAMITAL® provides well-tolerated and effective relief in patients with OM induced by chemo/radiotherapy in patients with head and neck tumours. These results have supported the approval by the FDA of an investigational new drug application (IND 104,011) for a large-scale, multicenter Phase II double-blind, placebo-controlled study to further investigate the effects of the botanical preparation SAMITAL® on the severity and duration of OM in 90 patients affected from head and neck cancer.
References
Peterson DE, Bensadoun RJ et al (2010) Management of oral and gastrointestinal mucositis: ESMO Clinical Practice Guidelines. Annals of Oncology 21(Suppl 5):v261–v265
Trotti A, Bellm LA, Epstein JB, Frame D, Fuchs HJ, Gwede CK, Komaroff E, Nalysnyk L, Zilberberg MD (2003) Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol 66(3):253–262
Trotti A, Garden A, Warde P, Symonds P, Langer C, Redman R et al (2004) A multinational, randomized Phase III trial of iseganan HCl oral solution for reducing the severity of oral mucositis in patients receiving radiotherapy for head-and-neck malignancy. Int J Radiation Oncology Biol Phys 58:674–681
Sonis ST (2009) Mucositis: the impact, biology and therapeutic opportunities of oral mucositis. Oral Oncol 45(12):1015–1020
Sonis ST (2007) Pathobiology of oral mucositis: novel insights and opportunities. J Support Oncol 5(9 suppl 4):3–11
Morazzoni P, Bombardelli E (1996) Vaccinium myrtillus L. Fitoterapia 67:1–28
Mackraj I, Govender T, Gathiram P (2008) Sanguinarine. Cardiovasc Ther 26:75–83
Fadeeva NI, Degtyariova IN, Gerasina SF, et al. (1987) Research of the mechanism of action of sanguiritrin conducted on some biochemical models. Vilar (Russion Institute of Medicinal and Aromatic Plants, Mosca). Unpublished data
Godowski KC (1989) Antimicrobial action of sanguinarine. J Clin Dent 1(4):96–101
Frankos VH, Brusick DJ, Johnson EM, Maibach HI, Munro I, Squire RA, Weil CS (1990) Safety of Sanguinaria extract as used in commercial toothpaste and oral rinse products. J Clin Dent 56(7 Suppl):41–47
Kopczyk RA, Abrams H, Brown AT, Matheny JL, Kaplan AL (1991) Clinical and microbiological effects of a sanguinaria-containing mouthrinse and dentifrice with and without fluoride during 6 months of use. J Periodontol 62(10):617–622
Chaturvedi MM, Kumar A, Darnay BG et al (1997) Sanguinarine (pseudochelerythrine) is a potent inhibitor of NF-κB activation, IκBα phosphorylation, and degradation. J Biol Chem 272:30129–30134
Raduner S, Majewska A, Chen JZ, Xie XQ, Hamon J, Faller B et al (2006) Alkylamides from Echinacea are a new class of cannabinomimetics. J Biol Chem 281(20):14192–14206
Guindon J, Hohmann AG (2008) Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharm 153:319–334
Birt DF, Widrlechner MP et al (2008) Echinacea in infection. Am J Clin Nutr 87(2):488S–492S
Sonis ST (2004) The pathobiology of mucositis. Nat Rev Cancer 4(4):277–284
Stiff PJ, Erder H, Bensinger WI, Emmanouilides C, Gentile T, Isitt J et al (2006) Reliability and validity of a patient self-administered daily questionnaire to assess impact of oral mucositis (OM) on pain and daily functioning in patients undergoing autologous hematopoietic stem cell transplantation (HSCT). Bone Marrow Transplant 37:392–401
Zhang XX, Ma L, Wang JL, Wu WM, Feng LC, Huang DL (2011) Management of oral mucositis in patients with head and neck cancer receiving chemoradiotherapy and/or molecular targeted therapy. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 46(6):505–508
Pawar D, Banavalikar M, Riva A, Bombardelli E, Petrangolini G, Morazzoni P (2011) Development and validation of an HPLC/MS/MS method for the quantitative determination of sanguinarine and its major metabolite dihydrosanguinarine in human plasma. Presented as a poster at the 40th ACCP Annual Meeting, September 2011
Federal Register (2003) 21 CFR Part 356 oral health care drug products for over-the- counter human use; antigingivitis/antiplaque drug products; establishment of a monograph; proposed rules; 68 FR 32231
Turner JA, Deyo RA, Loeser JD, Von Korff M, Fordyce WE et al (1995) The importance of placebo effect in pain treatment and research. JAMA 273(4):283
Rubenstein EB, Peterson DE, Schubert M et al (2004) Clinical practice guidelines for the prevention and treatment of cancer therapy-induced oral and gastrointestinal mucositis. Cancer 100(9 Suppl):2026–2046
Giacosa A, Pawar D, Bertoglio JC, Bombardelli E, Morazzoni P, Ronchi M, Petrangolini G, Riva A (2011) SAMITAL®: a new challenge for the treatment of oral mucositis induced by chemo/radiotherapy. Eur J Cancer 47(suppl 1):S242
Acknowledgments
This study was funded by Indena S.p.A, Milan, Italy. The authors acknowledge assistance from Luca Giacomelli, Ph.D., in the preparation of this manuscript.
Conflict of interest
AR, EB, MR, GP and PM are employees of Indena S.p.A. Other authors declare no conflict of interest. The authors of this manuscript have full control of all primary data and agree to allow the journal editor to review their data if requested.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pawar, D., Neve, R.S., Kalgane, S. et al. SAMITAL® improves chemo/radiotherapy-induced oral mucositis in patients with head and neck cancer: results of a randomized, placebo-controlled, single-blind Phase II study. Support Care Cancer 21, 827–834 (2013). https://doi.org/10.1007/s00520-012-1586-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-012-1586-5