
Abstract
Independent component analysis (ICA) is a powerful concept for reducing the dimension of big data in many applications. It has been used for the feature extraction of microarray gene expression data in numerous works. One of the merits of ICA is that a number of extracted features are always equal to the number of samples. When ICA is applied to microarray data, whenever, it faces the challenges of how to find the best subset of genes (features) from extracted features. To resolve this problem, in this paper, we propose a new (artificial bee colony) ABC-based feature selection approach for microarray data. Our approach is based on two stages: ICA-based extraction approach to reduce the size of data and ABC-based wrapper approach to optimize the reduced feature vectors. To validate our proposed approach, extensive experiments were conducted to compare the performance of ICA + ABC with the results obtained from recently published and other previously suggested methods of gene selection for Naïve Bayes (NB) classifier. To compare the performance of the proposed approach with other algorithms, a statistical hypothesis test was employed with six benchmark cancer classification datasets of the microarray. The experimental result shows that the proposed approach demonstrates an improvement over all the algorithms for NB classifier with a certain level of significance.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The field of machine learning provides an application of computer-based approach which is appropriate for the analysis of different types of datasets, and these approaches are developed and improved with experience (Ahmadi and Mahmoudi 2016; Ahmadi 2015b; Ahmadi and Bahadori 2016; Ahmadi et al. 2015d; Ahmadi and Shadizadeh 2012). Machine learning techniques solve the problem of clustering, classification, prediction and various other problems by using the application of supervised, unsupervised and semi-supervised method (Ahmadi et al. 2014b, c, d, e, f, g; Ahmadi and Ebadi 2014). One of the major applications of microarray data analysis is to perform sample classification for diagnostic and prognostic of disease. Some of the examples of machine learning techniques that have been used in cancer classification of microarray data include the decision tree, neural networks, support vector machine and the Naïve Bayesian classifier. However, small size of samples in comparison with high dimensionality is the main difficulty for most of the machine learning techniques. This problem is known as ‘curse of dimensionality.’ Dimension reduction is one of the main applications that plays an important role in the DNA microarray data classification (Lazar et al. 2012; Saeys et al. 2007). For dimension reduction, there are two important algorithms: feature extraction and feature selection. Feature extraction algorithm transforms the feature into the lower-dimensional space by using the combinations of the original features. The feature selection method selects the most relevant features from the entire features to construct the model for classification. Feature selection algorithms can be arranged into three types, namely filter, wrapper and embedded methods. Filter methods are the ones that select features as a preprocessing step and they select features without considering the classification accuracy, while the wrapper method is a repetitive search process in which the results of the learning algorithm in each repetition are used to guide the search process (Kohavi and John 1997) since wrapper methods continuously use the learning algorithm in the search process so that their computational cost is more, especially for high-dimensional datasets. The third type of feature selection algorithm is embedded method (Ahmadi and Golshadi 2012; Ahmadi 2011; Ahmadi et al. 2015h). The difference of embedded approach compared to other filter and wrapper approach is the search mechanism that is built into the classifier model. Recently, a hybrid search technique has been used to take the advantages of the extraction/filter and the wrapper approach. In the hybrid search algorithm, the first subset of features is selected or extracted based on the filter/extraction method, and after that, the wrapper method is used to select the final feature set. Therefore, the computational cost of the wrapper method becomes acceptable due to the use of reduced size features. Information gain and a memetic algorithm (Zibakhsh and Abadeh 2013), Fishers core with a GA and PSO (Zhao et al. 2011), mRMR with ABC algorithm (Alshamlan et al. 2015a) and independent component analysis with fuzzy algorithm (Aziz et al. 2016) are recently used hybrid methods to solve the problem of dimension reduction of microarray.
