
Abstract
The hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy characterized by the triad of thrombocytopenia, nonimmune microangiopathic hemolytic anemia and acute kidney injury. The most frequent form of HUS in children is secondary to Shiga toxin producing Escherichia coli and the term atypical HUS was initially used to designate any HUS not caused by these organisms. It is now clear that within the umbrella of atypical HUS are a number of specific causes of HUS (e.g., Streptococcus pneumoniae infection, cobalamin C defect and diacylglycerol kinase ε defect). Atypical HUS without these causes is mostly a disease of complement alternative pathway overactivation, due to hereditary mutations in complement genes or acquired autoantibodies against complement factor H. The prognosis of atypical HUS was poor, with high mortality, progression to permanent kidney failure and recurrence after transplantation. Since 2009, terminal complement blockade therapy by eculizumab has dramatically changed the hitherto dismal outcome of the disease. The aims of this chapter are to summarize the previous era of treatment, to review new knowledge in the domain of atypical HUS and to scan the horizon for future developments in the management of atypical HUS.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
The hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy (TMA) characterized by the triad of thrombocytopenia, nonimmune microangiopathic hemolytic anemia, and acute kidney injury [1]. The most frequent form of HUS in children is secondary to Shiga toxin (Stx)—producing Escherichia coli (STEC) and the term atypical HUS (aHUS) was initially used to designate any HUS not caused by STEC. It is now clear that within the umbrella of aHUS are a number of specific causes of HUS—for example Streptococcus pneumoniae infection, cobalamin C defect, Diacylglycerol kinase ε (DGKε) defect and various underlying conditions.
Atypical HUS without coexisting disease or specific infection is mostly a disease of complement alternative pathway (AP) overactivation, due to hereditary mutations in complement genes or acquired autoantibodies against complement factor H (FH). The clinical characteristics of patients, patient outcome and genotype-phenotype correlations were described [2,3,4,5,6,7]. Therefore, the term aHUS is today preferentially used to designate HUS without coexisting disease or specific infection [5, 6, 8,9,10,11]. Plasma exchange (PE) was the mainstay of treatment for aHUS until 2009, with considerable morbidity in children [12, 13]. Since 2009, terminal complement blockade therapy by eculizumab has dramatically changed the hitherto dismal outcome of the disease [14, 15]. The aims of this chapter are to summarize the previous era of treatment, to review new knowledge in the domain of atypical HUS and to scan the horizon for future developments in the management of atypical HUS.
Definition of Atypical HUS
Atypical HUS is one of a number of causes of TMA—life or organ threatening diseases characterized by microthrombi in small blood vessels which can be classified according to etiology and/or physiopathology [16,17,18,19] (Fig. 22.1). The two most important TMAs to exclude when suspecting aHUS are thrombotic thrombocytopenic purpura (TTP) and shigatoxin associated HUS (STEC HUS). The latter is the most common TMA affecting the kidneys in children. It is caused by intestinal infection by certain strains of E coli carrying a plasmid for producing shigatoxin, particularly serotypes O157:H7, O104:H4 and O26 and in rare cases by Shigella dysenteriae [20, 21]. This type of HUS was previously labeled as typical or D+, however this classification is now obsolete.

Thrombotic microangiopathies are classified into: Inherited or acquired primary; secondary; or infection associated TMAs. Current classifications define primary TMAs as hereditary (mutations in ADAMTS13, MMACHC (cb1c deficiency), or genes encoding complement proteins) or acquired (autoantibodies to ADAMTS13, or autoantibodies to complement FH, which is associated with homozygous CFHR3/1 deletion). TMA is associated with various infections: in STEC-HUS and pneumococcal HUS, distinct mechanisms result in TMA; in other infections, the processes are not defined and in some cases the infection may trigger manifestation of a primary TMA. Secondary TMAs occur in a spectrum of conditions, and in many cases the pathogenic mechanisms are multifactorial or unknown. The classification presented here is not unequivocal: in some secondary TMAs, for example pregnancy-associated TMA or de novo TMA after transplantation, a significant proportion of individuals will have a genetic predisposition to a primary TMA. AAV ANCA-associated vasculitis; ADAMTS13 a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; aHUS atypical hemolytic uremic syndrome; C3G C3 glomerulopathy; CAPS catastrophic antiphospholipid syndrome; cblC cobalamin C type; DGKE gene encoding diacylglycerol kinase ε; FH factor H; HELLP syndrome of hemolysis, elevated liver enzymes, and low platelets; HUS hemolytic uremic syndrome; IgAN IgA nephropathy; MN membranous nephropathy; MPGN membranoproliferative glomerulonephritis; SRC scleroderma renal crisis; STEC shiga toxin–producing Escherichia coli; TMA thrombotic microangiopathy; TTP thrombotic thrombocytopenic purpura. Reproduced from Brocklebank et al. [19]
TTP is an important cause of TMA that must be ruled out before making a diagnosis of aHUS. It is due to a severe deficiency (<10%) in ADAMTS13 (A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13) activity, either from a congenital absence of functional protein caused by homozygous or compound heterozygous mutations in the ADAMTS13 gene, or due to anti-ADAMTS13 antibodies [22].
TMA can also occur secondary to a coexisting disease or condition, such as malignancy or autoimmune disease. This is more common in adults than children, with the exception of post-hematopoietic stem cell transplant (HSCT) TMA.
As the classification of TMA has evolved with increasing understanding [23], there is general agreement that the term aHUS defines patients with HUS without a coexisting disease or specific infection [5, 6, 8,9,10,11]. This chapter is focused on aHUS according to this definition.
Incidence and Prevalence of Atypical HUS
Atypical HUS, defined as indicated above, is an ultra-rare disease. In the United States, aHUS is considered to have an annual incidence rate of two new pediatric cases per million total population [24]]. An incidence of approximately 0.11 new pediatric cases per million total population per year was also observed between July 2009 and December 2010 in an exhaustive cohort of children with aHUS from France, the United Kingdom, Spain, Netherlands and Canada [13]. A recent systematic review has reported an overview of global incidence and prevalence of aHUS [25]. Eight studies were reviewed from Europe, Australia, and New Zealand [5, 26,27,28,29,30,31,32]. In Europe the reported incidence (all ages) ranged between 0.23 and 1.9 per million annually [5, 32]. In Australia a pediatric study reported a calculated incidence of 0.44 per million annually [28]. Studies reporting incidence for individuals under 20 years of age ranged between 0.26 and 0.75 per million annually [27, 32]. A systematic review by Yan reported that in individuals under 20 years of age, the prevalence of aHUS ranged between 2.21 and 9.4 per million people [25].
The Alternative Pathway of Complement
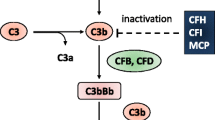
The alternative pathway (AP) of the complement system plays a predominant, though not exclusive role in aHUS (Fig. 22.2) [33,34,35,36,37,38,39,40,41,42,43,44,45,46]. The complement system is composed of plasma proteins that react with one another to opsonize microbes and induce a series of inflammatory responses that help the immune cells to fight infection. There is mounting evidence that complement participates not only in the defense against pathogens, but also in host homeostasis [47,48,49,50,51,52]. The complement cascade can be activated by three different pathways. While the activation of the classical and the lectin pathways occurs after binding to immune complexes or microorganisms respectively, the AP is continuously activated and generates C3b which binds indiscriminately to pathogens and host cells. On a foreign surface, C3b binds factor B (FB), which is then cleaved by Factor D to form the C3 convertase C3bBb. The C3 convertase, which is stabilized by its binding to properdin, induces exponential cleavage of C3b and the generation of C3bBbC3b complexes with C5 convertase activity. The C5 convertase cleaves C5 to generate C5a—the most potent anaphylatoxin—and C5b which initiates the formation of the membrane attack complex (MAC or C5b-9), able to lyse pathogens [52] (Fig. 22.2). The CAP amplification loop is normally strictly controlled at the surface of the host quiescent endothelium, which is protected from the local formation of the C3 convertase by complement regulatory proteins. These include regulators in serum, such as FH and Factor I (FI), as well as membrane bound CD46 (membrane cofactor protein (MCP), which cooperate locally to inactivate C3b. FH is the most important protein for the regulation of the CAP and consists of 20 short consensus repeats (SCRs) and contain two C3b-binding sites (Fig. 22.3). MCP is a widely expressed transmembrane glycoprotein that binds C3b and inhibits complement activation on host cells. The serine protease FI cleaves C3b in the presence of various cofactors including FH, complement receptor 1 (CR1, CD35) and MCP. Coagulation regulator thrombomodulin (THBD) enhances FI-mediated inactivation of C3b in the presence of FH [47, 52].

