
Abstract
Background
Steroid-dependent nephrotic syndrome (SDNS) carries a high risk of toxicity from steroids or steroid-sparing agents. This open-label, randomized controlled trial was designed to test whether the monoclonal antibody rituximab is non-inferior to steroids in maintaining remission in juvenile forms of SDNS and how long remission may last (EudraCT:2008-004486-26).
Methods
We enrolled 30 children 4–15 years who had developed SDNS 6–12 months before and were maintained in remission with low prednisone doses (0.1–0.4 mg/Kg/day). Participants were randomized following a non-inferiority design to continue prednisone alone (n 15, controls) or to add a single intravenous infusion of rituximab (375 mg/m2, n 15 intervention). Prednisone was tapered in both arms after 1 month. Children assigned to the control arm were allowed to receive rituximab to treat disease relapse.
Results
Proteinuria increased at 3 months in the prednisone group (from 0.14 to 1.5 g/day) (p < 0.001) and remained unchanged in the rituximab group (0.14 g/day). Fourteen children in the control arm relapsed within 6 months. Thirteen children assigned to rituximab (87%) were still in remission at 1 year and 8 (53%) at 4 years. Responses were similar in children of the control group who received rituximab to treat disease relapse. We did not record significant adverse events.
Conclusions
Rituximab was non-inferior to steroids for the treatment of juvenile SDNS. One in two children remains in remission at 4 years following a single infusion of rituximab, without significant adverse events. Further studies are needed to clarify the superiority of rituximab over low-dose corticosteroid as a treatment of SDNS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Idiopathic steroid-dependent nephrotic syndrome (SDNS) is the most frequent cause of proteinuria in children with an incidence of 2–10 children per 100,000 per year in Western countries and a prevalence of 16 cases [1]. As the denomination of the disease suggests [1,2,3], and practice guidelines indicate (KDIGO), these children need to take corticosteroids (generally prednisone) to maintain disease remission, and often treatment continues for years. Due the common and severe adverse effects of corticosteroids, including growth retardation, diabetes, cataract and osteoporosis, corticosteroid-sparing drugs have been investigated in the past (i.e. calcineurin inhibitors, cyclophosphamide, levamisole, mycophenolate) [3,4,5,6], and none of them were safer than steroids.
Rituximab, a chimeric monoclonal anti-CD20 antibody, is increasingly being used as a corticosteroid-sparing agent for children with nephrotic syndrome, especially in those who require corticosteroids and calcineurin inhibitors to maintain remission [7]. To date, only two randomized clinical trials studies have assessed the use of rituximab in SDNS dependent on corticosteroids alone, and both included children requiring high-dose prednisone (i.e. 0.5–0.7 mg/day/day). These trials showed a positive and lasting effect of rituximab in maintaining disease remission for 1 year [8, 9], without significant adverse effects in this short-term follow-up.
Although rituximab-associated side effects are uncommon and often clinically irrelevant [10], concerns still exist about consequences of longer follow-up. In this study, we tested the effects of rituximab vs. corticosteroids in children with SDNS treated with very low doses of steroids (< 0.4 mg/kg/day). We hypothesized that rituximab could prolong remission for years in these children making this drug an ideal corticosteroid-sparing agent if safe in the long term.
Materials and methods
Between August 2013 and October 2019, we conducted a randomized controlled trial to determine whether rituximab is non-inferior to steroids in maintaining SDNS in remission in children treated with low-dose prednisone (0.1–0.4 mg/kg/day). Potential participants underwent a 1-month run-in period during which instruction on urine collection and dipstick readings were carefully reviewed, proteinuria was monitored, adherence assessed, and oral prednisone dose reduced to the minimum dose required in the previous 6 months. This study was registered in the European Clinical Trial Database (EudraCT: 2008-004486-26).
Participants
Eligible participants were 1–16 years of age with an eGFR> 60 ml/min per 1.73 m2. They had to have idiopathic, steroid-dependent nephrotic syndrome for a minimum of 6 to a maximum of 12 months at the time of study entry. Participants must have been receiving low-dose prednisone (< 0.4 mg/kg per day) continuously to maintain remission for the 3 months preceding enrolment. Participants were excluded if they had received calcineurin inhibitors at any time or had received cyclophosphamide or mycophenolate in the 6 months before enrolment. Nephrotic syndrome was defined as proteinuria in the nephrotic range (> 1, 000 mg/m2 per day or protein-to-creatinine ratio > 4 mg/mg in a single urine specimen) or proteinuria ranging from 250 to 1000 mg/m2 per day in association with hypoalbuminemia (serum albumin < 2.5 g/dl) or dyslipidemia (total cholesterol > 250 mg/dl). Remission was defined as stable proteinuria < 150 mg/m2 per day. Genetic testing and renal biopsy were not required because juvenile nephrotic syndrome is diagnosed largely on the basis of a response to prednisone, as per Kidney Disease Improving Global Outcome guidelines. Cases were considered corticosteroid dependent if patient responded to full doses of prednisone (2 mg/kg) but experienced two consecutive relapses during prednisone tapering or within 2 week of prednisone withdrawal. Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers had to be avoided during the study. We excluded children with previous episodes of microhematuria, a history of hepatitis B or C, HIV infection, positivity of any markers of autoimmunity (antinuclear antibody nuclear DNA, ANCA), and low complement C3 levels. Children requiring diuretics albumin or anticoagulant were also excluded.