ICA is a multi-dimensional statistical method for finding the hidden information that is situated under a set of random variables (Hyvarinen et al. 2001). Nowadays, ICA technique has received growing attention as effective dimension reduction algorithm for NB classification of high-dimensional data (Kong et al. 2008). The reason for that is the conditional independence hypothesis rooted in the algorithm of NB classifier which could be successfully resolved as the components extracted by the ICA are statistically independent. There still exists an unsolved problem that is how one can choose a subset of independent component (IC) that improves the performance of base classifier. To solve this problem, different authors used different wrapper methods to choose best subset of the IC. For example, the sequential floating forward selection (SFFS) method is used to choose the ICA feature vector for SVM classification of microarray data (Zheng et al. 2006). Zheng et al. (2008) classified gene expression data with consensus independent component analysis (ICA) as a dimension reduction technique. A sequential feature extraction method was used to choose best genes set of independent component vector for NB classifier (Fan et al. 2009). Some of the authors used different filter methods for ranking of ICA feature vector to increase the classification accuracy of SVM and NB classifier (Rabia et al. 2015a, b). On the other hand. bio-inspired evolutionary techniques-based wrapper methods such as ant colony optimization (ACO) (Tabakhi et al. 2014), genetic algorithm (GA) (Huang and Wang 2006), particle swarm optimization (PSO) (Lin et al. 2008), bacterial foraging algorithm (BFA) and fish school search (FSS) are more relevant and provide a more exact solution than the other filter-based wrapper techniques because they have the ability to search and find the optimum or near-optimum solutions on high-dimensional solution space (Arqub and Abo-Hammour 2014; Ahmadi 2016; Ahmadi et al. 2015c, e, f, g; Ahmadi and Bahadori 2015; Baghban et al. 2015). These bio-inspired algorithms have been effectively applied for resolving the problem of dimension reduction in various applications such as financial domains, face recognition and text classification (Abo-Hammour et al. 2014). While on the other hand, the result of these bio-inspired techniques depends on the complexity of the search space, fitness function, the parameters used for the algorithm, convergence, etc. (Ali Ahmadi and Ahmadi 2016; Ahmadi 2015a; Ahmadi et al. 2014a, 2015a, b; Shafiei et al. 2014). All of these methods are classifier-based algorithms and have obtained satisfactory performance for dimension reduction in different types of fields, and they have not been frequently used for feature selection of DNA microarray data due to high computational cost. But the computational cost of the hybrid technique is lesser than only wrapper technique because of the use of the reduced number of features in its second step.
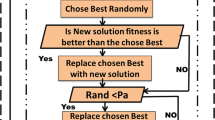
Since the ICA aims to address the issues arising from NB classification of microarray data, ABC-based wrapper approach with NB classifier is applied to optimize the ICA feature vectors for finding the smallest number of features that improved the classification accuracy of NB. In the research paper, we focus on the impact of the proposed algorithm ICA + ABC with NB classifier and how the performance of NB classifier improves using this combination. The proposed ICA + ABC hybrid algorithm is an iterative upgrading computational process where the population of agents chooses a different subset of features in each iteration. After that, the performance of the different subsets of features is estimated using the classification accuracy of NB (Fig. 1).

Schematic representation of the proposed methodology
2 Proposed approach
2.1 Feature extraction by ICA
Independent component analysis is a feature extraction technique, which was proposed by Hyvarinen to solve the typical problem of the non-Gaussian processes and has been applied successfully in different fields (Hyvarinen et al. 2001). The extraction process of ICA is very similar to the algorithm of principal component analysis (PCA). PCA maps the data into an another space with the help of principal component. In place of principal component, the ICA algorithm finds the linear representation of non-Gaussian data so that the extracted components are statistically independent. Theory of ICA algorithm can be found elsewhere (Aziz et al. 2016; Hsu et al. 2010).
2.2 Feature selection by ABC
Artificial bee colony (ABC) is an evolutionary feature selection algorithm that is used to select a best feature subset. The ABC algorithm reproduces the appearance of the intelligent foraging behavior of honey bee swarms for the optimization of feature subset, introduced by Dervis Karaboga in 2005 (Karaboga 2005). Garro and Beatriz applied ABC for selecting best genes set of microarray data with distance classifier for artificial neural network (ANN) classification. ABC algorithm is a combination of local search and global search method managed by three classes of bees (employed, onlooker and scouts bees) (Garro et al. 2016). These three classes of bees with different works in the search space (colony) find the convergence of the problem near to the optimal solution.
