
Abstract
Background
Hemolytic uremic syndrome (HUS) is a leading cause of acute kidney injury in children. Although international guidelines emphasize comprehensive evaluation and treatment with eculizumab, access to diagnostic and therapeutic facilities is limited in most developing countries. The burden of Shiga toxin-associated HUS in India is unclear; school-going children show high prevalence of anti-factor H (FH) antibodies. The aim of the consensus meeting was to formulate guidelines for the diagnosis and management of HUS in children, specific to the needs of the country.
Methods
Four workgroups performed literature review and graded research studies addressing (i) investigations, biopsy, genetics, and differential diagnosis; (ii) Shiga toxin, pneumococcal, and infection-associated HUS; (iii) atypical HUS; and (iv) complement blockade. Consensus statements developed by the workgroups were discussed during a consensus meeting in March 2017.
Results
An algorithm for classification and evaluation was developed. The management of Shiga toxin-associated HUS is supportive; prompt plasma exchanges (PEX) is the chief therapy in patients with atypical HUS. Experts recommend that patients with anti-FH-associated HUS be managed with a combination of PEX and immunosuppressive medications. Indications for eculizumab include incomplete remission with plasma therapy, life-threatening features, complications of PEX or vascular access, inherited defects in complement regulation, and recurrence of HUS in allografts. Priorities for capacity building in regional and national laboratories are highlighted.
Conclusions
Limited diagnostic capabilities and lack of access to eculizumab prevent the implementation of international guidelines for HUS in most developing countries. We propose practice guidelines for India, which will perhaps be applicable to other developing countries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Hemolytic uremic syndrome (HUS) is an important cause of severe acute kidney injury (AKI) in childhood. A significant proportion of patients require renal replacement therapy and about one third shows features of chronic kidney disease. Across the world, the majority of HUS follows gastrointestinal infection with Shiga toxin-producing organisms, chiefly Escherichia coli. HUS might be associated with systemic illnesses (secondary HUS), and with disorders of complement regulation (atypical HUS, aHUS; Table 1). Management of the disease is supportive, with attention to AKI and therapy of underlying disorders. The availability of eculizumab, a complement inhibitor, has had major impact on treatment of aHUS in the developed world.
There are differences in the epidemiology and management of HUS in India compared to developed countries. Improving standards of hygiene and healthcare have led to decline in incidence of Shigella-associated HUS, the chief form of Shiga toxin-associated disease in the region [1]. There is limited information on the epidemiology of enterohemorrhagic E. coli, with reports not suggesting a role in etiology of diarrheal or dysenteric illnesses [2,3,4]. The capacity for diagnosis of Shiga toxin-associated HUS and genetic screening is limited, posing hurdles in etiological classification. Unlike European cohorts, aHUS associated with factor H (FH) antibodies is more common, accounting for ~ 50% of pediatric patients [5]. Finally, in absence of access to eculizumab, plasma therapy forms the basis for managing aHUS in India and most developing countries, including patients with anti-FH-associated disease.
International guidelines on management of HUS [6,7,8,9,10,11,12,13] that emphasize comprehensive evaluation and specific therapy with eculizumab for patients with aHUS might not be suitable in resource constrained settings. Given these challenges, the Indian Society of Pediatric Nephrology has proposed guidelines for evaluation and management of HUS in Indian children. These guidelines are likely to be suitable for developing countries with less than optimal availability and access to diagnostic and therapeutic facilities.
Consensus development
Workgroups were constituted to address key issues in management: (i) investigations, biopsy, genetics, and differential diagnosis; (ii) Shiga toxin, pneumococcal, and other infection-associated HUS; (iii) atypical HUS; and (iv) complement blockade. Prior to the meeting on 31 March and 1 April 2017 in New Delhi, each workgroup searched databases for research articles, identified the state of knowledge and formulated questions though emails. During the meeting, the workgroups developed consensus statements through alternating breakout and plenary sessions. Research studies were rated from A to D using standard criteria [14].
-
A.
Systematic review, well-designed randomized controlled trials, or diagnostic studies without significant limitations
-
B.
Randomized controlled trials or diagnostic studies with methodological limitations; consistent evidence from observational studies
-
C.
Small cohorts or case control studies; case series
-
D.
Expert opinion; case reports
Each consensus statement was assigned one of two levels of recommendation, based on assessment of relative benefit versus harm, and relevance in context of available facilities.
-
Level 1.
Recommendation applicable to most subjects, based on consistent information confirming benefit over harm or vice versa
-
Level 2.
Suggestion or option based on equivocal or insufficient evidence, with unclear balance of benefit over harm
The final manuscript was circulated to participants for approval.
Guideline 1: Diagnosis of HUS
Rapid diagnosis and prompt management of HUS is essential to limit irreversible renal damage. Patients with suspected HUS require urgent evaluation by a specialist to initiate appropriate care.
-
1.1
We recommend making a diagnosis of HUS in presence of the following [1B]:
-
(i)
Microangiopathic hemolytic anemia (MAHA), defined by anemia (hemoglobin < 10 g/dl, hematocrit < 30%), and fragmented red cells on peripheral smear (schistocytes ≥ 2%) with either elevated lactate dehydrogenase (LDH > 450 IU/l) or undetectable haptoglobin
-
(ii)
Thrombocytopenia (platelet count < 150,000/μl)
-
(iii)
AKI, defined as increase in serum creatinine by 50% over baseline level [15]
-
(i)
-
1.2
We recommend evaluation and exclusion of disseminated intravascular coagulation (DIC) and thrombotic thrombocytopenic purpura (TTP), when clinically indicated (see below). [1B]
-
1.3
We suggest exclusion of common infections that mimic or trigger HUS, e.g., malaria, leptospirosis, and dengue. [2C]
Rationale
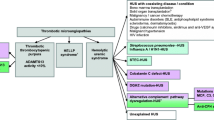
The diagnosis of HUS is based on presence of MAHA, thrombocytopenia, and AKI [6, 16]. The threshold for each of these parameters is not well defined. Patients may occasionally have a subacute presentation with AKI, systemic hypertension, and renal histological features of TMA, without thrombocytopenia and microangiopathic hemolysis [13, 17,18,19]. The diagnostic sensitivity of HUS is increased if platelet counts are screened at multiple time points [11]. Differentiating DIC from HUS is necessary, especially in the setting of sepsis or malignancy (Fig. 1). DIC is characterized by prolonged prothrombin time or activated partial thromboplastin time, low fibrinogen, elevated D-dimer and soluble fibrin monomers [20]; haptoglobin levels are normal (30–100 mg/dl) [11].
Online scoring criteria for diagnosis of DIC are available [21]: www.thecalculator.co/health/Disseminated-Intravascular-Coagulation-(DIC)-Score-Calculator-1048.html. Patients with Shigella-associated HUS may occasionally show features of DIC [1].

Evaluation of patients with hemolytic uremic syndrome (HUS). Patients with secondary and infection triggered HUS should also be screened for abnormalities of the alternate complement pathway. CD46 membrane cofactor protein; DIC, disseminated intravascular coagulation; LDH, lactate dehydrogenase; TTP, thrombotic thrombocytopenic purpura. *Consider renal biopsy if triad not present and/or unexplained acute kidney injury. **Also consider atypical HUS if positive non-synchronous family history, recurrent disease, or insidious disease
TTP is a rare disorder in children, with inherited and acquired TTP reported in 2.4% and 4.6% patients, respectively of a French cohort of patients with thrombotic microangiopathy (TMA) [22]. Confirmation is feasible, but assays for ADAMTS13 (A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13) activity are available at limited centers and have a long turnaround time.
Due to its rarity, a recent meeting of the KDIGO did not advise routine evaluation for ADAMTS13 activity in children with HUS [23]. TTP should be suspected in patients with severe, persistent thrombocytopenia (< 30,000/μl) [13, 24] and mild or no AKI [25, 26]. Severe AKI is, however, reported in 10–12% patients with idiopathic TTP [22, 25]. Congenital TTP (Upshaw Schulman syndrome) has varied phenotype, and may present in neonates with MAHA and jaundice, or in children with unexplained thrombocytopenia. It is characterized by ADAMTS13 activity < 5%, absent antibodies to ADAMTS13, and a homozygous or compound heterozygous mutation in ADAMTS13 [27].
Due to limited availability of the ADAMTS13 assay, we suggest storing blood samples (collected in 3.2% sodium citrate) at −20° to −70 °C in patients with suspected HUS. The possibility of TTP should be revisited if the etiology of TMA is unclear. The diagnosis is confirmed by demonstrating ADAMTS13 activity < 10% on fluorescence resonance energy transfer (FRET)-based assays and/or the presence of anti-ADAMTS13 antibodies.
HUS may at times mimic or occur in association with infections such as malaria [28], dengue [29], and leptospirosis [30]. These disorders might result in anemia, thrombocytopenia, and AKI that is not due to renal microangiopathy.