Randomization and masking
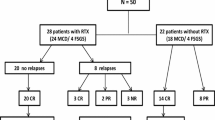
Participants were randomly assigned in a 1:1 ratio to continue oral prednisone alone (n 15, controls) or standard therapy plus a single infusion of rituximab (n 15, intervention). Relapses in children of the control group could be treated with rituximab using the same dose and protocol (see below). Randomization was done following a permuted-block design with block of variable sizes (Fig. 1). A distant site with no clinical involvement in the trial generated the randomization list and kept the allocation concealed. Assignments were notified electronically after signed consent was obtained (and assent for participants who were capable of assenting). Because of the nature of the intervention, the clinical investigators and study nurses were not blinded to group assignment, nor were the participants.

Scheme and flow-chart of the randomization process
Study treatments
In the intervention group, rituximab (MabThera/Rituxan; 375 mg/m2) was diluted in normal saline (1 mg/ml) and administered at a rate of 0.5–1.5 ml/min over approximately 6 h following the infusion of 2.5–5 mg of intravenous chlorphenamine maleate (based on the local protocol and patient tolerance), methylprednisolone (2 mg/kg) in normal saline and oral paracetamol (8 mg/kg). In both groups, prednisone was continued at the same doses as during the run-in, then tapered off by 0.3 mg/kg per week starting at 30 days and withdrawn if proteinuria levels were still < 1 g/m2 per day at the end of the taper. In both groups, prednisone 60 mg m2was restarted if proteinuria relapsed during the follow-up and tapered using the same scheme relapses in both groups could be treated with rituximab utilizing same doses and protocol.
Follow-up
Study visits occurred at baseline and every 3 months thereafter, unless complications or relapses occurred. Study coordinators maintained ongoing contact with the children, their families, and the family physician to collect clinical data, including BP and potential adverse events. Proteinuria was evaluated daily using a simple dipstick test and monthly using a 24-h urine collection. Because of the frequent false positivity of dipstick, we planned a 24-h urine collection for readings > = 1+. Kidney function, plasma proteins, and cholesterol values were measured monthly. White blood cell counts were monitored monthly in the rituximab group.
Outcomes
Participants randomly assigned to rituximab therapy were compared with those receiving standard care for the primary outcome of the percentage change in daily proteinuria at 6 months. The primary outcome was assessed at 3 months because most SDNS relapses occur within 2 weeks of prednisone withdrawal. We assessed the risk of relapse during the year following randomization as a secondary endpoint and collected data on repeated relapses beyond the initial year. Relapse was defined as proteinuria of 1000 mg/m2 per day (protein-to-creatinine ratio > 4 mg/mg) or proteinuria > 500 mg/m2 per day (protein-to-creatinine ratio 2–4 mg/mg). Long-term side effects after a single infusion of rituximab were recorded up to 4 years in all participants who received rituximab.
Statistical analyses
This study was designed to evaluate whether rituximab was non-inferior to standard care in maintaining proteinuria levels in remission with a non-inferiority margin of 3 for the geometric mean ratio. This margin was based on previous studies, which reported average proteinuria in children in remission as < 300 mg/m2 per day; a level three times this value remains below the 1000 mg/m2 per day threshold at which prednisone treatment is usually started to treat relapses. We assumed a log-normal distribution of proteinuria and used information from a prior study on its SD. With a coefficient of variation of 0.85 and a type I error rate of 0.01, we estimated that 30 participants would provide a power of 90% to detect a ratio of the geometric means of proteinuria between treatment and control groups < 3, after accounting for a 5% risk of withdrawals. We analysed outcome data according to the intention-to-treat principle, with no interim subgroup analyses. We modelled a 3-month log-transformed proteinuria using an analysis of covariance model with treatment as a factor and log-transformed baseline proteinuria as covariate (primary analysis). We used the Kaplan-Meier method to describe 1-year relapse-free survival and Cox regression to estimate the effect of treatment. We censored participants at the study end date if they were event free or at the time they left the study (main analyses). In sensitivity analyses, we assumed that the event occurred at the last observation time for participants who left the study before the planned 1-year follow-up. We used two-sided test with a significance level of 0.05 for all analyses. We used Stata software version 13.1 and R software version3.0.2 for all analyses.