Complement activation and its regulation. aHUS is the prototype of a disease resulting from inefficient protection of endothelial cells against complement activation. (a) Protection of cells surface. The AP is permanently active, with a continuous formation of small amounts of the C3 convertase C3bBb at the cell surface. To prevent unopposed complement activation resulting in cell damage, the complement system is tighly regulated. The glycocalyx is a multifunctional thick carbohydrate layer containing glycoaminoglycans (GAG) (heparin sulphate, sialic acid, polyanions) that covers all endothelial cells, in particular the glomerular endothelium in the kidney. FH binds to GAG and C3b. MCP is constitutionally anchored to endothelial membrane. Under normal conditions, the C3 convertase formation is stopped by the interaction of FH or MCP with C3b, which makes further binding of FB to C3b impossible. C3b is then cleaved by FI to iC3b, which cannot bind FB. (b) Activation of complement and covalent attachment of C3b to the microbial surfaces. The major function of complement is to act as a defense mechanism against microbes. Very small amounts of C3b are normally present in plasma due to low levels of spontaneous C3 cleavage but C3b can bind to bacteria. Once C3b is covalently bound to the surface of microorganisms, FB binds to it and becomes susceptible to cleavage by Factor D (FD). The resulting C3bBb complex is a C3 convertase that will continue to generate more C3b, thus amplifying C3b production. C3b attaches to bacterial surfaces for opsonization by phagocytes and simultaneous activation of the cytolytic terminal complement cascade. (c) In the case of aHUS, AP activation is uncontrolled and C3 convertase C3bBb and C5 convertase C3bC3bBb are formed. During complement activation, C5 is split into C5a and C5b. C5b together with complement proteins C6, C7, C8 and C9 form the C5b9 complex in sublytic quantities that activate endothelial cells to produce prothrombotic factors. AP alternative pathway, C3bBb C3 convertase; C3bC3bBb C5 convertase; FB complement factor B; FD complement factor D; FH complement factor H; FI complement factor I; MCP membrane cofactor protein (CD46)

Complement factor H. FH is a plasma protein consisting in 20 domains called short consensus repeats (SCRs) (numbered circles). FH has two C3b binding sites: one is localized within the N-terminal SCR 1–4, implicated in the cleavage of C3b by FI and the other in the C-terminal SCR 19–20, implicated in cell surface binding. FH regulates the formation, stability and decay of the C3 convertase C3bBb. 94 rare variants in CFH reported in the aHUS database https://www.complement-db.org are shown within columns. Blue squares indicate frameshift, deletion, nonsense and conserved cysteine affected variants (prediction of quantitative FH deficiency), orange squares indicate missense variants (with or without demonstrating functional consequences). 20% of all variants are located in the C-terminus SCR 20 and are mostly associated with normal FH plasma level. FH complement factor H; FI complement factor I; GAG glycoaminoglycans; SCR short consensus repeat
Over the past 20 years, genetic discoveries have substantially improved our understanding of the mechanisms responsible for aHUS and driven development of novel therapeutic strategies) [33,34,35,36,37,38,39,40,41,42,43,44,45,46] (Fig. 22.4). In a large genetic screen of 794 aHUS patients, rare variants in one the 5 genes (CFH, C3, CFI, CFB, or CD46) that encode proteins involved in the regulation of the alternative pathway of complement were identified in 41% of patients and combinations of mutations were noted in 3% of patients [53]. Predisposition to aHUS is inherited in an autosomal recessive or autosomal dominant manner with incomplete penetrance.

Discoveries that allowed a better understanding of the pathophysiology of aHUS during the last decades. This led to the approval of eculizumab for the treatment of patients with aHUS, to control the overactivation of complement. FB complement factor B; FH complement factor H; FI complement factor I; DGKE diacylglycerol kinase ε; MCP membrane cofactor protein (CD46); THBD thrombomodulin; RCA regulators of complement activation; CFHR complement factor H related protein
An updated database describing all rare variants identified in aHUS is available at https://www.complement-db.org [54, 55] and these genetic abnormalities are described in more detail in specific sections of this chapter. In addition, acquired autoantibodies to the FH protein (anti-FH) have been demonstrated in patients with aHUS, also described in more detail below.
Clinical Presentation
The majority of children with aHUS present with the complete triad of HUS; microangiopathic hemolytic anemia (with hemoglobin <10 g/dL, presence of schistocytes, high lactate dehydrogenase (LDH), decreased haptoglobin levels) thrombocytopenia with platelet count <150 × 109/L and acute kidney injury (serum creatinine above the upper limit of normal). Approximately 60% of them require dialysis at the first episode. Severe hypertension is common. However, the complete triad may be missing at admission and a gradual onset is possible. Particularly, platelet count may be >150 × 109/L (approximately 15% of patients) and hemoglobin may be >10 g/dL (approximately 5% of patients) [5]. Children may also have normal serum creatinine at presentation (approximately 15%) [56] and/or present with proteinuria/nephrotic syndrome/hematuria/hypertension as the only kidney manifestations. Thus, any association of two components of the triad with the third one missing can be a manifestation of HUS. While kidney biopsy is not required to establish the diagnosis when full-blown HUS is present, it is useful when hematology criteria are missing or incomplete and any time the diagnosis of HUS is uncertain, to document that the underlying histological lesion is TMA.
Age and Gender
In a cohort of French children with aHUS (66.2% of whom had a proven genetic or acquired complement abnormality), the mean age at onset was 1.5 years (0 to <15 years). 56% (50/89) of children had onset between birth and 2 years of age (28% between birth and 6 months, 28% between 6 months and 2 years) [5], similar to the proportion of 22% (10/45) of children having onset between 1 month and 1 year in another series [4] and 36.3% (53/146) less than 2 years and 19.8% (29/146) less than 1 year in another [57]. Atypical HUS in children is as frequent in females as in males (female-to-male ratio 0.9), in contrast with the female preponderance when the disease presents in adulthood (female-to-male ratio 3) [4, 5]. In a large series from the Global aHUS Registry, 387/851 (45%) of patients with aHUS presented before the age of 18 years (mean 3.8 years) and 166/387 (43%) of those with pediatric onset were female.
Age at onset in children varies according to the underlying genetic or acquired abnormality (more information about specific genetic/acquired abnormalities is given below). Onset between birth and 1 year of age has been reported in the majority of aHUS patients (37/50) reported to date with DGKE mutation [45, 46, 58,59,60,61] and all children with homozygous CFH mutation. It is also frequent in children with heterozygous CFH or CFI mutation-associated HUS. Conversely, MCP mutation-associated HUS in children exceptionally starts before the age of 1 year but most often between age 2 and 12 years. Anti factor H autoantibody associated HUS (anti-FH HUS) is also mostly a disease of late childhood and adolescence (onset between 5 and 12 years, mean age 7.6–9 years in five series including a total of over 500 patients with this form of aHUS [62,63,64,65,66,67]. C3 or CFB mutation-associated HUS and aHUS without complement mutation or anti-FH antibodies appears to start at any age [2, 5, 25].
Family History
As indicated above, despite aHUS being a genetic disease, a family history of HUS is present in only 20–30% of patients [2, 4, 5] due to incomplete penetrance. The diagnosis of HUS may be unknown in the family and questioning should ask about cases of acute or chronic kidney failure, thrombocytopenia, anemia, hypertension, dialysis and graft failure in the pedigree as well as about consanguinity, which is significant for homozygous mutations in CFH, MCP and DGKE. No familial case of anti-FH antibody-associated HUS has been reported [68].
Triggering Events
Atypical HUS episodes in children are frequently triggered by intercurrent infections, whatever the genetic background. Specific reported infections include varicella [69], influenza [70, 71], Bordetella pertussis [72] and recently SarsCov2 virus [73].
Diarrhea precedes the onset of aHUS in at least one third of children and upper respiratory tract infections in at least 10% [4, 5]. This frequency of diarrhea at onset of aHUS explains why the former “post-diarrheal” or “non post-diarrheal” (or D+/D-) criterion to differentiate STEC-HUS from aHUS was frequently misleading. It is, however, often unclear whether gastrointestinal symptoms in aHUS are linked to an infectious trigger or whether they are manifestations of intestinal TMA. Rare patients (approximately 1%) have been reported in whom the first episode of aHUS was caused by STEC gastroenteritis, with the diagnosis of aHUS being retained because the patient had subsequent relapses and a familial history of aHUS (one patient with MCP mutation) [5], a severe course possibly favored by the genetic complement abnormality (one patient with CFH mutation) [74] or recurrence after kidney transplantation (two patients with CFI or MCP mutation—the latter also in the mother who donated the kidney) [75] . In a cohort of 75 patients with proven STEC HUS, four patients (5%) were found to have pathogenic variants in complement genes, including one patient with severe outcome. In aHUS secondary to anti-FH antibodies, a gastrointestinal prodrome (such as diarrhea, vomiting and/or abdominal pain) has been reported in 27.7% [65, 76, 77]. This type of aHUS is more common in the Asian subcontinent where it comprises 56% of cases compared with 10–25% of European cohorts [66, 77]. A recent study looking for gastrointestinal pathogens in aHUS secondary to anti-FH antibodies showed that twice as many patients had evidence of gastrointestinal pathogens compared with those without anti-FH (62.5% compared with 31.5%) including Clostridium difficile, Giardia intestinalis, Salmonella, Shigella, Rotavirus, Norovirus and Entamoeba histolytica. No stool was positive for Shigatoxin [78]. However, STEC has been reported as the trigger for HUS in a couple of patients with anti-FH HUS [62, 65]. Interestingly, the association of homozygous MCP mutation with common variable immunodeficiency has been reported [41]. Therefore, patients with homozygous MCP mutation should be investigated for immunodeficiency that may require immunoglobulin therapy to prevent infections—and thus decrease the frequency of HUS relapses triggered by infections.
Lastly, pregnancy is the trigger for aHUS in 20% of adult women [5] and 86% of women with pregnancy—associated HUS (mostly in the post-partum period) have a complement mutation [79]. For this reason, pregnancy-associated HUS is now classified as aHUS.
Histology of Atypical HUS
The underlying histological lesion of aHUS is TMA involving afferent arterioles and glomerular capillaries. Characteristic features during the acute phase are platelet and fibrin thrombi within glomerular capillaries and the thickening of glomerular capillary walls related to endothelial cell swelling and detachment and the accumulation of flocculent material (proteins and cellular debris) between the endothelial cells and the basement membrane, with double contour appearance. Mesangiolysis (fluffy mesangial expansion) is also common. Bloodless and ischemic glomeruli related to the narrowing or occlusion of the capillary and arteriolar lumen can be observed. Arterial changes range from endothelial swelling and intramural fibrin to fibrinoid necrosis with occlusive thrombi and fragmented red blood cells. Immunofluorescence studies for immunoglobulin G or C3 deposits are generally negative. C5b-9 staining has been reported in microangiopathy attributed to complement abnormalities and other causes, however its presence is not reliable.
Chronic lesions are characterized by mesangial sclerosis, thickening of capillary walls with sparse or diffuse double contours, ischemic changes of glomeruli and mucoid intimal hyperplasia and narrowing of the arterial lumen (onion-skinning). The time course for histological resolution of TMA is unknown and therefore it is difficult to know if presence of chronic features points to an ongoing active TMA process or to a chronic sequel [80, 81].