Guideline 2: Shiga toxin-associated HUS
Shiga toxin-associated gut infection is one of the chief causes of HUS and confirmed by stool examination.
-
2.1
We recommend that diagnosis of Shiga toxin-associated HUS, either due to Shiga toxin-producing Escherichia coli (STEC) or Shigella dysenteriae, be based on the following criteria. [1A]
Confirmed case
HUS associated with infection with Shiga toxin-producing organisms confirmed by positive stool culture and either of the following:
-
(i)
Detection of virulence genes on fecal extracts or cultures (stx1, stx2, and eae by PCR)
-
(ii)
Free fecal Shiga toxin (detected on tissue culture or immunoassay)
-
(iii)
Serum IgM antibodies to serogroup specific lipopolysaccharide (by ELISA, passive hemagglutination assay) (Table 2)
Suspected case
HUS occurring within 2–3 weeks of bloody diarrhea and/or occurring during a known outbreak of STEC-HUS in patients above 6 months of age
-
2.2
We suggest screening for STEC infection in all suspected cases of STEC-HUS on stool samples collected at admission. [2A]
Rationale
Guidelines from the Center for Disease Control recommend stool culture and testing for Shiga toxin to detect serotypes of STEC [31]. Infection with STEC is the chief leading cause of HUS in developed countries [18], but fecal isolation is difficult if patients present after the first 7–10 days of illness and following antibiotic administration [32]. Direct screening of fecal extracts or cultures for Shiga toxin is possible using molecular biology, biochemical techniques, or Vero cell cultures [13, 31]. PCR analysis on primary fecal cultures is the most sensitive and specific investigation for screening for the toxin-producing organisms [33]. Isolation of the STEC strain and its characterization by serotype, phage type, eae genes, and subtypes of vt1/vt2 should be done if possible [31]. The presence of serum IgM antibodies against specific E. coli lipopolysaccharide is important evidence for recent STEC infection [34].
While circulation of S. dysenteriae is extremely low in recent years in India [35], there is limited data regarding the epidemiology of STEC. In a study from north India, STEC was isolated in 1.7% stool samples of acute non-bloody diarrhea and 1.6% samples of bloody diarrhea [2]. The authors found serogroup diversity of non-O157 STEC; O157 was not isolated, signifying either its absence or presence in low numbers [2]. Non-O157:H7 STEC was detected by PCR for stx and eae in 23% of 198 patients with acute diarrhea in southern India; only three isolates were identified as STEC by serotyping [3]. In the absence of epidemiological data of STEC-HUS in India, we suggest confirming the diagnosis by detection of fecal Shiga toxin as well as by culture. Since excretion of STEC is short-lasting (median 10–14 days; 92% detection rate within 6 days) [36, 37], delayed referral and prior antimicrobial use reduce the likelihood of detection. In such patients, presence of IgM antibodies against specific lipopolysaccharide enables the diagnosis [34]. Demonstration of elevated serum IgG antibodies against Shiga toxin, by ELISA, Western blot, or tissue culture cytotoxicity neutralization assay is chiefly an epidemiologic tool.
Five to 10% patients with sporadic STEC infection and ~20% patients during outbreaks develop HUS. Risk factors for occurrence of HUS are: young age (< 5 years), > 3 days of diarrhea, blood in stools, and high leukocyte count (> 15,000/μl) [38, 39]. While international guidelines state that patients with HUS should be screened for STEC infection, access to microbiological diagnosis is difficult in developing countries. In view of insufficient information on epidemiology, lack of surveillance of food borne pathogens, and limited facilities for microbiological confirmation, the diagnosis of STEC-HUS is usually clinical. The present guidelines suggest suspecting STEC-HUS in patients with prior history of bloody diarrhea, or if occurring during an outbreak of STEC infection. However, it is emphasized that ~ 30–40% cases of STEC-HUS may not have dysentery; the condition is also reported in patients without diarrhea and infants below 6 months of age [13].
Pneumococcal and other infection-associated HUS
HUS may also be associated with specific infections (Table 1). A diagnosis of probable pneumococcal HUS is made in patients, usually younger than 2 years, with sepsis, pneumonia or meningitis, and positive direct Coombs’ test without features of DIC [40, 41]. The diagnosis is confirmed by either: (i) S. pneumoniae isolated by bacterial culture, or detection of pneumococcal antigen by PCR or ELISA in appropriate body fluids or, (ii) positive peanut lectin (Arachis hypogaea) agglutination assay [40, 41]. Therapy is with parenteral cephalosporins; vancomycin is used if there is high prevalence of multi-drug resistance [42]. Some authors suggest avoiding plasma infusions or plasma exchanges (PEX) and using washed red cells and platelets, if necessary.
If indicated, serology for human immunodeficiency virus; polymerase chain reaction (PCR) for influenza virus (H1N1), cytomegalovirus, and Epstein-Barr virus; peripheral smear and quantitative buffy coat for malaria parasite; NS-1 antigen or dengue-IgM antibody; and investigations for leptospirosis and rickettsia are performed.
Guideline 3: Cobalamin deficiency-associated HUS
-
3.1
We suggest screening for cobalamin C (cblC) deficiency by estimating total homocysteine levels. [2C]
Probable cblC deficiency-related HUS
HUS and elevated plasma total homocysteine level (> 50–100 μM/l by chromatography, immunoassay) with normal levels of vitamin B12 and folate.
Confirmed cblC deficiency-related HUS
Homozygous or compound heterozygous mutation in the MMACHC gene in a patient with probable cblC deficiency.
-
3.2
We recommend that patients with probable or confirmed cblC deficiency should receive prompt therapy with parenteral hydroxycobalamin, oral betaine and folic acid. [1B]
Rationale
Cobalamin deficiency accounts for a small (~ 6–8%) proportion of patients with HUS. The condition may present at all ages, ranging from neonates to adults, and does not respond to PEX [18]. Features include failure to thrive, feeding difficulties, seizures, abnormal muscle tone, visual impairment, and developmental delay. Megaloblastic anemia is present in 10–25%. One-third patients do not have extrarenal features, although neurological (44%) or cardiopulmonary (39%) disease and pulmonary hypertension (17%) are frequent [43].
Elevated blood levels of total homocysteine are characteristic. Samples may be stored and processed later, if the assay is not immediately available. Plasma samples should be centrifuged within 30 min of collection and stored at − 20° to − 70 °C (Fig. 1). Free homocysteine levels are not required; vitamin B12 and folate deficiency also result in elevated total homocysteine [44]. Plasma and urine methylmalonic acid levels are high in cblC deficiency, but may be normal in cblD (MMADHC) deficiency [44].
The mortality in patients with untreated disease is high [18, 43]; early treatment is therefore recommended [44]. Patients respond to therapy with parenteral hydroxycobalamin (1 mg daily) to lower homocysteine levels < 40–60 μM/L; cyanocobalamin is not effective. Betaine (250 mg/kg/day in 3 divided doses) helps methylating homocysteine to methionine. Folate (5–30 mg divided in 2–3 doses per week) is a useful adjunct [44]. Specific therapy allows renal recovery and prevents relapses.
Guideline 4: Diagnosis of atypical HUS (aHUS)
-
4.1
We suggest the diagnosis of aHUS where suspected STEC and pneumococcal HUS, TTP, and secondary HUS are excluded based on clinical and laboratory features. [2D]
-
4.2
We also suggest a diagnosis of aHUS in patients with (i) negative results for infection by Shiga toxin-producing organism, (ii) positive non-synchronous family history, or (iii) recurrent disease. [2C]
Rationale
Many algorithms are proposed for evaluation of patients with TMA [7, 8, 11, 13]. In resource-limited settings, it is important to follow a pragmatic approach without compromising the principles of management. We endorse the etiology-based classification of HUS proposed by international guidelines, emphasizing the importance to differentiate between infection-associated illness, cblC deficiency, secondary HUS, and aHUS (Table 1, Fig. 1) [19, 45, 46]. Since TTP and cblC deficiency are relatively infrequent causes, units lacking facility to screen for these disorders might store samples for subsequent analysis. Confirmation of STEC-HUS is difficult due to limited diagnostic facilities, delayed analysis, and prior use of antibiotics. Plasma specimens may be stored for later serological investigations.