Results
Baseline clinical features
Median participant age was 6 (4–15) years in the control arm and 7.4 (4–14) years in the intervention arm; males were 80% in the control arm and 60% in the intervention arm; mean disease duration was variable from 5 to 109 months (Table 1). Before developing SDNS, participants experienced a median of 4 and 5 relapses in the control arm and in rituximab arm, respectively. Proteinuria levels were in the normal range and were similar in both groups at baseline, as were average prednisone doses, levels of baseline serum creatinine, cholesterol, and albumin. None had received other steroid-sparing agents before the study or had signs of steroid toxicity at enrolment.
Primary outcome
Proteinuria increased at 3 months in the control group, from 0.11 (median; range 0.04–0.4) to 1.9 g/day (0.05–4), and remained unchanged in the rituximab group, from 0.14 (0.03–0.3) to 0.1 (0.04–0.25). After we accounted for baseline proteinuria, 3-month proteinuria was 86% lower in the treatment group than in the control group (ratio of means, 0.14; 95% CI, 0.06 to 0.32). The upper CI was below the prespecified non-inferiority margin of 3 (primary hypothesis), and below the threshold for superiority of 1 (p < 0.01 accounting for multiple testing).
Secondary outcome
Figure 2 summarizes 1-year relapse-free survival by the treatment arm. Most patients in the control arm relapsed during tapering of prednisone (i.e., 12 at T3, 13 at T6), and all but one had nephrotic syndrome within 12 months of tapering. No child in the rituximab group relapsed within 6 months of randomization, and 13 were still in remission at 1 year with a relapse-free survival of 87% (95% confidence interval 71 to 100%).

Clinical outcome at 1 year from withdraw of Prednisone. Percent of patients free of relapse during the first year after randomization (Kaplan Meier analysis).Patients of the intervention arm (corticosteroid + rituximab) are marked in green and patients randomized to the control group (corticosteroid only) are marked in red. After 3 months, patients in both group tapered and eliminated steroids
Long-term outcome
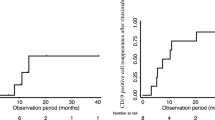
Figure 3 shows that 8 children assigned to rituximab (53%) were still in remission at 4 years. Of the children who were assigned to the control group, 10 were treated with rituximab following relapse. Six of them (60%) were still in remission 1 year following rituximab infusion, and 4(40%) were in remission at 4 years.

Number of patients in persistent remission after a single infusion of rituximab. Black dots are those patients originally treated with rituximab and grey dots are those patients of the control group who were treated with rituximab after that proteinuria had recurred following corticosteroids withdraw
Lymphocyte counts and immunoglobulin serum levels
CD20 counts were reduced to < 3% at 1 month in all participants treated with rituximab (Fig. 4). After 3 months, CD20 counts remained undetectable in all of these children. Median time to CD20 reconstitution was 5.8 months (range 4 to 12 months).

CD20 counts after rituximab in patients of the experimental cohorts after infusion of a single dose of drug. Each line corresponds to a single patient
Immunoglobulin levels over time are reported in Fig. 4. Medians and interquartile ranges demonstrate levels at 6 months from rituximab infusion comparable with those at baseline.
Adverse events
During rituximab infusion, few children had minimal skin rash that resolved by slowing the infusion rate. One child required higher dose of chlorphenamine. One participant had acute arthritis at the hip joint 52 days after rituximab infusion. Resolution was rapidly and completely achieved in 48 h with non-steroidal anti-inflammatory medications (Fig. 5).

IgG serum levels (gr/l) before and after rituximab in patients originally randomized to receive a single dose of drug. Results are expressed as median and interquartile ranges
Discussion
Rituximab has emerged as a main therapy for idiopathic nephrotic syndrome in children and adults [11, 12] and seems particularly effective in SDNS according to two randomized studies [8, 9]. This is the third randomized controlled trial designed to determine if rituximab is effective in maintaining remission of proteinuria in children with SDNS. As opposed to previous trials that tested the effects of rituximab in children with SDNS dependent on high doses of corticosteroids [8, 9], this trial included children dependent on very low steroid doses (0.1–0.4 mg Kg). This trial provides important information to assist clinical practice. Given the high rates of spontaneous remission in forms of SDNS dependent on very low-dose corticosteroids non-randomized studies are at high risk of selection bias. The results of this trial indicate that rituximab is non-inferior to corticosteroids in maintaining disease remission at 3 months, and likely superior both in the short and long term, although further trials with superiority design need to be completed to confirm our findings in other populations. These data are in line with other studies showing efficacy of rituximab in children with nephrotic syndrome dependent on high dose of steroids [8, 9].