There is a general consensus that it is not possible to determine the etiology of TMA from histological morphology [82].
Manifestations Outside the Kidneys
Although the TMA process predominates in the kidney vasculature, other organs may be involved. The most frequent manifestation outside the kidneys during acute episodes of aHUS is brain involvement, reported in 15–20% of children with aHUS [5, 83,84,85,86]. Symptoms can be seizures, altered mental status, altered consciousness, visual problems (diplopia, sudden visual loss), paresis and coma. Computed tomography scan is useful to rule out cerebral bleeding. Magnetic resonance imaging (MRI) shows hyperdensities of variable severity and extension. Focal cerebral infarction is possible. The prognostic significance of MRI abnormalities is generally uncertain. The frequency of cardiac involvement is poorly documented, but life threatening ischemic myocardiopathy may occur, which makes sequential troponin level assay, electrocardiography and echocardiography advisable during acute episodes [86,87,88,89] . Peripheral acute ischemia leading to gangrene of fingers/hands and toes/feet [90], skin necrosis [91,92,93] or retinal ischemia with sudden visual loss [94] have been reported in a few patients. Manifestations outside the kidneys may also include pancreatitis (increase in pancreatic enzymes with or without clinical/radiologic signs) and/or hepatitis (increase of hepatic enzymes) (5–10% of patients) and, exceptionally, intra-alveolar hemorrhage, severe gastro-intestinal manifestations including intestinal perforation or life-threatening multiorgan failure (2–3% of patients [5, 91]. Severe gastrointestinal symptoms (abdominal pain, vomiting, diarrhea, biochemical pancreatitis and hepatitis), myocardial and neurological manifestations appear to be particularly frequent in patients with anti-FH antibodies [62, 65, 77, 84, 90].
Four children with aHUS have been reported who developed cerebral ischemic events due to stenosis of cerebral arteries after several years on dialysis [95,96,97,98]. One of them also had stenosis of coronary, pulmonary and digestive arteries [96]. These observations have suggested that local complement activation during acute episodes and/or subclinically in the long term, may lead to such macrovascular complications, independently or as aggravating factors of the vascular consequences of long-term dialysis. Prospective studies are required to document whether aHUS patients have an increased risk of cardio-or cerebro-vascular events and of arterial disease due to the local complement activation [99].
Experience of eculizumab to treat manifestations of aHUS outside the kidneys is limited to case reports. However, eculizumab was impressively effective in two children with acute distal ischemia [90] or skin necrosis and intestinal perforation [91], respectively. Eculizumab may rescue central nervous system manifestations, as suggested by nine case reports, including four in children [85, 86, 88, 100, 101]. Eculizumab also appeared life-saving in four children with myocardial involvement [86,87,88,89]. Lastly, two children who had developed cerebral artery stenosis stopped having ischemic events under eculizumab therapy, with non-progression of arterial stenosis documented in one [95, 97].
Making the Diagnosis of Atypical Hemolytic Uremic Syndrome
When the features of HUS are present (as summarized in Table 22.1), careful assessment is required to exclude possible causes before a provisional diagnosis of aHUS can be reached [102, 103] (Fig. 22.5 and Table 22.2).

Suggested approach to making the diagnosis of atypical HUS. When the clinical triad of microangiopathic hemolytic anemia, acute kidney injury and thrombocytopenia are present without clinical evidence of STEC infection, further evidence should be sought for STEC and pneumococcal infection. TTP should be excluded, along with secondary causes of TMA. The triad of HUS may indicate sepsis, which should be sought and excluded. Evidence for cobalamin disorder should also be sought. *these investigations may take some time to return and treatment with anti-C5 therapy should not be delayed if results are not available and the diagnosis of aHUS is strongly suspected. In practice, if an alternative diagnosis is secured after commencement of anti-C5 therapy, it can be discontinued. STEC shiga-toxin producing Escherichia coli; PCR polymerase chain reaction; TTP thrombotic thrombocytopenic purpura; ADAMTS13 a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; pHUS pneumococcal haemolytic uremic syndrome; DAT direct antigen test; TMA thrombotic microangiopathy; HSCT hematopoietic stem cell transplant; VEGF vascular endothelial growth factor; DIC disseminated intravascular coagulation; CRP C-reactive protein; DGKE Diacylglycerol Kinase Epsilon; MMACHC methylmalonic aciduria and homocystinuria type C, FH factor H
Since the most common cause of HUS in children is STEC infection and because STEC infections may be asymptomatic, thorough microbiological assessment is required. This is reviewed in Chap. 24, but in brief requires stool culture (which commonly only detects E. coli serotype O157:H7) and stool polymerase chain reaction (PCR) for STEC virulence genes. Serological assessment may also be helpful. Medical history and physical examination usually eliminate HUS secondary to a coexisting condition—mostly HSCT- and generally suffice for the diagnosis of Streptococcus pneumoniae-HUS, that occurs mostly in children less than 2 years of age presenting with symptoms of invasive infection (pneumonia, meningitis, bacteremia) (see Chap. 2). Malignant hypertension can be difficult to distinguish from aHUS, and careful assessment for alternative causes of hypertension may help in this differential diagnosis. TTP must be excluded by urgent assessment of ADAMTS13 activity, since there is treatment dichotomy depending on the result. Evidence for a cobalamin C defect (raised plasma homocysteine and raised urinary methylmalonic acid) must also be sought since alternative treatment will be required [20, 102, 104,105,106,107,108,109,110,111,112,113,114].
Biological Assays to Support the Clinical Diagnosis of Atypical HUS
STEC infection should be ruled out as soon as possible when aHUS is suspected. Stools should be collected at admission or rectal swab performed if no stools are available, allowing stool culture and fecal PCR or immunologic assay for Stx (see Chap. 26). Negative results, mostly due to delayed stool collection or prior administration of antibiotics, are observed in at least 30% of cases classified clinically as STEC-HUS [21, 106]: in such cases, the clinical diagnosis should prevail. Congenital TTP requires urgent plasma infusion (PI) and acquired TTP requires urgent plasma exchange (PE) plus corticosteroids and rituximab. Blood samples collected before PE/PI are required for ADAMTS13 activity assays, which most commonly rely on the cleavage by plasma ADAMTS13 of the von Willebrand Factor (VWF) peptide containing the cleaving site of VWF (Fret-VWF 73). Results can be available within a few hours [115, 116]. A limitation is the interference of hyperbilirubinemia [117]. Results of commercial kits show reasonable though not full agreement (80–90% concordance) with Fret-VWF [116, 118, 119].
Lastly, assays to detect a cobalamin defect should be part of the initial biological sampling in any child suspected to have aHUS. Cobalamin defect—associated HUS, which can be rescued by hydroxocobalamine treatment, may occur in neonatal forms presenting with neurological, cardiac or multivisceral involvement, but at least as frequently in late-onset forms presenting with predominant or isolated HUS during childhood or early adulthood [110, 111, 120,121,122].
Complement Investigations in Patients Suspected to Have aHUS
All patients suspected of having aHUS should have blood sampling before PE/PI for measurement of C3, C4, FH, FI and FB plasma levels and screening for anti-FH antibodies. A recent publication from seven European laboratories reports the standardization of the enzyme linked immunosorbent assay technique for anti-FH antibodies [123], which could be developed in other countries. MCP surface expression on polymorphonuclear or mononuclear leucocytes is also required.
As indicated above, decreased C3 levels are observed in only 30% of children with aHUS [2, 4,5,6]. Therefore, a normal C3 level does not rule out the diagnosis of aHUS. Normal C4 concentration associated with decreased C3 level confirms activation of the AP as would a decreased FB concentration. As the C3 level is normal in patients with isolated MCP mutation and decreased in patients with high titer anti-FH antibodies, aHUS in a pre-adolescent or adolescent child—the age of onset in these two subgroups of complement-dependent HUS—is most likely MCP mutation-associated-HUS if C3 level is normal or anti-FH antibody-associated HUS if C3 level is low.
As indicated above, decreased FH or FI plasma levels are observed in approximately 50% and 30% of patients with mutations in CFH or CFI genes, respectively [5, 6]. Therefore, a normal FH or FI plasma level does not exclude a mutation in the corresponding gene.
Recent data suggest that levels of C5a and soluble C5b-9 (sC5b-9) are elevated during acute episodes of aHUS and may be biological markers to differentiate aHUS from TTP [124]. Increased C5a and sC5b-9 plasma levels have been confirmed in approximately half of aHUS patients during acute phases of the disease and also during remission [125]. However, a normalization of complement activation products levels after remission, including sC5b-9, has been reported by another group [126]. This, and the fact that sC5b-9 may be elevated in conditions which are not aHUS [127], means that the usefulness of these markers for routine clinical care remains to be determined.
Genetic Screening in Patients with Atypical HUS
Genetic screening results are not required for urgent therapeutic decisions but are necessary to establish whether the disease is complement-dependent or not, prognosis, risk of relapses and of progression to kidney failure, genetic counselling, decisions for complement blockade treatment duration and for kidney transplantation. The six complement genes identified as susceptibility factors for aHUS (CFH, CFI, MCP, C3, CFB and THBD) should be analysed by direct sequencing or next generation sequencing analysis. Multiplex ligation dependent probe amplification (MLPA) is required to detect hybrid CFH genes (5% of patients) and copy number variations in CFH and CFH Related (CFHR) protein genes. Because of the frequency of combined mutations indicated above, all six genes should be screened for mutations in all patients. Screening for DGKE mutations should be performed in children with onset of aHUS before the age of 1–2 years and maybe in older children if further reports indicate DGKE mutation-associated HUS may occur later in life. Sequence variants in complement genes have been identified in 5 of 13 patients with anti-FH antibody—associated HUS in one series [128], but none of 26 patients in another series [62]. Genetic analyses even when anti-FH antibodies are present may be justified. If the patient has anti-FH antibodies and a mutation, treatment should be decided according to the antibody titer and the functional consequences of the mutation [103]. Next-generation sequencing analysis allows the simultaneous study of all potentially relevant genes and should reduce the turnaround time for results and the cost of genetic analysis. Exome sequencing, which was successfully used to identify DGKE mutations [45], is still limited to research laboratories.