Guideline 5: Evaluation of atypical HUS (Table 2, Fig. 1)
-
5.1
We recommend the following evaluation in patients with aHUS: (i) Complement C3; (ii) test for anti-factor H (FH) antibodies; (iii) flow cytometry for expression of membrane cofactor protein (MCP, CD46) on neutrophils; and (iv) sequencing of CFH, CFI, CFB, C3, CD46, THBD, and DGKE. Blood specimens for C3 and anti-FH antibodies should be drawn prior to instituting therapy. [1B]
-
5.2
We recommend estimating anti-FH antibodies using enzyme-linked immunosorbent assay (ELISA). A positive threshold for the assay should be determined based on data in the local population. [1C]
-
5.3
We suggest that patients with anti-FH antibody-associated HUS do not routinely require screening for genetic variants. Genetic screening is advised with (i) early age of onset, (ii) relapsing course, (iii) family history of HUS, (iv) illness that is refractory to therapy with PEX, and (v) prior to renal transplantation. [2C]
Rationale
Atypical HUS is characterized by dysregulation of the alternative complement pathway, resulting in endothelial damage and microvascular thrombosis [47]. About 40% patients have a pathogenic variant in genes encoding regulatory proteins or factors of the alternative complement pathway (factor H, factor I, CD46, factor B, C3, and thrombomodulin) or rearrangements of genes encoding factor H (FH)-related proteins [48]. In addition to inherited defects, antibodies to FH (anti-FH), the chief regulatory protein of the alternative pathway, is an acquired cause of aHUS comprising a distinct subgroup of patients. Recent reports suggest that mutations in genes outside the complement pathway might be associated with aHUS phenotype. Recessive mutations in DGKE, which encodes diacylglycerol kinase-ε and is expressed in endothelial cells, platelets, and podocytes were identified in 2–3% patients with aHUS, presenting in the first 2 years of life [49]. Others report the association of aHUS with mutations in genes encoding plasminogen, inverted formin-2 and vitronectin [49,50,51]. The distinction between disease due to complement and non-complement genes is important for therapeutic purposes.
Evaluation of patients with aHUS is done in a step-wise manner, adapted to availability of resources (Table 2). Blood levels of C3 are reduced in 30–40% patients but are often normal. A low C3 level is not specific for aHUS, nor does it correlate with severity of disease. Flow cytometry for surface expression of CD46 is a useful screening test; labs are advised to simultaneously run specimens from healthy controls. Reduced expression that persists following hematological remission helps identify patients with possible CD46 mutation. Because these patients are unlikely to benefit from PEX, a precise diagnosis is important [52, 53].
Immune assays for FH, FI, and FB are expensive and require to be performed at specialized laboratories. Their normal levels do not rule out the possibility of normally expressed, but functionally impaired protein [9]. In our experience, measurement of blood levels of FH and FI has not been useful in screening children with aHUS. Estimation of levels is useful if a variant is found in the corresponding gene; if deficient, these levels may serve as a marker of efficacy of supplemental therapy.
Anti-FH antibody-associated illness constitutes ~ 50% of pediatric patients with aHUS in India, chiefly affecting children between the age of 5–15 years [5]. Most patients have high levels of antibodies (1000 to 20,000 AU/ml) [54]. Given the therapeutic implications, it is necessary to screen for anti-FH antibodies in all patients with aHUS. Methods for ELISA differ in approach to coating, blocking, incubation, and color development, with comparable results [55]. Sample-specific subtraction is done to rule out false positives and a reference arbitrary unit (AU/ml) scale, based on positive anti-FH serum is used for reporting. Using this method, a positive threshold of 150 AU/ml has been established for Indian controls [5]. There are efforts to establish an international standard that would enable comparison of data across centers. The current protocols are preferred over commercial ELISA kits, that apart from being expensive, may show false positive results and underestimate antibody titers, limiting their role during follow up; a positive threshold for these kits is also not defined [55].
Next-generation sequencing (NGS) allows rapid and simultaneous sequencing of large gene panels. Screening for the following genes using a NGS panel: CFH, CFI, CFB, C3, CD46, THBD, CFHR1–5, and DGKE and for rearrangements of CFH-CFHR5 by multiplex ligation-dependent probe amplification (MLPA) is recommended in patients with suspected aHUS to guide regarding prognosis, risk of relapses, transplantation, and genetic counseling. Homozygosity for risk haplotypes of CFH and MCP has been shown to increase disease penetrance and severity [56]. Variants identified on NGS should be validated by Sanger sequencing. Expert opinion is necessary for ascribing pathogenicity. A database of gene variants is available at: http://www.complement-db.org/home.php.
Anti-FH-associated aHUS is associated with an 84 kb homozygous deletion of the CFHR1 gene, present in ~ 5–10% healthy individuals across the world [54, 57]. MLPA or end-point PCR is useful in confirming the deletion, but not required in clinical practice [58]. Detection of deletion helps confirm antibody mediated disease, in patients with (i) low antibody titers, especially if received PEX before screening; (ii) onset of illness < 3 years of age; and (iii) atypical presentation.
The need for additional genetic screening in patients with anti-FH-associated aHUS is debated. Of 296 patients with anti-FH positive illness screened, 5% show change in complement regulatory genes, mostly polymorphisms or variants of unknown significance [59 unpublished data]. Thus the likelihood of detecting pathogenic mutations in complement genes in patients with anti-FH-associated aHUS is low. Evaluation for additional defects is suggested in patients with a refractory, relapsing or atypical illness and before renal transplant.
Guideline 6: Indications for renal biopsy
-
6.1
We recommend that renal biopsy be considered in patients with (i) unclear diagnosis of HUS, (ii) unsatisfactory clinical response, to determine extent of renal damage and help in prognosis, and (iii) distinguish between causes of allograft dysfunction, including recurrent HUS. [1B]
Rationale
The diagnosis of HUS is clinical. A renal biopsy is not required to demonstrate TMA and adds little to the etiology or prognostic information at onset. Histological diagnosis is required in patients with acute or subacute onset of unexplained renal dysfunction, proteinuria and/or hypertension, who might not show hemolysis or thrombocytopenia. A renal biopsy is important for confirming the diagnosis and establishing reliable renal prognosis in secondary HUS, particularly in context of lupus nephritis, antiphospholipid syndrome, associated malignancies and following hematopoietic stem cell, or solid organ transplant. Biopsy of the allograft may provide the first clue of TMA, especially in patients with aHUS undergoing renal transplantation. Adequate precautions are necessary before the biopsy, with attention to thrombocytopenia, coagulation parameters, and hypertension. The biopsy should be interpreted by a renal pathologist; histology findings of TMA have recently been reviewed [23].
Guideline 7: Therapy of Shiga toxin-associated HUS
-
7.1
We recommend maintaining hydration by early use of isotonic fluids in patients with dysentery, starting from onset of bloody diarrhea to the day of onset of HUS, and monitoring for fluid overload in patients with renal failure. [1B]
-
7.2
We recommend therapy with appropriate antibiotics for bloody diarrhea. [1A]
-
7.3
While we do not suggest the use of PEX in patients with Shiga toxin-associated HUS, therapy may be considered for patients with severe neurological or cardiac involvement. [2D]
-
7.4
We do not recommend the use of plasma infusions, heparin, urokinase, dipyridamole, antimotility agents, glucocorticoids, and Shiga toxin binders. [1B]
Rationale
Patients with HUS due to proven or presumed STEC infection show an association between dehydration and adverse outcomes [60]. A review of cohort studies in children with presumed or proven STEC infection showed that high hematocrit (> 23%) at presentation and lack of fluid administration before onset of HUS was associated with higher risk of oliguric AKI, need for renal replacement therapy, and neurological complications [60,61,62,63,64,65]. Ringer lactate or dextrose saline should be infused during bloody diarrhea and including the day of diagnosis of HUS to prevent dehydration. Since administration of intravenous fluids is not always feasible, the use of oral rehydration solution to maintain euvolemia is an alternative, but which has not been examined. Since fluid overload is an important predictor of mortality in AKI [15], we recommend judicious monitoring of fluid balance subsequently [7].
Patients with shigellosis require prompt antimicrobial treatment in order to reduce mortality, fecal shedding and complications [66]. While there is limited circulation of S. dysenteriae in India [35], bacteriological confirmation is often not feasible. The WHO recommends ciprofloxacin as first-line therapy for shigellosis [66]: ceftriaxone, azithromycin, and cefixime are alternate agents [67]. The role of antibiotics in STEC gastroenteritis is debated, since these might induce expression of Shiga toxin [68] and increase the risk of HUS [69]. Recent reviews and cohort studies do not show an increased risk of HUS with use of antibiotics [39, 68, 70,71,72,73,74]. Since clinical distinction between shigellosis and STEC infection may be difficult, we recommend that patients with bloody diarrhea receive treatment with oral ciprofloxacin, azithromycin or cefixime for 5 days.
The American Society of Apheresis does not recommend PEX for patients with Shiga toxin HUS [75]. Two multicenter randomized trials did not show benefit of plasma infusions over supportive care [76, 77]. Data in adults [72, 78] and children [79] from the recent German outbreak did not show benefit of PEX in reducing the need for dialysis and respiratory or neurological complications. While PEX reduced mortality in adults (31% versus 83% with or without PEX, respectively) [80], it did not improve outcomes of patients with severe neurological involvement [81]. Based on weak evidence, a recent systematic review of observational data suggests a possible improvement in outcomes of patients older than 60 years of age and children with severe systemic involvement administered PEX within 24–48 h of onset [82].