Employed bees: These bees search new neighborhood food around their hive. After that, they compare the new food source with the old food source by using Eq. (1):
where vij is a newly generated solution and k ≠ i. \( \phi_{i}^{j} \) is a random number between [− 1, 1]. If the fitness value \( v_{i}^{j} \) is better than \( x_{i}^{j} \), then \( x_{i}^{j} \) changed to \( v_{i}^{j} \), otherwise \( x_{i}^{j} \) unchanged.
Onlooker bees: Employed bees share this information of solution with onlooker bees. Then, by using the information of employed bees, onlooker bees find a food source with the probabilities related to their amount of nectar. This probability of finding the food source is calculated by using Eq. (2):
Scout bees: If the fitness value of the ith solution (fiti) cannot be improved longer during a predefined number called ‘limit,’ then this criterion is called ‘abandonment criteria.’ For this type of criterion, the scout bee creates new solutions to replace ith solution by using Eq. (3):
3 Classifier (NB)
Naïve Bayes is a simple supervised learning algorithm for machine learning classification. NB classifier used Bayes’ rule with strong independence assumption for mining of different types of data (Friedman et al. 1997; Hall 2007). Due to its simplicity, Naïve Bayes is an attractive classifier among the researchers for solving different classification problems, including microarray (Chen et al. 2009; Sandberg et al. 2001). It is found that its performance is more robust and efficient compared to other supervised machine learning classification algorithms. More detailed information on NB can be found elsewhere (Aziz et al. 2016; Fan et al. 2009).
4 Experimental setup
To evaluate the performance of ICA + ABC, six benchmark classification datasets of the microarray, including the colon cancer (Alon et al. 1999), acute leukemia (Golub et al. 1999), prostate cancer (Singh et al. 2002), lung cancer II (Gordon et al. 2002), high-grade glioma data (Nutt et al. 2003) of binary classification and leukemia 2 (Armstrong et al. 2002) of multi-classification, are used. Table 1 shows the detailed description (number of classes, number of features, etc.) of these datasets.
In this study, NB classifier with Gaussian distribution estimation is used for microarray data (Rabia et al. 2015b). The goodness for each training subset is estimated by leave-one-out cross-validation (LOOCV) classification accuracy of NB classifier that plays the role of fitness value of ABC. The performance of each feature selection algorithm is based on two criteria, the classification accuracy of NB and the smallest number of selected features that have been used for classification of data. Classification accuracy of NB is the overall correctness of the classifier, and it is calculated by the formula shown below:
where N is the total number of samples in the original microarray dataset and CC refers to correctly classified samples. For statistically validating the experimental results, each gene selection algorithm was implemented 30 times using the fitness value. On the other hand, parameters of ABC are selected on the basis of the studies of several related research articles concerned with the selection of ABC parameter (Akay and Karaboga 2009; Abu-Mouti and El-Hawary 2012; Alshamlan et al. 2015b; Garro et al. 2016). The parameter of ABC algorithm that was used in our experiments is given below:
-
Bee colony size = 100.
-
The maximum cycle = the maximum number of generations (100)
-
Number of runs = 30 runs
-
Limit = 5 iterations.
For implementations of ICA, MATLAB software package (R2014a) with the FastICA algorithm is used, and it can be found from the Internet (http://research.ics.aalto.fi/ica/fastica/code/dlcode.shtml). Codes for ABC feature selection are freely available on the Internet (http://mf.erciyes.edu.tr/abc/).
5 Experimental results and discussions
Tables 2, 3, 4, 5, 6 and 7 show the LOOCV estimations of the test set, classification accuracy rates of NB classifiers on the above six cancer datasets of microarray, with features selected by ICA + ABC and mRMR-ABC algorithms. The same parameters of ABC algorithm are used for ICA + ABC and for mRMR + ABC algorithms for the sake of fair comparison. The optimal results of all datasets (highest accuracy with minimum selected gene size) are highlighted using bold font. The ROC curve with a different value of threshold (Song et al. 2014) for six best selected subsets of genes obtained by ICA + ABC and mRMR + ABC methods for different binary datasets is shown in Figs. 2a, b, 3a, b, 4a, b, 5a, b and 6a, b. The ROC of multi-class dataset is represented in Fig. 7a, b with best subset of genes.