Rare Coding Variants in the CFH Gene
The role of CFH in aHUS was first suggested more than 40 years ago (Fig. 22.4). A decrease of plasma C3 level was first reported in 1973 in five patients with severe HUS [33] and low FH plasma levels were first reported in 1981 in a 8-month-old boy with HUS [34]. However, it is only in 1998 that Warwicker et al., by linkage analysis, could establish the link between aHUS and the Regulators of Complement Activation (RCA) region in chromosome 1q32, and the presence of mutations in CFH, mainly in the SCR 20, despite normal plasma levels of FH and C3 [37] .
During the last 15 years, at least 90 different rare variants of CFH with minor allele frequencies (MAF) <0.1% including missense or nonsense variants, short deletions or insertions, located everywhere in the gene, have been identified and referenced in the FH aHUS mutation database. The type I mutations, which induce a quantitative deficiency of the FH protein (low FH plasma levels), are located everywhere in the gene. By contrast, the mutations which induce a decreased ability of FH to bind to endothelial cells-bound C3b while plasma levels of FH are normal (namely type II mutations), are mostly located in SCR 20 (Fig. 22.3). More than 90% of reported mutations have been heterozygous and plasma C3 levels are decreased in approximately 50% of patients [2, 5, 45, 58, 65]. Less than 20 children (2–4% of reported children with aHUS), mostly from consanguineous families, carried a CFH homozygous variant or two heterozygous variants leading to complete FH deficiency, with permanently very low C3 levels. CFH pathogenic variants are the most common among aHUS patients, accounting for 20–30% of all aHUS cases in registries from the United States and Europe [1, 3,4,5] (Table 22.3).
The CFH gene is in close proximity to genes encoding for the five CFHR proteins that are thought to have arisen from several large genomic duplications. All CFHRs share a high degree of homology, which makes the region particularly prone to genomic rearrangement. The C-terminal SCR domains of CFHR1 proteins show a high level of amino acid sequence identity to the C-terminal recognition region of FH, representing the central combined cell surface anchoring- and C3b recognition region of FH. Using MLPA, genetic rearrangements between CFHR1 and FH, which result in a hybrid CFH/CFHR1 gene leading to the formation of hybrid CFH/CFHR1 protein have been reported in several unrelated aHUS patients from distinct geographic origins [129,130,131,132]. Two types of factor H/CFHR1 hybrid proteins have been described. One hybrid protein comprises the first 18 SCRs of FH linked to the C-terminal two SCRs of CFHR1. The second fusion protein has the first 19 SCRs of FH linked to SCR5 of CFHR1. Both hybrid factor H/CFHR1 proteins differ from their native C-terminal FH domain 20 by two amino acids only, at positions S1191L and V1197A [132]. They lack proper FH cell binding and protection from complement and are directly implicated in the disease pathogenesis [132].
Conversely, two types of hybrid CFHR1/CFH genes that encode a fusion protein with the first three short consensus repeats (SCRs) of FHR1 and the last two SCRs of FH or with the first four SCRs of FHR1 and the terminal SCR20 of FH have been identified in aHUS patients. Functional studies revealed that the hybrid protein causes complement dysregulation at the cell surface by acting as a competitive antagonist of FH.
Rare Coding Variants in the CFI Gene
To date more than 100 CFI distinct rare variants have been published, located everywhere in the gene. All but one variants are heterozygous [4]. CFI pathogenic variants induce a default of secretion of the mutant protein (de Jong) and less frequently disrupt its cofactor activity, with altered degradation of C3b in the fluid phase and on surfaces. However, 40% of CFI mutations have no identified functional consequences and their link with the disease remains unclear. Plasma C3 levels are below the normal range in approximately 50% of patients with CFI variants and FI levels are slightly decreased in 30% [5]. CFI rare variants account for 4–8% of aHUS cases [5].
Rare Coding Variants in the MCP Gene
More than 100 distinct rare variants in MCP gene have been reported in cohorts of patients with aHUS. Fifty percent of variants identified in MCP gene are splice site nonsense or frameshift variants. In the French aHUS cohort, one third of MCP mutations are homozygous and two-thirds are heterozygous [5]. Over 80% of the pathogenic variants induce a reduction in MCP expression on granulocytes. Plasma C3 level is normal in patients with isolated MCP mutations [5].
Rare Coding Variants in the C3 Gene
Screening the French aHUS cohort for mutations in the C3 gene led to the discovery of heterozygous pathogenic variants including a recurrent C3 variant (p.R139W) in aHUS patients. Functional studies showed that the nucleotide change induces either a defect in the ability of C3 to bind the regulatory proteins MCP and FH (indirect gain of function mutation) or an increase in the capacity of C3 to bind FB (direct gain of function mutation). In both cases, the genetic change induces enhanced C3bBb convertase formation and complement activation on cell surfaces [133]. There are now at least 90 distinct C3 mutations reported in hundreds of aHUS patients, however few functional studies have been reported. It is estimated that ~2–10% of incident aHUS patients will carry a C3 pathogenic mutation. The majority of these patients have persistently low plasma C3 levels [43].
Rare Coding Variants in the CFB Gene
Very few pathogenic variants of CFB with functional consequences have been identified. Therefore, CFB mutations account for only 1–2% of aHUS patients (Table 22.3). Functional analyses demonstrated that aHUS-associated CFB mutations are exclusively gain-of-function mutations that result in enhanced formation of the C3bBb convertase [134, 135]. CFB-mutated patients exhibit a permanent activation of the alternative pathway with very low C3.
Out of 9 CFB rare variants characterized using functional in vitro assays, only 5 revealed a gain-of-function phenotype; the other variants are classified of undetermined significance [134].
Combined Complement Gene Mutations
Only 8–10% of patients with mutations in CFH, C3, or CFB had combined mutations, whereas approximately 25% of patients with mutations in MCP or CFI had combined mutations [53].
Mode of Inheritance and Penetrance
Twenty to 30% of patients have a familial history of aHUS. More frequently the disease is sporadic with only one case per family. However, de novo mutations are exceptional [135]. Among pedigrees with familial aHUS, transmission of the disease is autosomal recessive in cases with homozygous or compound heterozygous mutations in CFH or MCP. Transmission is autosomal dominant in cases with a heterozygous mutation. Disease penetrance in family members who carry the heterozygous mutation has been evaluated to be approximately 50%, as only half of these subjects develop the disease. This may be an over-estimate, due to the issue of reporting bias, since pedigrees with more that one affected individual are more likely to be studied than those with just one patient. The identified mutation therefore appears to be a risk factor for the disease rather than its direct and unique cause and aHUS has to be regarded as a complex polygenic disease which results from a combination of genetic risk factors. Homozygous haplotypes (defined by five frequent genetic variants transmitted in block) in CFH (at risk CFH tgtgt), MCP at risk (MCP ggaac) [136] and CFHR1*B allele [137] have been demonstrated to be more frequent in patients with aHUS than in controls. In addition, precipitating events or triggers appear required for the disease to manifest in patients genetically at risk.
Complement Alternative Pathway: From Gene Change to TMA Lesion
Mutations in the genes CFH, MCP and CFI impair the mechanisms that regulate AP activation and gain of function variants in C3 and CFB increase AP activation. Whatever the pathogenic variants identified, endothelial cells are no longer protected from complement activation [138, 139]. The increased production of MAC at the endothelial cell surface induces alterations of these cells, which become procoagulant by producing high molecular weight multimers of von Willebrand Factor, thus triggering the formation of thrombi [140,141,142]. In addition, complement activation at the surface of platelets triggers platelet activation and aggregation and this contributes to the formation of thrombi within the microcirculation [143]. This physiopathological model is corroborated by transgenic animal models. Mice which express a FH variant lacking the C-terminal 16–20 domain responsible for the interaction of FH with C3b and the endothelium develop HUS similar to the human disease [144].
Variants of Unknown Significance in Complement Genes: Disease Relevant or Benign?
Over the coming decade, the challenge will be to optimize and to implement at scale, strategies that use human genetics to further the understanding of disease, and to maximize the clinical benefit of those discoveries. The modern genetic screening test to identify genetic abnormality in aHUS patients includes next generation sequencing (NGS) with at least a panel of 5 genes (CFH, CFI, MCP, C3 and CFB), Sanger sequencing and MLPA with an interpretation of the clinical consequences.
Not all detected complement gene variants have clinically relevant consequences. The standards and guidelines published in 2015 by the American College of Medical Genetics (ACMG) lay out an extensive framework of evidence for interpretation of sequence variants, including guidance for using population data and computational and predictive tools. The variants are classified along a gradient ranging from those that almost certainly have a pathogenic role to those that are very likely benign. However functional characterization of aHUS associated FH variants reveals limitations of routinely used variant classification methods. Access to resources that catalogue genetic variation across populations (such as gnomAD) has enabled the confident exclusion of genetic variants too common in population-level data to be plausible causes of rare diseases. As a general rule, variants with a MAF <0.1% might be considered relevant for the pathogenesis of aHUS or other complement-mediated disorders.
In 2021 this rule cannot be applied for variants in the complement genes. Genetic data are now available for >140,000 individuals from diverse populations in the Genome Aggregation Database (gnomAD). These data indicate that rare variants in the five complement genes with MAFs of <0.1% are present in 3.7% of healthy individuals and pathogenic variants can be found in 1% of samples of DNA from healthy blood donors. Only 9 out the 15 genetic changes in CFB identified in patients with aHUS led to functional activity changes compared to the wild-type protein [134]. Furthermore, only 29 of 79 rare variants in the CFH gene with a MAF <0.1% that have been identified in patients with aHUS are classified as pathogenic based upon the demonstration that they impair CFH regulatory activity [145]. The classification of complement gene variants relies on tools that help predict the potential pathogenicity of a variant [146].