Since there is evidence of activation of the complement pathway during acute STEC infection [83], complement blockade may have a role in treating patients with severe disease. While there was no proven benefit with use of eculizumab, a humanized monoclonal antibody against C5 that blocks the terminal complement pathway, in the German epidemic [72, 78], anecdotal reports suggest that patients with severe neurological and cardiac features might benefit from such therapy [84, 85]. A retrospective study on 28 patients treated with eculizumab for severe STEC-HUS showed neurological improvement in 67.9%, the majority improving after the first dose of the medication [86]. A review reported no benefit with plasma infusions, heparin, urokinase, dipyridamole, antimotility agents, steroids, and Shiga toxin binders [87].
Guideline 8: Managing atypical HUS without anti-FH antibodies
Inherited defects of the alternative pathway are the chief cause of aHUS in Europe and North America. The standard of care for patients is complement blockade with eculizumab, a humanized anti-C5 antibody [88]. However, eculizumab is expensive and unlikely to be available soon in India and other developing countries. Thus, PEX or plasma infusions remain the chief option for patients with aHUS, especially those with genetic defects in the complement pathway.
-
8.1
In the absence of eculizumab, we recommend prompt initiation of PEX in patients with aHUS. For initial therapy, we recommend that PEX be preferred to plasma infusions. [1C]
-
8.2
We suggest that PEX be administered daily until hematological remission and then tapered over 4–6 weeks (Table 3). [2D]
-
8.3
Patients on plasma therapy should be monitored for plasma or filter reactions, complications of catheter insertion, infection or thrombosis, and blood-borne infections. [1C]
-
8.4
We recommend efforts to enable therapy with eculizumab in the following: (i) lack of remission despite 7–10 days of PEX, (ii) life-threatening features (seizures, cardiac dysfunction), (iii) complications due to PEX or vascular access, and (iv) inherited defect in complement regulation. [1C]
Rationale
Prior to approval and use of eculizumab, the 2009 European guidelines recommended prompt therapy of aHUS with PEX within 24-h of presentation [9]. PEX may be performed by membrane filtration or by centrifugation based methods [89] (Table 3). PEX is preferred to plasma infusions for initial therapy, since large volumes of plasma can be infused even in patients with oliguria. PEX also enables removal of dysfunctional proteins and antibodies, which might be present in a proportion of children with aHUS.
An audit on the safety of PEX from centers in Europe and North America showed procedure-related complications and hypersensitivity to plasma in one-third patients [90]. While there were risks of securing vascular access in infants, a multicenter study confirmed the safety of the procedure [91]. An audit of 2024 PEX sessions (n = 109 patients) in New Delhi showed adverse events in ~ 10%, including chills, abdominal pain, hypotension, urticaria, seizures, and hypocalcemia (unpublished data). Plasma hypersensitivity was rare, and rate of catheter-related infections was 1.45/1000 catheter-days. Parents should be counseled about the risk of blood-borne infections with repeated therapies. While use of Octaplas, sterile pooled human plasma treated by solvent detergent process, reduces the risk of hypersensitivity reactions and blood-borne infections [92], the product is not available in most developing countries.
Eculizumab
The outcome of aHUS from large cohorts managed with plasma therapy alone is unsatisfactory, with 30–50% patients progressing to end-stage renal disease at 1–5 years follow up [19, 93]. Complement blockade with eculizumab has emerged as specific therapy for aHUS with inherited defects in complement regulation. The evidence for its benefit is derived from prospective single arm industry sponsored trials in adults and children [88, 94,95,96]. In a trial on 17 adults refractory to plasma therapy, hematological remission was achieved by 1–2 weeks and sustained in 88% [88]; renal functions improved with 12% patients on chronic dialysis after 2 years therapy [95]. Another trial on 20 plasma-dependent adults with chronic kidney disease showed hematological remission in 90% [88] and increase in glomerular filtration rate over the next 2 years [95]. Two subsequent trials underscored the medium term efficacy of eculizumab as first-line treatment of aHUS in children [96] and adults [94]. Progression to end-stage renal disease or death occurred in fewer patients (6–15%), compared to those managed with plasma therapy [19, 93].
Despite lack of availability of eculizumab in developing countries, all efforts must be made to make the medication accessible for patients with aHUS who fail to respond adequately to PEX. Standard guidelines should be followed while instituting therapy with eculizumab including monitoring for complement blockade, dose interval, and duration of therapy (Supplementary Table 1). Hematological remission is expected at median of 7–8 days; patients lacking response by 10–14 days of therapy should be reviewed for: (i) inadequate dose, suggested by trough level < 50 μg/ml and CH50 > 20%; (ii) losses (nephrotic range proteinuria, or PEX); (iii) resistance due to ongoing infection, inflammation or surgery; and (iv) C5 variant with impaired binding to eculizumab [97].
Eculizumab is not indicated for therapy of non-complement disorders, e.g., DGKE or MMACHC mutations [98]. Information on efficacy of eculizumab for extrarenal manifestations is limited. Therapy was useful in a patient with digital gangrene and another with skin necrosis and intestinal perforation [99,100,101]. The medication has variable efficacy in managing neurological or myocardial manifestations [102,103,104]. The duration of therapy is not clear. Discontinuation of treatment results in relapses in 60–72% patients with CFH mutations, 43% with C3 defects, 37–50% with MCP mutations and ~ 10% with no identified defect [97].
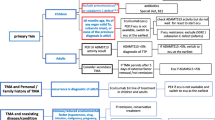
Guideline 9: Managing anti-FH antibody-associated HUS (Fig. 2)
-
9.1
We recommend a combination of prompt PEX (with fresh frozen plasma as replacement fluid) and immunosuppressive therapy for patients with anti-FH antibodies (Fig. 2). [1B]
-
9.2
We do not recommend use of immunosuppressive medications without confirming the presence of anti-FH antibodies. [1D]
-
9.3
We suggest daily PEX until hematological remission and then taper over 3–5 weeks. We do not recommend plasma infusions as a substitute for PEX. [2D]
-
9.4
Since high anti-FH levels might predict a relapse, we recommend monitoring antibody titers frequently during the first 12–24 months. [1C]
-
9.5
We suggest therapy with eculizumab in the following: (i) lack of remission despite 7–10 PEX; (ii) life-threatening features (seizures, cardiac dysfunction); (iii) complications due to PEX or vascular access; and (iv) inherited defect in complement regulation. [2C]

Management of patients with anti-factor H (FH)-associated atypical hemolytic uremic syndrome (aHUS). Initial therapy with plasma exchanges (PEX) is continued until hematological remission, followed by tapering over next 4–6 weeks (Table 3). Modification of therapy may be required in specific instances. *Prednisolone is given at a dose of 1 mg/kg/day for 4 weeks, then on alternate days for next 4 weeks, followed by taper every 2 weeks to 0.2–0.3 mg/kg/alternate days for 10–12 months. AU, arbitrary units; IV, intravenous; PEX, plasma exchange
Rationale
The aim of therapy for patients with anti-FH-associated HUS is reduction of antibody titers. The American Society for Apheresis assigns level I category to anti-FH-associated HUS, implying that PEX is a primary therapeutic intervention [75]. Since antibodies are chiefly IgG, 5–7 PEX achieve 80% reduction in antibody titers [105]. Prompt initiation and continued PEX for at least 3–5 weeks, is advised. Blockade of the complement pathway by eculizumab is effective in inducing hematological remission, but has limited impact on antibody titers.
We endorse guidelines that emphasize combined therapy with PEX and immunosuppressive agents for patients with anti-FH antibodies (Fig. 2) [13, 18]. Induction therapy with oral corticosteroids and IV cyclophosphamide is preferred initially. Immunosuppression inhibits further production of antibodies, especially following PEX, with improved short and medium term outcomes [54]. Outcomes were similar for patients managed by rituximab (n = 14) and intravenous cyclophosphamide (n = 31) [106].
Antibody titers are monitored closely between days 7–28 and then every 3–6 months. Elevated titer (> 1500 AU/ml) during the first 12–24 months is associated with an increased risk of relapse [54]. Relapses follow minor infectious illnesses in the initial 2 years. Therapy with azathioprine or mycophenolate mofetil and tapering doses of prednisolone is useful in significant reduction of this risk [5].
Many centers in the developed world initiate empiric therapy with eculizumab, pending anti-FH antibody results. Once high titer of anti-FH antibodies is detected, there is controversy whether eculizumab therapy be continued, or replaced by PEX and immunosuppression. Treatment with eculizumab does not affect generation of antibodies and immunosuppression might be required. The use of eculizumab has been reported in 18 patients with anti-FH-associated disease, either after failing PEX or as first-line therapy [97]. Since relapses were uncommon, the efficacy of concomitant immunosuppression in reducing antibody titers and the risk of relapse could not be determined. Prospective studies are required to determine the comparative efficacy of eculizumab to PEX and immunosuppression in these patients. Until then, we recommend the use of PEX with immunosuppression for management of these patients. Therapy with eculizumab should be considered in patients who are refractory to PEX, show life-threatening features, or have concomitant defect(s) in complement regulation [54].