ROC curve with six best selected subsets of genes with ICA + ABC and mRMR + ABC algorithms for NB classifier of colon dataset

ROC curve with six best selected subsets of genes with ICA + ABC and mRMR + ABC algorithms for NB classifier of acute leukemia dataset

ROC curve with six best selected subsets of genes with ICA + ABC and mRMR +ABC algorithms for NB classifier of prostate cancer dataset

ROC curve with six best selected subsets of genes with ICA + ABC and mRMR + ABC algorithms for NB classifier of high-grade glioma dataset

ROC curve with six best selected subsets of genes with ICA + ABC and mRMR + ABC algorithms for NB classifier of lung cancer II dataset

ROC curve with the best selected subset of genes with ICA + ABC and mRMR + ABC algorithms for NB classifier of leukemia 2 dataset
Now, the following observations can be made from Tables 2, 3, 4, 5, 6 and 7 and Figs. 2a, b, 3a, b, 4a, b, 5a, b, 6a, b and 7a, b
-
1.
As can be seen from Tables 2, 3, 4, 5, 6 and 7, the proposed algorithm, ICA + ABC, was able to predict the best gene subset for resolving the classification problem of different datasets. To obtain best gene subsets, initially ICA chooses an average of 50 to 180 genes from a set of 2000–12,500 genes from a different training dataset of the microarray. After that, ABC selects a different subset of genes from the set of ICA feature vectors of different datasets, then finds the classification accuracy of NB with these selected gene subsets and obtains a smallest gene set that gives better accuracy of NB classifier and finally finds the classification accuracy of test data. By the repetition of this process, the original thousands of genes of different datasets were reduced to within an average of 15.4 genes for highest classification accuracy.
-
2.
The positive outcome with the smallest number of genes selected by ABC from ICA feature vectors is clearly noticeable with respect to classification accuracy. For the colon cancer data, the mean classification accuracy with all ICA features was 70.71%, while with the ABC wrapper approach it increases 91.12% with 16 genes. In the same manner, ABC obtained an average classification accuracy of 96.55% for acute leukemia with 12 genes, while the average classification accuracy had been less than 66.82% with all 72 ICA feature vectors. Same situations can also be found in all other remained datasets with the ICA + ABC algorithm. Therefore, ABC has a powerful optimization algorithm for providing best genes subset with ICA for NB classification of microarray.
-
3.
We can observe that the AUC (area under the ROC curve) values of NB classifier varied with different sizes of gene subsets, and the best subset of genes for different datasets is different that gives greatest AUC value. The best AUC value of colon data is 95.05 with 16 selected genes for the 0.3 threshold value, and the number of 12 genes gives the highest AUC for acute leukemia data with the value of 97.11 for the 0.7 threshold value. The highest AUC of the prostate and high-grade glioma with 0.5 threshold value is found at 94.21 and 93.65 with 16 and nine genes, respectively. For the lung cancer data II, the best value of AUC is at 92.87 with 24 selected genes for the 0.2 threshold value. We also plot ROC for leukemia 2 data for the best subset of genes obtained by ICA + ABC and mRMR + ABC. The black curve depicts the ROC when the class one is separated from class two and three, while the red curve shows the ROC when class two is separated from class three and one, and similarly, the blue curve shows the ROC when the class 3 is separated from the class one and two. The average AUC value with proposed approach is 95.97 and 94.79 for mRMR + ABC algorithm. It can be seen also from Figs. 2a, b, 3a, b, 4a, b, 5a, b, 6a, b and 7a, b that ICA + ABC produces the best AUC scores with the smallest number of genes compared to the mRMR + ABC algorithms.
-
4.
Furthermore, the results produced by ICA with a conventional feature selection method such that signal-to-noise ratio (SNR), fuzzy algorithm and also with other similar bio-inspired algorithm such that PSO and GA shown in Table 8. Result shows that the proposed ICA + ABC method obtained the best classification accuracy among the other gene selection methods with three datasets of binary classification and one dataset of multi-classification. For the other two cancer data, ICA + GA algorithm obtained the highest accuracy for high-grade glioma data, while ICA + fuzzy algorithm obtained the highest accuracy for lung cancer II data. Also, it was noticeable that ICA + GA and ICA + fuzzy algorithms obtained the best classification accuracy, but ICA + ABC chooses the smallest number of genes for all six cancer datasets of a microarray for best classification accuracy.