In clinical practice, analysis of functional alterations in complement proteins takes into account the level of expression of the encoded protein (in plasma for CFH and CFI and at the granulocyte surface for CD46), the impact of the variant on the function of the encoded protein (assessed using in vitro assays and prediction of the pathogenicity of a variant based on functional domains) and in silico analyses. Establishing a causal relationship is difficult with the lack of experimental data. According to ACMG guidelines, more frequently the variants have only moderate evidence for pathogenicity and the variant will be classified as a variant of undetermined significance VUS). The current classification of complement gene variants is not optimal and the clinical relevance of individual variants should therefore be regularly re-evaluated.
In summary, not all detected gene variants have clinically relevant consequences. In practice, where a VUS is found in a complement gene of a patient with aHUS, it is important that other causes of a TMA are still excluded, rather than attributing causality. In addition, it is important not to screen family members for the presence of a VUS, since this could attribute risk where none exists or conversely, falsely reassure when risk still exists.
Genetic Abnormalities in Genes Not Related to Complement
Diacylglycerol Kinase ε Mutations
Diacylglycerol kinase ε (DGKε) is an intracellular lipid kinase highly expressed in glomerular capillaries, podocytes and platelets of healthy mice and humans. DGKs are enzymes that phosphorylate diacylglycerol molecules to phosphatidic acid. Using exome sequencing, deficiency in DGKε was established as a novel cause of pediatric onset aHUS in 2013 [45]. Subsequent to the first publication of 13 aHUS children from 9 kindreds, 6 new cases from 4 kindreds have been identified [58, 59], followed by a third cohort with clinical information on 15 patients based on data from the UK National Renal Complement Therapeutics Centre including patients from UK, United Arab Emirates and New Zealand [46]. This cohort also established the presumed prevalence and incidence of DGKε-aHUS in the UK population at 0.009 per million per year, when the incidence rate of complement-mediated aHUS was 0.47 per million per year [46]. The phenotypic spectrum and outcome of DGKE disease was reviewed by Azukaitis et al. in a global cohort of 44 (including 10 previously unpublished) cases [60].
Transmission of DGKε mutations follows an autosomal recessive pattern and all patients reported to date carry homozygous or compound heterozygous nonsense, splice site or frameshift mutations. A likely explanation for the pathogenesis of DGKε mutations is that the loss of DGKε enhances protein kinase C activation in endothelial cells, platelets and podocytes, which may result in upregulation of prothrombic factors and platelet activation and altered podocyte function [45, 147]. However, the pathophysiological mechanisms of DGKε nephropathy have not yet been fully understood.
The aHUS relapses are clustered in early life and appear to be less prevalent later. In addition to presentation with aHUS, patients carrying DGKε mutations can also present with proteinuria without aHUS or steroid resistant nephrotic syndrome with MPGN pattern on biopsy. The symptoms can overlap in individuals, when in early life patients present with nephrotic range proteinuria and relapsing course of aHUS progressing further to chronic kidney disease (CKD) later in life with proteinuria, microscopic hematuria and hypertension. Neither the UK nor the global cohort found predictors that increase the risk of reaching end stage kidney disease or evidence of a significant role for complement activation on progression and relapses of DGKε-aHUS [46, 60]. These resolve regardless of therapy including complement blockade by eculizumab. Moreover, there were aHUS episodes or relapses during treatment with complement blocking therapy. Therefore, complement blocking therapy is probably not beneficial in this specific cohort of aHUS and patients should be managed supportively and in those already on eculizumab, withdrawal should be considered. DGKE nephropathy appears to take a slowly progressive course; only 20% of patients reach ESRD within 10 years of diagnosis [60]. There are no reports of DGKE nephropathy recurrence after transplantation (6 transplant cases reported as of October 2019) [46, 60]; therefore, individuals who progress to end-stage kidney disease (ESKD) should undergo kidney transplantation without the need for pre-emptive eculizumab [46, 60].
Cobalamin C Metabolism Defect Related HUS
A defect in the remethylation pathway caused by cobalamin C deficiency can lead to a clinical presentation very similar to HUS. It can present fulminantly in the neonatal period or later in life. The triad of HUS is also accompanied by other metabolic symptoms like delayed development and growth, seizures, hypo or hypertension and leucopenia. The inheritance is autosomal recessive, and it is usually caused by a defect in the MMCHC gene.
The major markers are elevated homocysteine and methylmalonic acid levels in plasma. Levels of homocysteine over 50 μM/L with normal levels of vitamin B12 and folate are pathognomonic. TMA in cobalamin C deficiency is believed to be caused by the endothelial toxicity of high plasma homocysteine levels.
Treatment is by loading dose of intramuscular vitamin B12 (hydroxycobalamin) followed by lifelong supplementation [110, 121]. Although rare, Cblc deficiency should not be missed since the prognosis of undiagnosed patients is dismal, it can easily be treated once detected. Hence, plasma homocysteine should be included in the routine diagnostic panel of aHUS.
Rare Variants in Genes with Debatable Clinical Relevance
Genetic defects in THBD, which encodes thrombomodulin, a protein that interconnects the coagulation cascade and complement system, have been suggested to contribute to the pathogenesis of aHUS. Few mutations in THBD affecting the functions of the protein have been identified to date, with a frequency varying from 0 to 5% of all aHUS cases [2, 3, 44] (Table 22.3). Burden or aggregate association tests, in which all rare variants affecting the same gene are combined into one test, are used to increase the statistical power for rare variant association. Although rare variants in THBD have been reported in 41 patients with aHUS, their frequency is not significantly higher in these patients than in controls the general population [148]. Therefore, the link of THBD with aHUS remains debatable.
The potential clinical relevance of rare variants in genes such as PLG (which encodes plasminogen) [149], INF2 (which encodes inverted formin 2) [150], VTN (which encodes vitronectin) [148] and CLU (which encodes clusterin) [151] identified in patients presented with some features of HUS warrant further assessment.
Acquired Complement Abnormalities in Atypical HUS
Anti-factor H Autoantibodies
Anti-factor H (anti-FH) autoantibodies are identified in 5–11% in European aHUS cohorts and in about 20% in Asian aHUS cohorts [65, 76, 79]. Interestingly, they are identified in more than 56% cases from India [80]. A Czech cohort showed a rather outlying large proportion of anti FH antibodies in comparison to other European cohorts of aHUS at 61% which could be due to small sample size and sampling method [67].
Anti-factor H autoantibodies associated HUS (anti-FH HUS) usually manifests later than genetic types of aHUS caused by factor H mutations, usually between 5 and 15 years of age [77]. An international aHUS registry reported a median of age of 13.1 (6.1–31.3) years at presentation [81] for this group of patients. However, the youngest reported aHUS patient with anti-FH antibodies identified was younger than 1 year and the oldest reported patient was over 75 years old. Most of the published series show a slightly higher prevalence of anti-FH HUS in males.
Anti-FH antibodies bind mostly to SCR 19 and 20 of FH but also to other epitopes of FH and thus inhibit the majority of regulatory functions of FH at cell surfaces [153]. Plasma C3 level is decreased in 40–60% of patients with anti-FH antibodies during the acute phase, while FH levels are decreased in only approximately 20% of patients [62]. C3 levels are significantly lower in patients with very high anti-FH antibody titer.
Ninety percent of patients with anti-FH antibodies have a complete deficiency of CFHR1 and CFHR3 due to a homozygous deletion of CFHR1-R3, a polymorphism carried by 2–9% of European, 16% of African and ≤2% of Chinese healthy controls [128, 154]. The reason why individuals with CFHR1-R3 deletion develop anti-FH antibodies is uncertain. The current theory linking the deletion in CFHR1 with the generation of antibodies is based upon the interaction of the FH protein that is used by pathogens for immune evasion. In individuals with CFHR1 gene deletion, CFHR1 protein is recognized as foreign by their immune system. When a CFHR1-deficient individual is infected by an organism that can bind CFHR1, CFHR3 and FH proteins, the FH protein is changed by the infectious organism to resemble CFHR1 and host immunity mounts a response, leading to production of anti-FH inhibiting FH, thus leading to endothelial dysfunction and symptoms of aHUS. This is corroborated by structural differences found between CFHR1 and FH [155].
Fujisawa et al. described three aHUS patients where anti-FH antibodies affected platelets directly [82]. Washed platelets aggregated more when in contact with plasma from these patients compared to plasma from healthy controls or from aHUS patients with complement genetic variants.
Anti-factor I Antibodies
Two cases of anti-FI antibody-associated HUS have been reported to date [156]. The clinical relevance of these antibodies is difficult to establish, also given their extreme rarity.
Outcome of Atypical HUS Prior to the Availability of C5 Inhibition Therapy
In the era before eculizumab became available the death rate in children with aHUS was 8% [5], 9% [4] and 14% [2] in three pediatric series at average follow-up times of 3.8, 7.5 and 3 years, respectively. Most deaths occurred in children less than 1 year of age and at first episode or during the first year after onset. Approximately 20% of children progressed to ESKD or died at first episode or within <1 month after onset, 30% within 1 year and 40% at 5 years follow-up. The most severe outcome was in children with CFH mutations, of whom one third progressed to ESKD or died at first episode, half at 1 year and two thirds at 5 years follow-up. The prognosis of CFI and C3 mutation-associated HUS was hardly less severe than that of CFH mutation-associated HUS. MCP mutation-associated HUS in children had the best prognosis, with an ESKD risk of 25% at median follow-up of 18 years. aHUS in children with no complement mutation identified also had a relatively favorable outcome [5].
Lastly, the outcome of anti-FH antibodies associated HUS was poor when treatment was limited to PE, including death in 10% of patients, CKD in 40% and ESKD in one third at mean follow-up 39 months [62, 152]. Early treatment with a combination of PE, corticosteroids and immunosuppressants allowed a more favorable outcome, similar to that of MCP mutation—associated HUS [5, 64].