Supportive care and monitoring
Standard recommendations should be followed for AKI with attention to fluid and electrolytes, avoiding radiocontrast and nephrotoxic agents, and timely institution of dialysis [15]. The choice of dialytic modality is based on feasibility and physician expertise. We suggest avoiding platelet transfusions unless count is < 10,000/μl, or to enable vascular catheter insertion. Blood transfusion is recommended for patients with hemoglobin < 6 g/dl or hemodynamic instability. Standard measures are instituted to retard CKD progression, especially control of hypertension and proteinuria [107]. Complications such as malignant hypertension, seizures, bowel infarction, stenosis or perforation, pancreatitis, and cardiomyopathy require appropriate management. All patients require follow up for at least 5 years for hypertension, proteinuria, and estimated GFR. Patients and their families should be counseled regarding the risk and recognition of relapses, and complications of the disease.
Transplantation
Patients with aHUS show variable risk of recurrent disease in the allograft. Donors and recipients need careful evaluation and peri-transplant management (Supplementary Table 2). Screening of recipients for mutations in relevant genes and for anti-FH antibodies is recommended. While the risk of recurrence is high in patients with dysregulation of the alternate pathway, those with STEC-HUS and abnormalities in membrane anchored (CD46) and intracellular (DGKE) proteins have low risk [56, 98, 108]. Families should be counseled about the risk of recurrence, based on presence and type of mutations and titer of anti-FH antibodies.
Live-related donation should be avoided in patients at high risk of disease recurrence [109]. Such patients require either combined liver kidney transplantation or therapy with eculizumab (peri- and post-transplant) [109], at an appropriate center. Patients with high titer of anti-FH antibodies and no additional defects are managed by PEX prior to and following transplantation, and use of rituximab. Live donors should be tested by MLPA for copy number variations in CFHR1/3. Since homozygous CFHR1 deletion is a risk factor for anti-FH-associated aHUS, it is unclear whether healthy live-related donors with a homozygous deletion should be accepted as donors.
Patients should be monitored for recurrence following transplantation [54, 110]. If recurrence of HUS is confirmed, all efforts should be made to procure and institute therapy with eculizumab.
Conclusions and perspectives
Guidelines from developed countries recommend comprehensive diagnostic assessment of patients with HUS, especially those with atypical disease. Compared to PEX, while terminal complement blockade with eculizumab has significantly improved outcomes in patients with aHUS, patients with anti-FH antibody-associated aHUS are satisfactorily managed with PEX and immunosuppression. Given limited diagnostic capabilities and lack of access to eculizumab, the international guidelines are not likely to be implemented in most developing countries in the near future. Table 4 summarizes practice guidelines for patients with HUS in India and other developing countries and compares them to international recommendations for the disease. We believe that it is desirable to invest resources in our country for setting up facilities for diagnosis and therapy (Table 5). Changing epidemiology of the disease and availability of novel agents that block the complement cascade might necessitate revision of these guidelines.
References
Srivastava RN, Moudgil A, Bagga A, Vasudev AS (1991) Hemolytic uremic syndrome in children in northern India. Pediatr Nephrol 5:284–288
Kumar A, Taneja N, Sharma M (2014) An epidemiological and environmental study of Shiga toxin-producing Escherichia coli in India. Foodborne Pathog Dis 11:439–446
Purwar S, Roy S, Metgud S (2016) Non-O157:H7 Shiga toxin producing diarrhoeagenic Escherichia coli (STEC) in southern India: a tinderbox for starting epidemic. J Clin Diagn Res 10:DC11–DC15
Hegde A, Ballal M, Shenoy S (2012) Detection of diarrheagenic Escherichia coli by multiplex PCR. Indian J Med Microbiol 30:279–284
Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, Saini H, Kotresh ST, Ali U, Bhatia D, Ohri A, Kumar M, Agarwal I, Gulati S, Anand K, Vijayakumar M, Sinha R, Sethi S, Salmona M, George A, Bal V, Singh G, Dinda AK, Hari P, Rath S, Dragon-Durey MA, Bagga A, Indian HUS Registry (2014) Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int 85:1151–1160
Sawai T, Nangaku M, Ashida A, Fujimaru R, Hataya H, Hidaka Y, Kaname S, Okada H, Sato W, Yasuda T, Yoshida Y, Fujimura Y, Hattori M, Kagami S (2014) Diagnostic criteria for atypical hemolytic uremic syndrome proposed by the Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society. Pediatr Int 56:1–5
Kato H, Nangaku M, Hataya H, Sawai T, Ashida A, Fujimaru R, Hidaka Y, Kaname S, Maruyama S, Yasuda T, Yoshida Y, Ito S, Hattori M, Miyakawa Y, Fujimura Y, Okada H, Kagami S, Joint Committee for the Revision of Clinical Guides of Atypical Hemolytic Uremic Syndrome in Japan (2016) Clinical guides for atypical hemolytic uremic syndrome in Japan. Clin Exp Nephrol 20:536–543
Cheong HI, Jo SK, Yoon SS, Cho H, Kim JS, Kim YO, Koo JR, Park Y, Park YS, Shin JI, Yoo KH, Oh D (2016) Clinical practice guidelines for the management of atypical hemolytic uremic syndrome in Korea. J Korean Med Sci 31:1516–1528
Ariceta G, Besbas N, Johnson S, Karpman D, Landau D, Licht C, Loirat C, Pecoraro C, Taylor CM, Van de Kar N, Vandewalle J, Zimmerhackl LB, European Paediatric Study Group for HUS (2009) Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol 24:687–696
Taylor CM, Machin S, Wigmore SJ, Goodship TH, on behalf of a working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society (2010) Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 148:37–47
Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, Grinyo JM, Macia M, Mendizabal S, Praga M, Roman E, Torra R, Valdes F, Vilalta R, Rodriguez de Cordoba S (2015) An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia 35:421–447
Fox LC, Cohney SJ, Kausman JY, Shortt J, Hughes PD, Wood EM, Isbel NM, de Malmanche T, Durkan A, Hissaria P, Blombery P, Barbour TD (2018) Consensus opinion on diagnosis and management of thrombotic microangiopathy in Australia and New Zealand. Intern Med J 48:624–636
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, Coppo R, Emma F, Johnson S, Karpman D, Landau D, Langman CB, Lapeyraque AL, Licht C, Nester C, Pecoraro C, Riedl M, van de Kar NC, Van de Walle J, Vivarelli M, Fremeaux-Bacchi V, HUS International (2016) An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 31:15–39
Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, Schunemann HJ, Grade Working Group (2008) GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 336:924–926
Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group (2012) KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl 2:1–138
Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G, International Registry of Recurrent and Familial HUS/TTP (2006) Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108:1267–1279
Daram SR, Philipneri M, Puri N, Bastani B (2005) Thrombotic thrombocytopenic purpura without schistocytes on the peripheral blood smear. South Med J 98:392–395
Fakhouri F, Zuber J, Fremeaux-Bacchi V, Loirat C (2017) Haemolytic uraemic syndrome. Lancet 390:681–696
Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, Burtey S, Niaudet P, Deschenes G, Lebranchu Y, Zuber J, Loirat C (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8:554–562
Di Nisio M, Baudo F, Cosmi B, D’Angelo A, De Gasperi A, Malato A, Schiavoni M, Squizzato A, Italian Society for Haemostasis and Thrombosis (2012) Diagnosis and treatment of disseminated intravascular coagulation: guidelines of the Italian Society for Haemostasis and Thrombosis (SISET). Thromb Res 129:e177–e184
Bakhtiari K, Meijers JC, de Jonge E, Levi M (2004) Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med 32:2416–2421