Table 8 Classification performance of the proposed algorithm compared with some conventional and bio-inspired algorithms when combined with ICA for NB classifier on six microarray datasets -
5.
SVM is also a popular supervised machine learning algorithm of data classification, and various studies showed that SVM outperforms others for microarray data classification. Therefore, we also implement the proposed approach with SVM classifier and compare the result of SVM and NB classifier for six datasets. It can be seen from Table 9 that NB classifier gives slightly better result than SVM classifier with proposed approach but for lung cancer data II SVM gives better classification accuracy than NB classifier.
Table 9 Classification performance of SVM and NB classifiers with the proposed algorithm on six microarray datasets
To compare the performance of ICA + ABC and mRMR + ABC algorithms, a statistical hypothesis test was employed to determine with a certain level of confidence whether there exists a significant difference between them. A parametric paired t test was applied with α = 0.05 to check whether the average difference in their performance over the problems is significantly different from zero (Derrac et al. 2011). A paired t test carries out a pairwise comparison of the performance of two algorithms. This test evaluates the statistical significance of the null hypothesis; i.e., if the results of two algorithms come from the same observation, hypothesis is rejected when the p value reported by the test is smaller than the significance level (α). If calculated t value is greater than the tabulated value of the distribution of the t test for (n-1) degrees of freedom, the null hypothesis is rejected which means that a given algorithm outperforms the other one with the associated p value (Zar 1999).
From Table 10, it is clear that the proposed approach ICA + ABC not only shows an improvement over mRMR + ABC but also with the other four methods, i.e., ICA + fuzzy, ICA + SNR, ICA + PSO and ICA + GA algorithms with a level of significance α = 0.05. Here, the value of h = 1 represents that the null hypothesis is rejected while h = 0 shows that null hypothesis is accepted. Statistical significance difference between ICA + ABC algorithm other five algorithms is easily seen by the rejection of null hypothesis, as the reported p value in all the cases is less than 0.05 that is depicted in Fig. 8. MATLAB software version (R2014a) is used for the computation of the p value and t value for t tests.

T test comparing results of the proposed algorithm over other algorithms. The red line marks the significance level α = 0.05 (color figure online)
Significantly, it can be said that ABC wrapper approach in ICA feature vector improves the accuracy of NB classifiers by discarding the inappropriate genes while mRMR + ABC picked a bit larger gene subset that gives improved classification accuracy compared to ICA + ABC for all the six datasets of the microarray. There are two main reasons that the performance of ICA + ABC was better than mRMR + ABC feature selection algorithms, because ICA is capable of accurately extracting independent component in the first stage that satisfied the classification criteria of NB classifier. The second cause is the novel hybrid search method (extraction approach with the random search wrapper approach) integrated in ICA + ABC. The other researchers have not used such a hybrid search method for NB classification. The benefit of adopting such type of the hybrid search technique can clearly be seen in Fig. 9. Therefore, ICA + ABC had a significant ability to generate improved classification accuracy of a NB classifier for different microarray datasets, using the smaller number of genes.

Average error rate of NB classifier for the six datasets with different gene selection methods when combined with ICA
6 Conclusion
To improve the classification accuracy of NB classifier, an improved gene selection method based on ABC and ICA is proposed in this paper. Comparative performance shows, with smaller subsets of genes selected by the proposed method, NB classifier achieves higher classification accuracy on six benchmark cancer classification datasets of the microarray compared to other previously proposed methods, which shows the efficiency and effectiveness of the proposed gene selection method. Therefore, this work suggests two-stage gene selection method (ICA + ABC) that can select best gene subsets to achieve higher classification accuracy of a NB classifier for classification of the microarray.