Less than 10% of children with DGKE-aHUS progress to ESKD in the first year after onset, but patients with this form of aHUS develop proteinuria and nephrotic syndrome, severe hypertension and progress to CKD stages 4–5 (eGFR 15–29 mL/min/1.73 m2 or ESKD) between the age of 20 and 25 years [45, 46, 60].
In the pre-eculizumab era, several series suggested that approximately half of children with aHUS experienced relapses [4, 5]. Among children who had not died or reached ESKD at first episode or at 1 year follow-up, 25% had relapses during the first year and 47% after the first year. However, a high relapse rate after the first year was mostly in patients with DGKE (83% during the first year, 50% up to 5 years) or MCP mutations (25% during the first year, 92% after the first year), while relapse rate after the first year was 20–30% in other genetic subgroups [5, 45, 46, 60]. Despite this risk of relapses in the long term, MCP mutation-associated HUS has the best prognosis in children, as indicated above. Last, anti-FH HUS had a relapsing course in two third of patients when untreated or treated only with PE/PI [62, 65], which was reduced to approximately 10% by early treatment combining PE + immunosuppressants + corticosteroids [5, 64].
Treatment of Atypical HUS Prior to the Availability of C5 Inhibition Therapy
Plasma Therapy
PE (or PI when PE was not possible) was first line treatment for aHUS until recent year [12] Approximately 15 case reports, mostly in children with CFH mutation, showed that early, intensive plasmatherapy, followed by maintenance PE/PI, could prevent relapses and preserve kidney function at follow-up up to 6 years [8, 9, 157]. However, although plasmatherapy was associated with complete or partial remission (hematologic remission with kidney sequels) in approximately 80% of aHUS episodes in children, half of them had died or reached ESKD at 3 years follow-up [2]. In addition, plasmatherapy carried significant morbidity. An audit of complications in children receiving PE for aHUS revealed 31% developed catheter-related complications (including infection, thrombosis and hemorrhage) and 11% developed plasma hypersensitivity [13]. The benefit of PE/PI is uncertain in DGKE mutation-associated HUS, as proteinuria, the main marker of a progressing course in DGKE mutation–associated HUS, persisted in 9 of the 12 patients who received plasmatherapy [45, 46, 60].
Kidney Transplantation
The risk of post-transplant recurrence of aHUS was 60% in the pre-complement blockade era [2, 104]. Forty percent of recurrences occurred during the first month after transplantation and 70% during the first year. Graft survival was 30% at 5 years follow-up in patients with recurrence, compared to 68% in those without recurrence [104]. Eighty percent of patients who had lost a prior graft from recurrence had recurrence after retransplantation. The main independent risk factor for recurrence was the presence of a complement mutation. The highest risk (approximately 80%) was in patients with CFH and C3 or CFB gain of function mutation, the risk in patients with CFI mutation was approximately 50% and patients with no complement mutation identified had the lowest risk (approximately 20%) [104]. The risk of post-transplant recurrence in patients with MCP mutation has been shown to be low (<10%) if the mutation is isolated (the graft brings the non-mutated MCP protein), while it is approximately 30% if the MCP mutation is associated with a mutation in CFH, CFI or C3 [53]. No post-transplant recurrence was observed in four patients with DGKE mutations [45, 46, 60]. The risk is low in anti-FH HUS if the antibody titer is low at the time of transplantation, while substantial if elevated [62, 65, 66] [128, 158]. One patient with THBD mutation has been reported to have had post-transplant recurrence [159].
This shows that genetic screening is necessary before listing a patient for kidney transplantation to predict the risk of post-transplant recurrence and guide decisions for the choice of the donor and the prevention of recurrence.
PE/PI for post-transplant recurrence generally failed to avoid graft loss [2, 104]. Therefore, prophylactic PE/PI was recommended [160]. The efficacy of this strategy is poorly documented. However, graft survival rate free of recurrence was significantly higher in nine patients who received prophylactic PE/PI than in 62 patients without prophylactic PE/PI [104]. Interestingly, calcineurin inhibitors (CNI) did not significantly increase the risk of recurrence in a recent series, while mTOR inhibitor use was an independent significant risk factor for recurrence, possibly related to the anti-VEGF (vascular endothelium growth factor) action of these drugs [104]. The current consensus is that aHUS is not per se a contraindication to CNI. Strict monitoring of blood levels and overdosage avoidance is recommended, while CNI-free mTOR based immunosuppressive regimens should be avoided [161].
Treatment Recommendations
During the initial manifestation of HUS, unless it is a relapse in a patient already know to carry a risk variant in genes associated to aHUS, diagnosis is challenging, and aHUS cannot be excluded purely on clinical grounds or complement markers. Children with suspected aHUS should ideally be transferred to a children’s kidney center capable of kidney replacement therapy and intensive care. In contrast to adults, and because the incidence of acquired TTP in children is very low, immediate PE whilst awaiting ADAMTS13 results is not routinely recommended. Children in whom complement driven aHUS is strongly suspected or proven should receive eculizumab (see below) as first line treatment, to avoid PE and the complications of central catheters [103]. Confirmation of a complement mutation is not required for the decision of treatment initiation in such cases. As treatment delay may affect recovery of kidney function, eculizumab treatment should be initiated as soon as possible, ideally within 24–48 h of onset or admission. In addition to targeted treatment with eculizumab, symptomatic care is based on general recommendations for AKI [162] and on consensus from observational studies. The cornerstone is appropriate fluid management, kidney replacement therapy in patients with high urea or unsafe electrolyte profile and stopping of nephrotoxic drugs. Red blood cell transfusions should be given to patients who have symptoms of severe anemia or when hemoglobin falls rapidly. Platelet transfusions are not advised unless patient has a life threating bleeding or requires invasive procedure like placement of vascular catheter for adequate dialysis.
Eculizumab
Eculizumab, a monoclonal humanized anti-C5 antibody, prevents C5 cleavage and the formation of C5a and C5b-9, thus blocking the C5a pro-inflammatory and the C5b-9 pro-thrombotic consequences of complement activation. It has been the accepted treatment of paroxysmal nocturnal hemoglobinuria (PNH), another complement dependent disease, for more than 15 years [163].
The rationale for treatment with complement C5 blockade was first proposed based on first two aHUS patients treated successfully with eculizumab in 2009 [164, 165], followed by plethora of successful cases [15, 85, 87, 89,90,91, 168,169,170,171,170].
Four observational prospective single-arm non randomized multinational trials [14, 171, 172] demonstrated the efficacy of eculizumab to stop the TMA process, allowing sustained remission of aHUS with improved or preserved kidney function in the majority of patients. One of these trials [172] specifically studied eculizumab in children with aHUS. Figure 22.6 shows the remarkable efficacy of eculizumab in these patients. Data from the trials also suggested that an early switch from PE/PI to eculizumab or the use of eculizumab as first line therapy gave patients the best chance of full recovery of kidney function. Treatment was well tolerated, with no increase of adverse events over time. However, two of the 100 patients who entered these trials developed meningococcal meningitis. Eculizumab is administered by intravenous infusion according to the weight-directed dose and schedule shown in Table 22.4.

(a) Improvement in platelet count over 27 weeks of eculizumab treatment in 22 children with aHUS. N values <5 were not included. Bars represent standard error of the mean (SEM). (b) Improvement in estimated glomerular filtration rate (eGFR) over 27 weeks of eculizumab treatment. N values <5 were not included. Bars represent SEM. Arrows denote administration of eculizumab
Eculizumab is currently approved by both the Food and Drug Administration and the European Medicines Agency for the treatment of atypical HUS. The cost of the drug and the presumption that patients should receive lifelong treatment played a major role in approving the cover of the costs by healthcare systems. Access and funding are achievable in USA, Australia, United Kingdom and in most states of the European Union, with exception of Bulgaria and Romania. Other countries with availability include Israel and Japan. African and Asian countries, including China and India, have no access to the drug. The availability generally corresponds to the countries’ economic position (apart from New Zealand where access is very limited).
Ravulizumab
A second generation complement inhibitor, ravulizumab, has recently been developed by targeted re-engineering of eculizumab. Two structural changes were incorporated, aimed at extending the terminal half-life. The first change enhanced the dissociation rate of the mAb:C5 complex at pH 6.0, eliminating the target mediated antibody clearance. The second change enhanced the affinity of the antibody to human neonatal Fc receptor [173].
Ravulizumab has not been directly compared to eculizumab in a clinical trial. However, efficacy and safety were confirmed in a phase 3 single-arm trial in adult patients (n = 56) with aHUS naïve to complement inhibitor treatment [174]. Complete TMA response, defined as normalization of platelet count and LDH and ≥25% improvement in serum creatinine, was achieved in 53.6% of patients in the 26 week study period. This lower response rate than reported within the eculizumab trials may have been due to broader eligibility criteria for recruitment (only 20.5% had genetic complement mutations or anti-FH identified compared with 76% in the equivalent eculizumab trial). There were no severe treatment-related events reported. Four deaths were reported, none of which were considered treatment-related by the study investigator [175].
Two trials tested the efficacy and safety of ravulizumab in children under 18 years of age with aHUS. Fourteen of 18 complement-inhibitor naïve patients with aHUS (78%), achieved complete TMA response with ravulizumab. Ten aHUS patients who were already receiving eculizumab, switched to ravulizumab for a period of 26 weeks without significant safety issues and showed unchanged kidney function and hematological remission of aHUS even after extended observation of one year. No unexpected adverse events, deaths, or meningococcal infections occurred. There were not enough data to demonstrate the effectiveness of ravulizumab in children weighing less than 10 kg [176, 177].
Taken together, these trials indicate that ravulizumab, is effective for the long-term treatment of patients with aHUS. Current evidence suggests an acceptable safety profile, although longer-term surveillance will be required. The main risk of ravulizumab treatment is similar to that of eculizumab arising from the same principle of C5 blockade. Therefore, all patients must strictly adhere to the same prevention protocols against meningococcal infection as with eculizumab. Dose and schedule information for ravulizumab are shown in Table 22.5.