Joly BS, Stepanian A, Leblanc T, Hajage D, Chambost H, Harambat J, Fouyssac F, Guigonis V, Leverger G, Ulinski T, Kwon T, Loirat C, Coppo P, Veyradier A, French Reference Center for Thrombotic Microangiopathies (2016) Child-onset and adolescent-onset acquired thrombotic thrombocytopenic purpura with severe ADAMTS13 deficiency: a cohort study of the French national registry for thrombotic microangiopathy. Lancet Haematol 3:e537–e546
Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, Nester CM, Noris M, Pickering MC, Rodriguez de Cordoba S, Roumenina LT, Sethi S, Smith RJ, Conference Participants (2017) Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 91:539–551
Scully M, Cataland S, Coppo P, de la Rubia J, Friedman KD, Kremer Hovinga J, Lammle B, Matsumoto M, Pavenski K, Sadler E, Sarode R, Wu H, International Working Group for Thrombotic Thrombocytopenic Purpura (2017) Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost 15:312–322
Cataland SR, Wu HM (2013) Atypical hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: clinically differentiating the thrombotic microangiopathies. Eur J Intern Med 24:486–491
Coppo P, Schwarzinger M, Buffet M, Wynckel A, Clabault K, Presne C, Poullin P, Malot S, Vanhille P, Azoulay E, Galicier L, Lemiale V, Mira JP, Ridel C, Rondeau E, Pourrat J, Girault S, Bordessoule D, Saheb S, Ramakers M, Hamidou M, Vernant JP, Guidet B, Wolf M, Veyradier A, French Reference Center for Thrombotic Microangiopathies (2010) Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One 5:e10208
Scully M, Hunt BJ, Benjamin S, Liesner R, Rose P, Peyvandi F, Cheung B, Machin SJ, British Committee for Standards in Haematology (2012) Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 158:323–335
Sinha A, Singh G, Bhat AS, Mohapatra S, Gulati A, Hari P, Samantaray JC, Dinda AK, Agarwal SK, Bagga A (2013) Thrombotic microangiopathy and acute kidney injury following vivax malaria. Clin Exp Nephrol 17:66–72
Bhatia D, Khandelwal P, Sinha A, Hari P, Cheong HI, Bagga A (2015) Incomplete penetrance of CD46 mutation causing familial atypical hemolytic uremic syndrome. Pediatr Nephrol 30:2215–2220
Muthuppalaniappan VM, Rajakariar R, Blunden MJ (2018) Leptospirosis presenting as haemolytic uraemic syndrome: a case report. BMC Nephrol 19:20
Gould LH, Bopp C, Strockbine N, Atkinson R, Baselski V, Body B, Carey R, Crandall C, Hurd S, Kaplan R, Neill M, Shea S, Somsel P, Tobin-D'Angelo M, Griffin PM, Gerner-Smidt P, Centers for Disease Control and Prevention (CDC) (2009) Recommendations for diagnosis of Shiga toxin-producing Escherichia coli infections by clinical laboratories. MMWR Recomm Rep 58:1–14
Igarashi T, Ito S, Sako M, Saitoh A, Hataya H, Mizuguchi M, Morishima T, Ohnishi K, Kawamura N, Kitayama H, Ashida A, Kaname S, Taneichi H, Tang J, Ohnishi M, Study group for establishing guidelines for the diagnosis and therapy of hemolytic uremic syndrome (2014) Guidelines for the management and investigation of hemolytic uremic syndrome. Clin Exp Nephrol 18:525–557
Qin X, Klein EJ, Galanakis E, Thomas AA, Stapp JR, Rich S, Buccat AM, Tarr PI (2015) Real-time PCR assay for detection and differentiation of Shiga toxin-producing Escherichia coli from clinical samples. J Clin Microbiol 53:2148–2153
Wijnsma KL, van Bommel SA, van der Velden T, Volokhina E, Schreuder MF, van den Heuvel LP, van de Kar NC (2016) Fecal diagnostics in combination with serology: best test to establish STEC-HUS. Pediatr Nephrol 31:2163–2170
Taneja N, Mewara A (2016) Shigellosis: epidemiology in India. Indian J Med Res 143:565–576
Tarr PI, Neill MA, Clausen CR, Watkins SL, Christie DL, Hickman RO (1990) Escherichia coli O157:H7 and the hemolytic uremic syndrome: importance of early cultures in establishing the etiology. J Infect Dis 162:553–556
Vonberg RP, Hohle M, Aepfelbacher M, Bange FC, Belmar Campos C, Claussen K, Christner M, Cramer JP, Haller H, Hornef M, Fickenscher H, Fraedrich K, Knobloch JK, Kuhbacher T, Manns MP, Nitschke M, Peters G, Pulz M, Rohde H, Roseland RT, Sayk F, Schaumburg F, Schocklmann HO, Schubert S, Solbach W, Karch H, Suerbaum S (2013) Duration of fecal shedding of Shiga toxin-producing Escherichia coli O104:H4 in patients infected during the 2011 outbreak in Germany: a multicenter study. Clin Infect Dis 56:1132–1140
Bell BP, Griffin PM, Lozano P, Christie DL, Kobayashi JM, Tarr PI (1997) Predictors of hemolytic uremic syndrome in children during a large outbreak of Escherichia coli O157:H7 infections. Pediatrics 100:E12
Mody RK, Gu W, Griffin PM, Jones TF, Rounds J, Shiferaw B, Tobin-D'Angelo M, Smith G, Spina N, Hurd S, Lathrop S, Palmer A, Boothe E, Luna-Gierke RE, Hoekstra RM (2015) Postdiarrheal hemolytic uremic syndrome in United States children: clinical spectrum and predictors of in-hospital death. J Pediatr 166:1022–1029
Loupiac A, Elayan A, Cailliez M, Adra AL, Decramer S, Thouret MC, Harambat J, Guigonis V (2013) Diagnosis of Streptococcus pneumoniae-associated hemolytic uremic syndrome. Pediatr Infect Dis J 32:1045–1049
Spinale JM, Ruebner RL, Kaplan BS, Copelovitch L (2013) Update on Streptococcus pneumoniae associated hemolytic uremic syndrome. Curr Opin Pediatr 25:203–208
Manoharan A, Manchanda V, Balasubramanian S, Lalwani S, Modak M, Bai S, Vijayan A, Shet A, Nagaraj S, Karande S, Nataraj G, Yewale VN, Joshi SA, Iyer RN, Santosham M, Kahn GD, Knoll MD, Alliance for Surveillance of Invasive Pneumococci Study Group (2017) Invasive pneumococcal disease in children aged younger than 5 years in India: a surveillance study. Lancet Infect Dis 17:305–312
Beck BB, van Spronsen F, Diepstra A, Berger RM, Komhoff M (2017) Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr Nephrol 32:733–741
Huemer M, Diodato D, Schwahn B, Schiff M, Bandeira A, Benoist JF, Burlina A, Cerone R, Couce ML, Garcia-Cazorla A, la Marca G, Pasquini E, Vilarinho L, Weisfeld-Adams JD, Kozich V, Blom H, Baumgartner MR, Dionisi-Vici C (2017) Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis 40:21–48
Kavanagh D, Goodship TH (2011) Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program 2011:15–20
Bouts AH, Roofthooft MT, Salomons GS, Davin JC (2010) CD46-associated atypical hemolytic uremic syndrome with uncommon course caused by cblC deficiency. Pediatr Nephrol 25:2547–2548
Riedl M, Fakhouri F, Le Quintrec M, Noone DG, Jungraithmayr TC, Fremeaux-Bacchi V, Licht C (2014) Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost 40:444–464
Thergaonkar RW, Narang A, Gurjar BS, Tiwari P, Puraswani M, Saini H, Sinha A, Varma B, Mukerji M, Hari P, Bagga A (2018) Targeted exome sequencing in anti-factor H antibody negative HUS reveals multiple variations. Clin Exp Nephrol 22:653–660
Bu F, Zhang Y, Wang K, Borsa NG, Jones MB, Taylor AO, Takanami E, Meyer NC, Frees K, Thomas CP, Nester C, Smith RJH (2018) Genetic analysis of 400 patients refines understanding and implicates a new gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol 29:2809–2819
Challis RC, Ring T, Xu Y, Wong EK, Flossmann O, Roberts IS, Ahmed S, Wetherall M, Salkus G, Brocklebank V, Fester J, Strain L, Wilson V, Wood KM, Marchbank KJ, Santibanez-Koref M, Goodship TH, Kavanagh D (2017) Thrombotic microangiopathy in inverted formin 2-mediated renal disease. J Am Soc Nephrol 28:1084–1091
Osborne AJ, Breno M, Borsa NG, Bu F, Fremeaux-Bacchi V, Gale DP, van den Heuvel LP, Kavanagh D, Noris M, Pinto S, Rallapalli PM, Remuzzi G, Rodriguez de Cordoba S, Ruiz A, Smith RJH, Vieira-Martins P, Volokhina E, Wilson V, Goodship THJ, Perkins SJ (2018) Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol 200:2464–2478
Khandelwal P, Birla S, Bhatia D, Puraswani M, Saini H, Sinha A, Hari P, Sharma A, Bagga A (2018) Mutations in membrane cofactor protein (CD46) gene in Indian children with hemolytic uremic syndrome. Clin Kidney J 11:198–203
Fremeaux-Bacchi V, Moulton EA, Kavanagh D, Dragon-Durey MA, Blouin J, Caudy A, Arzouk N, Cleper R, Francois M, Guest G, Pourrat J, Seligman R, Fridman WH, Loirat C, Atkinson JP (2006) Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol 17:2017–2025