A secondary aim of our study was to assess the long-term effects of a single dose of rituximab. Most patients of the experimental arm and 10 controls who received rituximab after they relapsed had 4-year follow-up data, which exceeds follow-up duration of previous studies [8, 9, 11,12,13]. These data suggest rituximab treatment may have extremely favourable long-term effects, in terms of benefits and possible harms. We found that one in two children is still in remission at 4 years from rituximab infusion with the need of corticosteroid treatment. This trial indicates that rituximab may maintain far longer corticosteroid-free remission in children with the mildest form of SDNS than other children with low sensitivity to steroids (i.e., dependent on high-dose corticosteroids alone) or those who require other drugs including mycophenolate and calcineurin inhibitors. Data from the collaborative international multicentre study that collectively evaluated the outcome in 511 children treated with rituximab for multidrug dependence [14] indicate that the mean duration of remittance after one rituximab infusion in the absence of immune suppression is 8.5 months and increases to 12.7 and 14.7 months in patients treated with 2 and 4 pulses, respectively. Besides differences in design (randomized vs non-randomized design) and in population characteristics (SDNS vs. multidrug-dependent forms of nephrotic syndrome), in our protocol, we gave rituximab in a single dose as opposed to 2–4 doses as in the majority of previous studies. Considering that the effect duration (relapse free time) may vary in individual patients, the use of a single-dose regimen has the advantage of lower cumulative doses and opens to personalized therapeutic load of the drug. This approach allows minimizing possible adverse effects both in the short and in the long period. For example, we had only minimal adverse effects over time and did not find cases of hypogammaglobulinemia at 6–24 months. To our knowledge, this is the first study reporting immunoglobulin levels in patients with SDNS not treated previously with any adjunctive immunosuppressant and who received a single rituximab infusion. Other studies reported hypogammaglobulinemia in patients treated for at least 6 months with calcineurin inhibitors of mycophenolate [15] or who received more than one dose of rituximab [10, 16]. We should take into account this important finding in considering risks linked with rituximab. Given the favourable clinical outcome of a single dose, clinicians should consider the utility of rituximab in this particular category of patients balancing positive and negative effects compared with corticosteroids.
Conclusion
In conclusion, our study shows that a single dose of rituximab is non-inferior, and may be superior, to corticosteroid treatment in maintaining remission of proteinuria in children with SDNS at 3 months. According to our study, rituximab may also be effective and safe in the longer term. These data open the question of whether, in children with SDNS, rituximab may replace steroid treatment earlier in the course of the disease.
References
Radhakrishnan J, Cattran DC (2012) The KDIGO practice guideline on glomerulonephritis: reading between the (guide)lines--application to the individual patient. Kidney Int 82:840–856
Hodson EM, Knight JF, Willis NS, Craig JC (2005) Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst Rev CD001533
Filler G (2003) Treatment of nephrotic syndrome in children and controlled trials. Nephrology, Dialysis, Transplantation : Official Publication of the European Dialysis and Transplant Association - European Renal Association 18 Suppl 6:vi75-78
Ghiggeri GM, Catarsi P, Scolari F, Caridi G, Bertelli R, Carrea A, Sanna-Cherchi S, Emma F, Allegri L, Cancarini G, Rizzoni GF, Perfumo F (2004) Cyclosporine in patients with steroid-resistant nephrotic syndrome: an open-label, nonrandomized, retrospective study. Clin Ther 26:1411–1418
Sinha A, Puraswani M, Kalaivani M, Goyal P, Hari P, Bagga A (2019) Efficacy and safety of mycophenolate mofetil versus levamisole in frequently relapsing nephrotic syndrome: an open-label randomized controlled trial. Kidney Int 95:210–218