There are now two licensed treatments for patients with aHUS. Clinicians and patients now have a choice between short-acting and long-acting C5 inhibition. Since the diagnosis of aHUS is complex and not all patients initiated on C5 inhibition will continue with long-term therapy, it may be an option to commence initial treatment with eculizumab, with a switch to long-acting therapy once a need for long-term treatment is established (Fig. 22.7). This approach would minimise the risk of prolonged C5 blockade for those in whom treatment is discontinued due to an alternative diagnosis (for example, a subsequent positive STEC result). However, commencing with ravulizumab at time of initial presentation is also an option.

Recommended management approach when atypical HUS is suspected
A subcutaneous ravulizumab formulation is currently undergoing evaluation in a phase 3 trial in adult patients with PNH [177] and may be a future option also for patients with aHUS.
Risk of Meningococcal Infection on C5 Inhibition Therapy and Its Prevention
The host defense against Neisseria meningitis depends on the lytic terminal complement complex C5b-9. The incidence of meningococcal infections in patients with hereditary complete deficiency of terminal complement factors is 0.5% per year, corresponding to a 1000 to 2000-fold relative risk increase compared to the normal population [178]. Patients undergoing eculizumab or ravulizumab treatment have the same risk as these patients. Therefore, prevention of meningococcal infection is crucial, relying on vaccination and antibiotic prophylaxis. Tetravalent conjugated vaccines protect against serogroups A, C, W135 and Y. Polysaccharide vaccines against serogroup B are also available and recommended for all patients receiving eculizumab.
The frequency of invasive meningococcal infection has been approximately 0.5/100 patient years in patients with PNH treated with eculizumab, despite meningococcal vaccination (not anti-B) [179].
Two of the 100 aHUS patients treated within trial protocols and one among approximately 80 case reports developed invasive meningococcal infection despite being vaccinated [180]. The more recent analysis of eculizumab safety from the Global Atypical HUS registry showed meningococcal infection in two adult cases (0.17 per 100 patients years) and one pediatric patient (0.11 in 100 patient years) out of 1321 patients ever treated with eculizumab. Two patients recovered completely, and one case was fatal [181].
Whilst long-term antibiotic prophylaxis for patients on C5 inhibition is not mandated by the manufacturer, many clinicians add this to their patients’ treatment (phenoxymethylpenicillin or erythromycin if penicillin allergic). This is strongly recommended for children and in some countries, this is obligatory [103]. Neither vaccines nor antibiotic prophylaxis guarantee full protection, hence the importance of patient and family education on signs of meningococcal infection and the provision of alert cards to be carried by the patient or their caregiver to present to their health care provider when unwell, in order to minimize delay in recognition.
Treatment of Anti-factor H Antibody Associated HUS
The first large series of patients with anti-FH HUS (treated mostly with PE without immunosuppressants) suggested that many of these patients suffer a relapsing course, leading to end stage kidney disease [62]. A much more favorable outcome, similar to that of MCP mutation-associated HUS, has been reported in children with this form of HUS, treated early with PE, immunosuppressants and corticosteroids [5].
The largest experience with the approach to decrease anti-FH antibodies with immunosuppressive treatment comes from India, where 55.8% of their 781 aHUS patients presented with anti-FH antibodies [66].
Combined Plasma exchange (PE) and immunosuppression improved long-term outcomes (HR 0.37; P = 0.026); maintenance therapy with corticosteroids and MMF reduced the relapse risk (HR 0.11; P < 0.001) [66].
Maintenance treatment with corticosteroids and mycophenolate mofetil (MMF) or azathioprine significantly decreased the risk of relapses, from 87% to 46% at last follow-up [64, 157].
Eculizumab was documented as an effective treatment of HUS symptoms in anti-FH HUS in several cases [88, 182].
In a cohort of 17 patients with anti-CFH antibody associated aHUS, four patients were treated with first-line eculizumab rather than PE. Patients treated with eculizumab achieved remission in 100% of cases, whereas treatment with PE and immunosuppression was associated with a poor rate of renal recovery in 7 of 11 treated. Therefore, treatment with eculizumab did not appear inferior to PE and immunosuppression [183]. This gives patients and clinicians different treatment options with different adverse effect profiles—broad immunosuppression with rituximab or cyclophosphamide, or more targeted treatment with eculizumab [103]. The National Renal Complement Therapeutics Centre in the UK recommends initial treatment with eculizumab for anti-FH HUS based upon a more favorable adverse effect profile .
Global Variations in Atypical HUS Treatment Recommendations
Eculizumab has revolutionized the management of aHUS. The current best recommendations and guidelines are challenging to implement globally, particularly in resource-limited healthcare settings due to the prohibitive cost of eculizumab and its successor ravulizumab. In response to this, the Indian Society of Pediatric Nephrology have published consensus guidelines for countries lacking access to eculizumab and complex diagnostic facilities [157].
These pragmatic guidelines are mirroring the best recommendations from the pre-eculizumab era [64]. (See also Fig. 22.7).
They recommend treating aHUS where anti-FH antibodies are not suspected with prompt initiation of PE using fresh frozen plasma (The dose is recommended as 1.5 times of the plasma volume or 60–75 mL/kg) and repeated daily until hematological remission is achieved (platelets over 100 × 109/L, schistocytes under 2% and LDH in normal range). The tapering follows after 5 days of daily PE or when remission is achieved followed by alternate days PE and 2–3 weeks of twice a week PE.
In case of positive anti-FH antibodies, additional immunosuppression is administered, starting with prednisolone 1 mg/kg daily for 4 weeks followed by alternate day dosing for another 4 weeks and tapering down for 1 year. Cyclophosphamide (500 mg/m2 q 4 weeks, 3–5 doses) or Rituximab (500 mg/m2 q 7 day, 2 doses) are given to further decrease the production of antibodies. Mycophenolate mofetil (500–750 mg/m2/day) or azathioprine (1–2 mg/kg/day) are used as additional long-term immunosuppression [157].
C5 Inhibition Therapy Monitoring
Data from the prospective studies show that complete complement blockade is obtained within 1 h after the first dose of eculizumab and is maintained long-term in patients receiving the recommended treatment schedule [184]. The role of assessment of complement blockade during routine use of eculizumab is uncertain. If there are features of inadequate disease control, this should be assessed. Incomplete blockade may be due to insufficient dose (especially in children with a weight just below a weight threshold requiring a higher dose) or heavy proteinuria with nephrotic syndrome (leakage of the drug in the urine). A genetic cause might also have to be considered in aHUS patients of Japanese or Asian origin with poor response to eculizumab and/or complement non-blockade, such as the C5 variant which impairs the binding of eculizumab to C5 [185]. For the long term, complement blockade assessment is mostly required in cases of apparent resistance to eculizumab, including relapse of HUS but also isolated abnormalities in platelet count, LDH and/or haptoglobin levels, appearance or increase of proteinuria or serum creatinine, especially if kidney biopsy suggests ongoing TMA.
Currently available assays of complement blockade under eculizumab are a CH50 or other hemolytic-based assays or the Wieslab Complement System [186]. Due to the site of action of eculizumab, low C3 levels as seen in some mutations are not expected to normalize under eculizumab, and this has been observed [125]. Soluble C5b-9 remains detectable or increased in aHUS patients under eculizumab and therefore cannot be recommended to monitor the efficacy of eculizumab [125, 187]. Most aHUS patients treated within the prospective trials who received the protocol schedule had suppression of CH50 activity and eculizumab trough levels ≥150 μg/ml [14]. However, the correlation between drug levels and complement activity in aHUS patients is not fully established and the availability of plasma eculizumab measurement is currently limited. An additional consideration is that hemolytic assay monitoring may be less accurate for ravulizumab than for eculizumab. One recent study showed that full inhibition of CH50 was not achieved despite high levels of ravulizumab present in plasma [188].
Some authors have reported using assays of hemolytic activity as a tool to lengthen the interval between doses, in an attempt to reduce cost and infusion burden [189]. This approach is not recommended by the manufacturer. It has been recommended that increasing the interval between doses should be considered only in patients who maintain CH50 activity <10% despite longer intervals or lower doses [186].
Duration of C5 Inhibition Therapy in Children with aHUS in Their Native Kidneys
Lifelong treatment with inhibition of C5 for patients with aHUS has been the accepted paradigm. However, there is no definitive evidence for persisting use. The recommendation of not stopping C5 blockade is being reconsidered for some patients with aHUS in their native kidneys based on estimating the risk of relapse by genetic testing.
Having a safe strategy for discontinuation of treatment would be beneficial not only for reducing the significant cost of the therapy but also for safety of the patients and the long-term quality of their life.
Even in the pre-eculizumab era, not all patients relapsed after successful PE. A report of 214 patients, of which 146 were treated with PE, showed that 42% of children were relapse free after 5 years of observation [5]. A Dutch cohort reported sustained remission in 25% of patients [4] and even a cohort of patients with aHUS with changes in the CFH gene had normalized kidney function in 22.5% [190]. Following several case reports and series of successful eculizumab discontinuation [170, 189, 193,194,193], more evidence to support the safety of eculizumab withdrawal appeared from larger studies. The report on this topic from the Global aHUS Registry showed that 33/151 (22%) patients experienced TMA recurrence, particularly in those with pathogenic variants in genes associated with aHUS or with a family history of aHUS. Eight percent then progressed to end-stage kidney disease and 5% required subsequent kidney transplant [194]. During an unprecedented event between 2016 and 2019 when distribution of eculizumab supply was disrupted in Brazil, the effect of discontinuation without stratifying patients based on genetic risk was demonstrated. There were 11 relapses in a cohort of 24 patients, of which 8 occurred in patients with a CFH gene variant [195].