Durey MA, Sinha A, Togarsimalemath SK, Bagga A (2016) Anti-complement-factor H-associated glomerulopathies. Nat Rev Nephrol 12:563–578
Watson R, Lindner S, Bordereau P, Hunze EM, Tak F, Ngo S, Zipfel PF, Skerka C, Dragon-Durey MA, Marchbank KJ (2014) Standardisation of the factor H autoantibody assay. Immunobiology 219:9–16
Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, Pinto S, Goodship TH, Alberti M, Ribes D, Valoti E, Remuzzi G, Noris M, European Working Party on Complement Genetics in Renal Diseases (2013) Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 24:475–486
Holmes LV, Strain L, Staniforth SJ, Moore I, Marchbank K, Kavanagh D, Goodship JA, Cordell HJ, Goodship TH (2013) Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS One 8:e60352
Gurjar BS, Manikanta Sriharsha T, Bhasym A, Prabhu S, Puraswani M, Khandelwal P, Saini H, Saini S, Verma AK, Chatterjee P, Guchhait P, Bal V, George A, Rath S, Sahu A, Sharma A, Hari P, Sinha A, Bagga A (2018) Characterization of genetic predisposition and autoantibody profile in atypical haemolytic-uraemic syndrome. Immunology. https://doi.org/10.1111/imm.12916
Khandelwal P, Faruq M, Puraswani M, Sinha A, Bagga A (2018) Coexisting variations in complement regulatory genes increase risk of relapse in anti-Factor H antibody associated atypical hemolytic uremic syndrome. https://www.asn-online.org/education/kidneyweek/2018/program-abstract.aspx?controlId=3024369
Grisaru S, Xie J, Samuel S, Hartling L, Tarr PI, Schnadower D, Freedman SB, Alberta Provincial Pediatric Enteric Infection Team (2017) Associations between hydration status, intravenous fluid administration, and outcomes of patients infected with Shiga toxin-producing Escherichia coli: a systematic review and meta-analysis. JAMA Pediatr 171:68–76
Ake JA, Jelacic S, Ciol MA, Watkins SL, Murray KF, Christie DL, Klein EJ, Tarr PI (2005) Relative nephroprotection during Escherichia coli O157:H7 infections: association with intravenous volume expansion. Pediatrics 115:e673–e680
Ardissino G, Dacco V, Testa S, Civitillo CF, Tel F, Possenti I, Belingheri M, Castorina P, Bolsa-Ghiringhelli N, Tedeschi S, Paglialonga F, Salardi S, Consonni D, Zoia E, Salice P, Chidini G (2015) Hemoconcentration: a major risk factor for neurological involvement in hemolytic uremic syndrome. Pediatr Nephrol 30:345–352
Ardissino G, Tel F, Possenti I, Testa S, Consonni D, Paglialonga F, Salardi S, Borsa-Ghiringhelli N, Salice P, Tedeschi S, Castorina P, Colombo RM, Arghittu M, Daprai L, Monzani A, Tozzoli R, Brigotti M, Torresani E (2016) Early volume expansion and outcomes of hemolytic uremic syndrome. Pediatrics 137
Hickey CA, Beattie TJ, Cowieson J, Miyashita Y, Strife CF, Frem JC, Peterson JM, Butani L, Jones DP, Havens PL, Patel HP, Wong CS, Andreoli SP, Rothbaum RJ, Beck AM, Tarr PI (2011) Early volume expansion during diarrhea and relative nephroprotection during subsequent hemolytic uremic syndrome. Arch Pediatr Adolesc Med 165:884–889
Balestracci A, Martin SM, Toledo I, Alvarado C, Wainsztein RE (2012) Dehydration at admission increased the need for dialysis in hemolytic uremic syndrome children. Pediatr Nephrol 27:1407–1410
WHO (2017) The selection and use of essential medicines. www.who.int/medicines/publications/essentialmedicines/en/
Williams PCM, Berkley JA (2018) Guidelines for the treatment of dysentery (shigellosis): a systematic review of the evidence. Paediatr Int Child Health 38:S50–S65
Agger M, Scheutz F, Villumsen S, Mølbak K, Petersen AM (2015) Antibiotic treatment of verocytotoxin-producing Escherichia coli (VTEC) infection: a systematic review and a proposal. J Antimicrob Chemother 70:2440–2446
Freedman SB, Xie J, Neufeld MS, Hamilton WL, Hartling L, Tarr PI, Alberta Provincial Pediatric Enteric Infection T, Nettel-Aguirre A, Chuck A, Lee B, Johnson D, Currie G, Talbot J, Jiang J, Dickinson J, Kellner J, MacDonald J, Svenson L, Chui L, Louie M, Lavoie M, Eltorki M, Vanderkooi O, Tellier R, Ali S, Drews S, Graham T, Pang XL (2016) Shiga toxin-producing Escherichia coli infection, antibiotics, and risk of developing hemolytic uremic syndrome: a meta-analysis. Clin Infect Dis 62:1251–1258
Safdar N, Said A, Gangnon RE, Maki DG (2002) Risk of hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 enteritis: a meta-analysis. JAMA 288:996–1001
Rosales A, Hofer J, Zimmerhackl L-B, Jungraithmayr TC, Riedl M, Giner T, Strasak A, Orth-Höller D, Würzner R, Karch H (2012) Need for long-term follow-up in enterohemorrhagic Escherichia coli–associated hemolytic uremic syndrome due to late-emerging sequelae. Clin Infect Dis 54:1413–1421
Menne J, Nitschke M, Stingele R, Abu-Tair M, Beneke J, Bramstedt J, Bremer JP, Brunkhorst R, Busch V, Dengler R, Deuschl G, Fellermann K, Fickenscher H, Gerigk C, Goettsche A, Greeve J, Hafer C, Hagenmuller F, Haller H, Herget-Rosenthal S, Hertenstein B, Hofmann C, Lang M, Kielstein JT, Klostermeier UC, Knobloch J, Kuehbacher M, Kunzendorf U, Lehnert H, Manns MP, Menne TF, Meyer TN, Michael C, Munte T, Neumann-Grutzeck C, Nuernberger J, Pavenstaedt H, Ramazan L, Renders L, Repenthin J, Ries W, Rohr A, Rump LC, Samuelsson O, Sayk F, Schmidt BM, Schnatter S, Schocklmann H, Schreiber S, von Seydewitz CU, Steinhoff J, Stracke S, Suerbaum S, van de Loo A, Vischedyk M, Weissenborn K, Wellhoner P, Wiesner M, Zeissig S, Buning J, Schiffer M, Kuehbacher T, EHEC-HUS consortium (2012) Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: case-control study. BMJ 345:e4565
Corogeanu D, Willmes R, Wolke M, Plum G, Utermohlen O, Kronke M (2012) Therapeutic concentrations of antibiotics inhibit Shiga toxin release from enterohemorrhagic E. coli O104:H4 from the 2011 German outbreak. BMC Microbiol 12:160
Nitschke M, Sayk F, Hartel C, Roseland RT, Hauswaldt S, Steinhoff J, Fellermann K, Derad I, Wellhoner P, Buning J, Tiemer B, Katalinic A, Rupp J, Lehnert H, Solbach W, Knobloch JK (2012) Association between azithromycin therapy and duration of bacterial shedding among patients with Shiga toxin-producing enteroaggregative Escherichia coli O104:H4. JAMA 307:1046–1052
Schwartz J, Padmanabhan A, Aqui N, Balogun RA, Connelly-Smith L, Delaney M, Dunbar NM, Witt V, Wu Y, Shaz BH (2016) Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the writing committee of the American Society for Apheresis: the seventh special issue. J Clin Apher 31:149–162
Rizzoni G, Claris-Appiani A, Edefonti A, Facchin P, Franchini F, Gusmano R, Imbasciati E, Pavanello L, Perfumo F, Remuzzi G (1988) Plasma infusion for hemolytic-uremic syndrome in children: results of a multicenter controlled trial. J Pediatr 112:284–290
Loirat C, Sonsino E, Hinglais N, Jais JP, Landais P, Fermanian J (1988) Treatment of the childhood haemolytic uraemic syndrome with plasma. A multicentre randomized controlled trial. The French Society of Paediatric Nephrology. Pediatr Nephrol 2:279–285
Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, Kuhlmann U, Bramstedt J, Panzer U, Vischedyk M, Busch V, Ries W, Mitzner S, Mees S, Stracke S, Nurnberger J, Gerke P, Wiesner M, Sucke B, Abu-Tair M, Kribben A, Klause N, Schindler R, Merkel F, Schnatter S, Dorresteijn EM, Samuelsson O, Brunkhorst R, Collaborators of the DGfN STEC-HUS registry (2012) Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant 27:3807–3815
Loos S, Ahlenstiel T, Kranz B, Staude H, Pape L, Hartel C, Vester U, Buchtala L, Benz K, Hoppe B, Beringer O, Krause M, Muller D, Pohl M, Lemke J, Hillebrand G, Kreuzer M, Konig J, Wigger M, Konrad M, Haffner D, Oh J, Kemper MJ (2012) An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis 55:753–759
Dundas S, Murphy J, Soutar RL, Jones GA, Hutchinson SJ, Todd WT (1999) Effectiveness of therapeutic plasma exchange in the 1996 Lanarkshire Escherichia coli O157:H7 outbreak. Lancet 354:1327–1330
Nathanson S, Kwon T, Elmaleh M, Charbit M, Launay EA, Harambat J, Brun M, Ranchin B, Bandin F, Cloarec S, Bourdat-Michel G, Pietrement C, Champion G, Ulinski T, Deschenes G (2010) Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol 5:1218–1228
Keenswijk W, Raes A, De Clerck M, Vande Walle J (2018) Is plasma exchange efficacious in Shiga toxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Ther Apher Dial. https://doi.org/10.1111/1744-9987.12768