References
Abo-Hammour Z, Abu Arqub O, Mohammad Momani S, Shawagfeh N (2014) Optimization solution of Troesch’s and Bratu’s problems of ordinary type using novel continuous genetic algorithm. Discrete Dyn Nat Soc 2014. https://doi.org/10.1155/2014/401696
Abu-Mouti FS, El-Hawary ME (2012) Overview of artificial bee colony (ABC) algorithm and its applications. In: Systems conference (SysCon), 2012 IEEE international. IEEE, pp 1–6
Ahmadi MA (2011) Prediction of asphaltene precipitation using artificial neural network optimized by imperialist competitive algorithm. J Pet Explor Prod Technol 1(2–4):99–106
Ahmadi MA (2015a) Connectionist approach estimates gas–oil relative permeability in petroleum reservoirs: application to reservoir simulation. Fuel 140:429–439
Ahmadi MA (2015b) Developing a robust surrogate model of chemical flooding based on the artificial neural network for enhanced oil recovery implications. Math Probl Eng
Ahmadi MA (2016) Toward reliable model for prediction drilling fluid density at wellbore conditions: a LSSVM model. Neurocomputing 211:143–149
Ahmadi M-A, Bahadori A (2015) A LSSVM approach for determining well placement and conning phenomena in horizontal wells. Fuel 153:276–283
Ahmadi MA, Bahadori A (2016) Prediction performance of natural gas dehydration units for water removal efficiency using a least-square support vector machine. Int J Ambient Energy 37(5):486–494
Ahmadi MA, Ebadi M (2014) Evolving smart approach for determination dew point pressure through condensate gas reservoirs. Fuel 117:1074–1084
Ahmadi MA, Golshadi M (2012) Neural network based swarm concept for prediction asphaltene precipitation due to natural depletion. J Pet Sci Eng 98:40–49
Ahmadi MA, Mahmoudi B (2016) Development of robust model to estimate gas–oil interfacial tension using least square support vector machine: experimental and modeling study. J Supercrit Fluids 107:122–128
Ahmadi MA, Shadizadeh SR (2012) New approach for prediction of asphaltene precipitation due to natural depletion by using evolutionary algorithm concept. Fuel 102:716–723
Ahmadi M-A, Ahmadi MR, Hosseini SM, Ebadi M (2014a) Connectionist model predicts the porosity and permeability of petroleum reservoirs by means of petro-physical logs: application of artificial intelligence. J Pet Sci Eng 123:183–200
Ahmadi MA, Ebadi M, Hosseini SM (2014b) Prediction breakthrough time of water coning in the fractured reservoirs by implementing low parameter support vector machine approach. Fuel 117:579–589
Ahmadi MA, Ebadi M, Marghmaleki PS, Fouladi MM (2014c) Evolving predictive model to determine condensate-to-gas ratio in retrograded condensate gas reservoirs. Fuel 124:241–257
Ahmadi MA, Ebadi M, Yazdanpanah A (2014d) Robust intelligent tool for estimating dew point pressure in retrograded condensate gas reservoirs: application of particle swarm optimization. J Pet Sci Eng 123:7–19
Ahmadi MA, Masoumi M, Askarinezhad R (2014e) Evolving connectionist model to monitor the efficiency of an in situ combustion process: application to heavy oil recovery. Energy Technol 2(9–10):811–818
Ahmadi MA, Masumi M, Kharrat R, Mohammadi AH (2014f) Gas analysis by in situ combustion in heavy-oil recovery process: experimental and modeling studies. Chem Eng Technol 37(3):409–418
Ahmadi MA, Soleimani R, Bahadori A (2014g) A computational intelligence scheme for prediction equilibrium water dew point of natural gas in TEG dehydration systems. Fuel 137:145–154
Ahmadi M-A, Bahadori A, Shadizadeh SR (2015a) A rigorous model to predict the amount of dissolved calcium carbonate concentration throughout oil field brines: side effect of pressure and temperature. Fuel 139:154–159
Ahmadi M-A, Pouladi B, Javvi Y, Alfkhani S, Soleimani R (2015b) Connectionist technique estimates H2S solubility in ionic liquids through a low parameter approach. J Supercrit Fluids 97:81–87