Gruppen MP, Bouts AH, Jansen-van der Weide MC, Merkus MP, Zurowska A, Maternik M, Massella L, Emma F, Niaudet P, Cornelissen EAM, Schurmans T, Raes A, van de Walle J, van Dyck M, Gulati A, Bagga A, Davin JC, all members of the Levamisole Study G (2018) A randomized clinical trial indicates that levamisole increases the time to relapse in children with steroid-sensitive idiopathic nephrotic syndrome. Kidney Int 93:510–518
Ravani P, Bonanni A, Rossi R, Caridi G, Ghiggeri GM (2016) Anti-CD20 antibodies for idiopathic nephrotic syndrome in children. Clinical Journal of the American Society of Nephrology : CJASN 11:710–720
Ravani P, Rossi R, Bonanni A, Quinn RR, Sica F, Bodria M, Pasini A, Montini G, Edefonti A, Belingheri M, De Giovanni D, Barbano G, Degl'Innocenti L, Scolari F, Murer L, Reiser J, Fornoni A, Ghiggeri GM (2015) Rituximab in children with steroid-dependent Nephrotic syndrome: a multicenter, open-label, noninferiority, randomized controlled trial. J Am Soc Nephrol 26:2259–2266
Basu B, Sander A, Roy B, Preussler S, Barua S, Mahapatra TKS, Schaefer F (2018) Efficacy of rituximab vs tacrolimus in pediatric corticosteroid-dependent nephrotic syndrome: a randomized clinical trial. JAMA Pediatr 172:757–764
Bonanni A, Calatroni M, D'Alessandro M, Signa S, Bertelli E, Cioni M, Di Marco E, Biassoni R, Caridi G, Ingrasciotta G, Bertelli R, Di Donato A, Bruschi M, Canepa A, Piaggio G, Ravani P, Ghiggeri GM (2018) Adverse events linked with the use of chimeric and humanized anti-CD20 antibodies in children with idiopathic nephrotic syndrome. Br J Clin Pharmacol 84:1238–1249
Iijima K, Sako M, Nozu K, Mori R, Tuchida N, Kamei K, Miura K, Aya K, Nakanishi K, Ohtomo Y, Takahashi S, Tanaka R, Kaito H, Nakamura H, Ishikura K, Ito S, Ohashi Y, Rituximab for Childhood-onset Refractory Nephrotic Syndrome Study G (2014) Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet 384:1273–1281
Ravani P, Magnasco A, Edefonti A, Murer L, Rossi R, Ghio L, Benetti E, Scozzola F, Pasini A, Dallera N, Sica F, Belingheri M, Scolari F, Ghiggeri GM (2011) Short-term effects of rituximab in children with steroid- and calcineurin-dependent nephrotic syndrome: a randomized controlled trial. Clinical Journal of the American Society of Nephrology : CJASN 6:1308–1315
Kemper MJ, Gellermann J, Habbig S, Krmar RT, Dittrich K, Jungraithmayr T, Pape L, Patzer L, Billing H, Weber L, Pohl M, Rosenthal K, Rosahl A, Mueller-Wiefel DE, Dotsch J (2012) Long-term follow-up after rituximab for steroid-dependent idiopathic nephrotic syndrome. Nephrology, Dialysis, Transplantation : Official Publication of the European Dialysis and Transplant Association - European Renal Association 27:1910–1915
Chan EY, Webb H, Yu E, Ghiggeri GM, Kemper MJ, Ma AL, Yamamura T, Sinha A, Bagga A, Hogan J, Dossier C, Vivarelli M, Liu ID, Kamei K, Ishikura K, Saini P, Tullus K (2020) Both the rituximab dose and maintenance immunosuppression in steroid-dependent/frequently-relapsing nephrotic syndrome have important effects on outcomes. Kidney Int 97:393–401
Fujinaga S, Ozawa K, Sakuraya K, Yamada A, Shimizu T (2016) Late-onset adverse events after a single dose of rituximab in children with complicated steroid-dependent nephrotic syndrome. Clin Nephrol 85:340–345
Parmentier C, Delbet JD, Decramer S, Boyer O, Hogan J, Ulinski T (2020) Immunoglobulin serum levels in rituximab-treated patients with steroid-dependent nephrotic syndrome. Pediatr Nephrol 35:455–462
Funding
This study was supported by Istituto Giannina Gaslini deriving from ‘Cinque per mille of IRPEF-Finanziamento della ricerca sanitaria’, the Italian Ministry of Health, The Renal Child Foundation, and the ‘Fondazione La Nuova Speranza’ (‘Progetto integrato per la definizione dei meccanismi implicati nella glomerulosclerosi focale’). GMG received a grant from Compagnia San Paolo (ROL-9849).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organization that might have an interest in the submitted work in the previous 3 years; and no other relationships or activities that could appear to have influenced the submitted work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ravani, P., Lugani, F., Pisani, I. et al. Rituximab for very low dose steroid-dependent nephrotic syndrome in children: a randomized controlled study. Pediatr Nephrol 35, 1437–1444 (2020). https://doi.org/10.1007/s00467-020-04540-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-020-04540-4