Bruyand M, Mariani-Kurkdjian P, Gouali M, de Valk H, King LA, Le Hello S, Bonacorsi S, Loirat C (2018) Hemolytic uremic syndrome due to Shiga toxin-producing Escherichia coli infection. Med Mal Infect 48:167–174
Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T (2015) Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore) 94:e1000
Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, Helou S, Bordes C, Reffet A, Llanas B, Skopinski S, Rolland P, Gruson D, Combe C (2014) Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant 29:565–572
Percheron L, Gramada R, Tellier S, Salomon R, Harambat J, Llanas B, Fila M, Allain-Launay E, Lapeyraque AL, Leroy V, Adra AL, Berard E, Bourdat-Michel G, Chehade H, Eckart P, Merieau E, Pietrement C, Sellier-Leclerc AL, Fremeaux-Bacchi V, Dimeglio C, Garnier A (2018) Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol 33:1385–1394
Michael M, Elliott EJ, Ridley GF, Hodson EM, Craig JC (2009) Interventions for haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura. Cochrane Database Syst Rev:CD003595
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nurnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368:2169–2181
Hafer C, Golla P, Gericke M, Eden G, Beutel G, Schmidt JJ, Schmidt BM, De Reys S, Kielstein JT (2016) Membrane versus centrifuge-based therapeutic plasma exchange: a randomized prospective crossover study. Int Urol Nephrol 48:133–138
Johnson S, Stojanovic J, Ariceta G, Bitzan M, Besbas N, Frieling M, Karpman D, Landau D, Langman C, Licht C, Pecoraro C, Riedl M, Siomou E, van de Kar N, Walle JV, Loirat C, Taylor CM (2014) An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr Nephrol 29:1967–1978
Paglialonga F, Schmitt CP, Shroff R, Vondrak K, Aufricht C, Watson AR, Ariceta G, Fischbach M, Klaus G, Holtta T, Bakkaloglu SA, Zurowska A, Jankauskiene A, Vande Walle J, Schaefer B, Wright E, Connell R, Edefonti A (2015) Indications, technique, and outcome of therapeutic apheresis in European pediatric nephrology units. Pediatr Nephrol 30:103–111
Davin JC, Groothoff J, Gracchi V, Bouts A (2011) Long-term renal function under plasma exchange in atypical hemolytic uremic syndrome. Pediatr Nephrol 26:1915–1916
Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, Piras R, Donadelli R, Maranta R, van der Meer I, Conway EM, Zipfel PF, Goodship TH, Remuzzi G (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5:1844–1859
Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, Menne J, Minetti EE, Provot F, Rondeau E, Ruggenenti P, Weekers LE, Ogawa M, Bedrosian CL, Legendre CM (2016) Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis 68:84–93
Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, Delmas Y, Douglas K, Furman RR, Gaber OA, Goodship T, Herthelius M, Hourmant M, Legendre CM, Remuzzi G, Sheerin N, Trivelli A, Loirat C (2015) Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 87:1061–1073
Greenbaum LA, Fila M, Ardissino G, Al-Akash SI, Evans J, Henning P, Lieberman KV, Maringhini S, Pape L, Rees L, van de Kar NC, Vande Walle J, Ogawa M, Bedrosian CL, Licht C (2016) Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int 89:701–711
Fakhouri F, Loirat C (2018) Anticomplement treatment in atypical and typical hemolytic uremic syndrome. Semin Hematol 55:150–158
Azukaitis K, Simkova E, Majid MA, Galiano M, Benz K, Amann K, Bockmeyer C, Gajjar R, Meyers KE, Cheong HI, Lange-Sperandio B, Jungraithmayr T, Fremeaux-Bacchi V, Bergmann C, Bereczki C, Miklaszewska M, Csuka D, Prohaszka Z, Gipson P, Sampson MG, Lemaire M, Schaefer F (2017) The phenotypic spectrum of nephropathies associated with mutations in diacylglycerol kinase epsilon. J Am Soc Nephrol 28:3066–3075
Malina M, Gulati A, Bagga A, Majid MA, Simkova E, Schaefer F (2013) Peripheral gangrene in children with atypical hemolytic uremic syndrome. Pediatrics 131:e331–e335
Ariceta G, Arrizabalaga B, Aguirre M, Morteruel E, Lopez-Trascasa M (2012) Eculizumab in the treatment of atypical hemolytic uremic syndrome in infants. Am J Kidney Dis 59:707–710
Ardissino G, Tel F, Testa S, Marzano AV, Lazzari R, Salardi S, Edefonti A (2014) Skin involvement in atypical hemolytic uremic syndrome. Am J Kidney Dis 63:652–655
Krishnappa V, Gupta M, Elrifai M, Moftakhar B, Ensley MJ, Vachharajani TJ, Sethi SK, Raina R (2018) Atypical hemolytic uremic syndrome: a meta-analysis of case reports confirms the prevalence of genetic mutations and the shift of treatment regimens. Ther Apher Dial 22:178–188
Gulleroglu K, Fidan K, Hancer VS, Bayrakci U, Baskin E, Soylemezoglu O (2013) Neurologic involvement in atypical hemolytic uremic syndrome and successful treatment with eculizumab. Pediatr Nephrol 28:827–830
Hu H, Nagra A, Haq MR, Gilbert RD (2014) Eculizumab in atypical haemolytic uraemic syndrome with severe cardiac and neurological involvement. Pediatr Nephrol 29:1103–1106
Williams ME, Balogun RA (2014) Principles of separation: indications and therapeutic targets for plasma exchange. Clin J Am Soc Nephrol 9:181–190
Khandelwal P, Gupta A, Sinha A, Saini S, Hari P, Dragon Durey MA, Bagga A (2015) Effect of plasma exchange and immunosuppressive medications on antibody titers and outcome in anti-complement factor H antibody-associated hemolytic uremic syndrome. Pediatr Nephrol 30:451–457
Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group (2013) KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 3:1–150
Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, Chatelet V, Mousson C, Mourad G, Bridoux F, Cassuto E, Loirat C, Rondeau E, Delahousse M, Fremeaux-Bacchi V (2013) Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant 13:663–675
Bacchetta J, Cochat P (2015) Primary disease recurrence-effects on paediatric renal transplantation outcomes. Nat Rev Nephrol 11:371–384
Khandelwal P, Sinha A, Hari P, Bansal VK, Dinda AK, Bagga A (2014) Outcomes of renal transplant in patients with anti-complement factor H antibody-associated hemolytic uremic syndrome. Pediatr Transplant 18:E134–E139
Acknowledgments
Funding support for collaborative research studies and consensus development by Indo-French Centre for the Promotion of Advanced Research (CEFIPRA) [IFC/A/4703-1/2015/1562; IFC/Network 1/2185]; Department of Biotechnology, Government of India [BT/PR14651/MED/30/566/2010]; Indian Council of Medical Research [Advanced Centre for Research in Pediatric Kidney Diseases; 5/7/1090/2013-RHN]; Department of Science and Technology, Government of India [EMR12016/002781]; and All India Institute of Medical Sciences, New Delhi [A-386].
Working group members
Ranjeet Thergaonkar, Vishakapatnam (Group Chair)
Kirtisudha Mishra, New Delhi (Group Chair)
Sushmita Banerjee, Kolkata
Kamran Afzal, Aligarh
Sriram Krishnamurthy, Puducherry
Girish Bhatt, Bhopal
Manish Kumar, New Delhi
Mamta Puraswani, New Delhi
Anil Vasudevan, Bangalore (Group Chair)
Jyoti Sharma, Pune (Group Chair)
Indira Agarwal, Vellore
OP Mishra, Varanasi
Karalanglin Tiewsoh, Chandigarh
Aditi Sinha, New Delhi (Group Chair)
Saroj K Patnaik, New Delhi (Group Chair)
Amarjeet Mehta, Jaipur (President, ISPN)
Susan Uthup, Thiruvanthapuram
Rajiv Sinha, Kolkata
Sudha Ekambaram, Chennai
Pankaj Hari, New Delhi (Group Chair)
Sidharth Sethi, Gurugram (Group Chair and Secretary, ISPN)
Abhijeet Saha, New Delhi
Swati Bhardwaj, New Delhi
Priyanka Khandelwal, New Delhi (Rapporteur)
Arvind Bagga, New Delhi (Coordinator)
Experts
Marie-Agnes Dragon-Durey, Hopital Europeen Georges Pompidou, INSERM UMRS 1138, Paris, France
Amit K Dinda, All India Institute of Medical Sciences, New Delhi
Neelam Taneja, Postgraduate Institute of Medical Education and Research, Chandigarh
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 27 kb)
Rights and permissions
About this article

Cite this article
Bagga, A., Khandelwal, P., Mishra, K. et al. Hemolytic uremic syndrome in a developing country: Consensus guidelines. Pediatr Nephrol 34, 1465–1482 (2019). https://doi.org/10.1007/s00467-019-04233-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-019-04233-7