Ahmadi M, Hasanvand MZ, Bahadori A (2015c) A LSSVM approach to predict temperature drop accompanying a given pressure drop for the natural gas production and processing systems. Int J Ambient Energy 38:122–129
Ahmadi MA, Ebadi M, Samadi A, Siuki MZ (2015d) Phase equilibrium modeling of clathrate hydrates of carbon dioxide + 1,4-dioxine using intelligent approaches. J Dispers Sci Technol 36(2):236–244
Ahmadi MA, Lee M, Bahadori A (2015e) Prediction of a solid desiccant dehydrator performance using least squares support vector machines algorithm. J Taiwan Inst Chem Eng 50:115–122
Ahmadi MA, Masoumi M, Askarinezhad R (2015f) Evolving smart model to predict the combustion front velocity for in situ combustion. Energy Technol 3(2):128–135
Ahmadi MA, Zahedzadeh M, Shadizadeh SR, Abbassi R (2015g) Connectionist model for predicting minimum gas miscibility pressure: application to gas injection process. Fuel 148:202–211
Ahmadi MH, Ahmadi MA, Sadatsakkak SA, Feidt M (2015h) Connectionist intelligent model estimates output power and torque of stirling engine. Renew Sustain Energy Rev 50:871–883
Akay B, Karaboga D (2009) Parameter tuning for the artificial bee colony algorithm. In: International conference on computational collective intelligence. Springer, pp 608–619
Ali Ahmadi M, Ahmadi A (2016) Applying a sophisticated approach to predict CO2 solubility in brines: application to CO2 sequestration. Int J Low-Carbon Technol 11(3):325–332
Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ (1999) Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci 96(12):6745–6750
Alshamlan H, Badr G, Alohali Y (2015a) mRMR-ABC: a hybrid gene selection algorithm for cancer classification using microarray gene expression profiling. BioMed Res Int 2015. https://doi.org/10.1155/2015/604910
Alshamlan HM, Badr GH, Alohali YA (2015b) Genetic bee colony (GBC) algorithm: a new gene selection method for microarray cancer classification. Comput Biol Chem 56:49–60
Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ (2002) MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 30(1):41–47
Arqub OA, Abo-Hammour Z (2014) Numerical solution of systems of second-order boundary value problems using continuous genetic algorithm. Inf Sci 279:396–415
Aziz R, Verma C, Srivastava N (2016) A fuzzy based feature selection from independent component subspace for machine learning classification of microarray data. Genomics Data 8:4–15
Baghban A, Ahmadi MA, Pouladi B, Amanna B (2015) Phase equilibrium modeling of semi-clathrate hydrates of seven commonly gases in the presence of TBAB ionic liquid promoter based on a low parameter connectionist technique. J Supercrit Fluids 101:184–192
Chen J, Huang H, Tian S, Qu Y (2009) Feature selection for text classification with Naïve Bayes. Expert Syst Appl 36(3):5432–5435
Derrac J, García S, Molina D, Herrera F (2011) A practical tutorial on the use of nonparametric statistical tests as a methodology for comparing evolutionary and swarm intelligence algorithms. Swarm Evol Comput 1(1):3–18
Fan L, Poh K-L, Zhou P (2009) A sequential feature extraction approach for Naïve Bayes classification of microarray data. Expert Syst Appl 36(6):9919–9923
Friedman N, Geiger D, Goldszmidt M (1997) Bayesian network classifiers. Mach Lear 29(2–3):131–163
Garro BA, Rodríguez K, Vázquez RA (2016) Classification of DNA microarrays using artificial neural networks and ABC algorithm. Appl Soft Comput 38:548–560
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286(5439):531–537
Gordon GJ, Jensen RV, Hsiao L-L, Gullans SR, Blumenstock JE, Ramaswamy S, Richards WG, Sugarbaker DJ, Bueno R (2002) Translation of microarray data into clinically relevant cancer diagnostic tests using gene expression ratios in lung cancer and mesothelioma. Cancer Res 62(17):4963–4967
Hall M (2007) A decision tree-based attribute weighting filter for naive Bayes. Knowl-Based Syst 20(2):120–126
Hsu C-C, Chen M-C, Chen L-S (2010) Integrating independent component analysis and support vector machine for multivariate process monitoring. Comput Ind Eng 59(1):145–156
Huang C-L, Wang C-J (2006) A GA-based feature selection and parameters optimization for support vector machines. Expert Syst Appl 31(2):231–240
Hyvarinen A, Karhunen J, Oja E (2001) Independent component analysis. Wiley, London
Karaboga D (2005) An idea based on honey bee swarm for numerical optimization. Technical Report-tr06, Erciyes University, Engineering Faculty, Computer Engineering Department,
Kohavi R, John GH (1997) Wrappers for feature subset selection. Artif Intell 97(1):273–324
Kong W, Vanderburg CR, Gunshin H, Rogers JT, Huang X (2008) A review of independent component analysis application to microarray gene expression data. Biotechniques 45(5):501
Lazar C, Taminau J, Meganck S, Steenhoff D, Coletta A, Molter C, De Schaetzen V, Duque R, Bersini H, Nowe A (2012) A survey on filter techniques for feature selection in gene expression microarray analysis. IEEE/ACM Trans Comput Biol Bioinform (TCBB) 9(4):1106–1119
Lin S-W, Ying K-C, Chen S-C, Lee Z-J (2008) Particle swarm optimization for parameter determination and feature selection of support vector machines. Expert Syst Appl 35(4):1817–1824
Nutt CL, Mani D, Betensky RA, Tamayo P, Cairncross JG, Ladd C, Pohl U, Hartmann C, McLaughlin ME, Batchelor TT (2003) Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 63(7):1602–1607
Rabia A, Namita S, Chandan KV (2015a) t-Independent component analysis For SVM classification of DNA-microarray data. Int J Bioinform Res 6(1):305–312
Rabia A, Namita S, Chandan KV (2015b) A weighted-SNR feature selection from independent component subspace for NB classification of microarray data. Int J Adv Biotechnol Res 6(2):245–255
Saeys Y, Inza I, Larrañaga P (2007) A review of feature selection techniques in bioinformatics. Bioinformatics 23(19):2507–2517
Sandberg R, Winberg G, Bränden C-I, Kaske A, Ernberg I, Cöster J (2001) Capturing whole-genome characteristics in short sequences using a naive Bayesian classifier. Genome Res 11(8):1404–1409
Shafiei A, Ahmadi MA, Zaheri SH, Baghban A, Amirfakhrian A, Soleimani R (2014) Estimating hydrogen sulfide solubility in ionic liquids using a machine learning approach. J Supercrit Fluids 95:525–534
Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP (2002) Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 1(2):203–209
Song B, Zhang G, Zhu W, Liang Z (2014) ROC operating point selection for classification of imbalanced data with application to computer-aided polyp detection in CT colonography. Int J Comput Assist Radiol Surg 9(1):79–89
Tabakhi S, Moradi P, Akhlaghian F (2014) An unsupervised feature selection algorithm based on ant colony optimization. Eng Appl Artif Intell 32:112–123
Zar JH (1999) Biostatistical analysis. Pearson Education India, New Delhi
Zhao W, Wang G, H-b Wang, H-l Chen, Dong H, Z-d Zhao (2011) A novel framework for gene selection. Int J Adv Comput Technol 3:184–191
Zheng C-H, Huang D-S, Shang L (2006) Feature selection in independent component subspace for microarray data classification. Neurocomputing 69(16):2407–2410
Zheng C-H, Huang D-S, Kong X-Z, Zhao X-M (2008) Gene expression data classification using consensus independent component analysis. Genomics Proteomics Bioinform 6(2):74–82
Zibakhsh A, Abadeh MS (2013) Gene selection for cancer tumor detection using a novel memetic algorithm with a multi-view fitness function. Eng Appl Artif Intell 26(4):1274–1281
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Rabia Aziz, C. K. Verma, Namita Srivastava declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by V. Loia.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Musheer, R.A., Verma, C.K. & Srivastava, N. Novel machine learning approach for classification of high-dimensional microarray data. Soft Comput 23, 13409–13421 (2019). https://doi.org/10.1007/s00500-019-03879-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00500-019-03879-7