A prospective, single-arm study conducted in France stopped eculizumab after at least 6 months of treatment in 55 patients of which 19 were children and showed an overall aHUS relapse rate of 23% (n = 13). Twenty-eight patients (51%) had rare variants in complement genes (MCP n = 12, CFH n = 6, CFI n = 6, FH antibodies, n = 4). Six out of 19 children (32%) relapsed [196]. All 13 patients who relapsed were carrying a variant in genes associated with aHUS except one that was subsequently found to have hereditary ADAMTS13 deficiency (and therefore did not have aHUS). C5 blocking therapy was restarted in all 13 patients. Eleven returned to their baseline kidney function and two remained with decreased function, one progressing to ESKD. Therefore, prompt identification of relapse in aHUS patients who stopped C5 blocking therapy seems to be crucial for preventing long term kidney damage [196].
The overall evidence suggests that patients with no mutation in known complement genes who achieve stable remission for at least 6 months are at low risk of relapse after stopping the treatment. Patients with mutations relapse at much higher rate and in this situation C5 blocking therapy must be restarted promptly to prevent loss of kidney function.
Further safety data for the withdrawal of C5 inhibition in patients with aHUS are required, particularly for patients with mutations. An ongoing trial of stopping eculizumab in patients with aHUS in UK (SETS, EudraCT Number: 2017-003916-37) [197] aims to corroborate the current evidence for safe stopping. If all evidence supports safe withdrawal of C5 inhibition in patients with aHUS, the challenge for patients and clinicians will be to implement individualized care plans to enable rapid detection of signs of relapse and facilitate immediate access to C5 inhibition when a relapse occurs. Without this in place, there is a risk that aHUS will cause ESKD.
Current evidence suggests that eculizumab can be withdrawn in a significant proportion of patients and pragmatic recommendations can be drawn from the experience and protocols used in the described trials. The prerequisite is that patient has achieved remission of HUS with no signs of TMA, normalization of platelets, LDH and stable kidney function. The duration from initiation of treatment to its discontinuation is not established, yet the arbitrary duration of 6 months chosen for the trials could be used as base recommendation. Genetic testing and testing for anti-FH antibodies could help stratifying risk of possible relapse. Patients with no mutation and no anti-FH antibodies (or low titers) are at lowest risk of relapse. Patients with variants of unknown significance and those with pathogenic variants in CFI and CD46 are making a middle tier with slight risk of relapse. Patients with anti-FH antibodies in high titres, pathogenic FH, FB and C3 mutations are at highest risk and the discontinuation of treatment can likely lead to relapse. Most of the relapses would occur in the first weeks after discontinuation and patients need to be reviewed frequently, ideally weekly, with testing eGFR and blood count to identify relapse quickly and restart the treatment.
Further lifelong specialist follow-up of patients who discontinued complement blocking treatment, their education and providing them with straight pathway for review and restarting treatment in case of possible relapse is essential part of the process. There is limited data on another try to stop in case of relapse and restart and it is likely these patients will need lifelong treatment, ideally with long-acting C5 complement blocker, ravulizumab.
Specific patient group are the patients after kidney transplant, where approach needs to be individualized, based on weighing the benefits of continuous or restrictive therapy.
Kidney Transplantation for aHUS Patients Today
Complement blockade therapy has transformed the approach to kidney transplantation for aHUS patients and there are international consensus guidelines regarding the use of eculizumab in this situation (KDIGO 2017). The guidelines state:
-
1.
Kidney transplantation should be deferred until at least 6 months after the start of dialysis because limited kidney recovery may occur several months after starting eculizumab
-
2.
The decision to use anti-complement therapy during transplantation should be based upon recurrence risk
-
(a)
Those with a high risk (50–100%) of recurrence include patients with previous recurrence, pathogenic mutation or gain of function mutation. They should be treated with prophylactic eculizumab to start on the day of transplantation and to continue indefinitely
-
(b)
Those with moderate risk of recurrence include patients with no mutation identified, isolated CFI mutations, complement gene variants of unknown significance or persistently low titer anti-FH antibodies. They can be treated with prophylactic eculizumab or PE or without preventative strategy (left to the discretion of the clinician).
-
(c)
Those with low recurrence risk of recurrence (<10%) include isolated MCP mutation or persistently negative anti-FH antibodies. These patients do not require prophylactic treatment with PE or eculizumab. NB with subsequent data, DGKE HUS can also be viewed as low recurrence risk [46].
-
(a)
-
3.
Living donor transplantation can be considered, with the decision relying on a careful assessment of the risk of aHUS in the donor after kidney donation. A risk assessment for potential living related donation requires full genetic screening of the recipient and the donor, leading to the following possible outcomes:
-
(a)
If the mutation found in the recipient is undeniably responsible for the occurrence of HUS (e.g., CFH mutation in C terminus SCR 19 or 20) and is not found in the donor, the risk of HUS is low for the donor and living-related donor transplantation can proceed.
-
(b)
If the donor has the same mutation as the recipient, the risk of HUS is present for the donor and living-related donor transplantation should not proceed.
-
(c)
If the role of the variant found in the recipient is uncertain (not reported in databases, unknown functional consequences) the risk of HUS is intermediate for the donor, who may share with the recipient an unknown risk factor, and living-related donor transplantation is not advised.
-
(d)
If no mutation is identified in the recipient and the donor, the risk to the donor is not quantified. KDIGO advises that if there is no evidence of alternate complement pathway activation in the donor, donation is feasible.
-
(a)
In general, additional endothelial damaging factors should be avoided, such as delayed graft function (prolonged ischemia time, non-heart-beating donor), cytomegalovirus infection, high levels of CNI and the association of CNI + mTOR inhibitors and hypertension.
Prior to the availability of eculizumab, isolated liver transplantation and combined liver-kidney transplantation were recognized treatment approaches for patients with mutations in FH, C3 and FB (all synthesized in the liver). These had relatively high morbidity and mortality (for example, 20% of patients who received combined liver-kidney transplant for aHUS died) [103, 198, 199]. Eculizumab represents a safer treatment option, but liver transplantation may still have a role when eculizumab is not available [161].
Potential Future Therapies
Current therapies are limited to monoclonal antibodies against C5. Numerous drugs that inhibit complement via different targets are undergoing clinical development, some of which may be suitable for use in aHUS. OMS721 (Narsoplimab) is a monoclonal antibody targeting mannan-binding lectin associated serine protease-2, the effector enzyme of the lectin pathway of the complement system and is currently undergoing a phase 3 clinical trial in adult patients with aHUS (NCT03205995. LMP023 (Iptacopan) is an oral FB inhibitor also undergoing a phase 3 clinical trial in adult patients with aHUS (NCT04889430) [102].
Conclusion
The journey to an effective treatment for aHUS is a striking demonstration that understanding the pathophysiology of a disease opens the way to new therapies. The demonstration that aHUS was mostly a disease of complement dysregulation paralleled the development of the anti-C5 monoclonal antibody eculizumab. Prospective trials as well as off-label clinical experience confirmed the efficacy of complement blockade to prevent progression to ESKD in aHUS patients and allowed successful kidney transplantation in those on dialysis. This new era in the field of aHUS emphasizes the need for an etiology/pathophysiology-based classification of the various forms of TMAs. Hopefully, new discoveries will identify the etiology of the 30% of aHUS cases that remain unexplained today, and the aHUS denomination will then disappear.
An important current question is that of the duration of eculizumab treatment, and prospective studies are underway to define whether discontinuation can be considered, in which patients, and when. Should withdrawal be shown as feasible in some patients, this would decrease the burden and risks of continuous treatment for patients, but also the cost for health care systems.
The addition of long acting ravulizumab to the treatment armamentarium for aHUS brings choices for patients and clinicians that may improve quality of life. Over the next decade, it is possible that further agents will be shown to be effective for the treatment of aHUS. Thus, future treatment of aHUS might comprise a decision about specific treatment and mode of delivery, followed by a decision regarding continuation or discontinuation of therapy with appropriate monitoring. Such options were not imaginable by aHUS patients 20 years ago.
References
Nester CM, Barbour T, de Cordoba SR, et al. Atypical aHUS: state of the art. Mol Immunol. 2015;67:31–42.
Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–59.
Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31:E1445–60.
Geerdink LM, Westra D, van Wijk JA, et al. Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol. 2012;27:1283–91.
Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8:554–62.
Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33:508–30.
Rodriguez de Cordoba S, Hidalgo MS, Pinto S, Tortajada A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost. 2014;40:422–30.
Nester CM, Thomas CP. Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? Hematology Am Soc Hematol Educ Program. 2012;2012:617–25.
Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrología (English Edition). 2015;35:421–47.
Scully M, Goodship T. How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol. 2014. https://doi.org/10.1111/bjh.12718.
Cataland SR, Wu HM. How I treat: the clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. Blood. 2014. https://doi.org/10.1182/blood-2013-11-516237.
Ariceta G, Besbas N, Johnson S, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol. 2009;24:687–96.
Johnson S, Stojanovic J, Ariceta G, et al. An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr Nephrol. 2014. https://doi.org/10.1007/s00467-014-2817-4.
Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–81.
Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–57.
Besbas N, Karpman D, Landau D, Loirat C, Proesmans W, Remuzzi G, Rizzoni G, Taylor CM, van de Kar N, Zimmerhackl LB. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006;70:423–31.
Aigner C, Schmidt A, Gaggl M, Sunder-Plassmann G. An updated classification of thrombotic microangiopathies and treatment of complement gene variant-mediated thrombotic microangiopathy. Clin Kidney J. 2019;12:333–7.
George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:654–66.
Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol. 2018;13:300–17.
Joseph A, Cointe A, Mariani Kurkdjian P, Rafat C, Hertig A. Shiga toxin-associated hemolytic uremic syndrome: a narrative review. Toxins. 2020. https://doi.org/10.3390/toxins12020067.
Jenkins C, Byrne L, Vishram B, Sawyer C, Balasegaram S, Ahyow L, Johnson S. Shiga toxin-producing Escherichia coli haemolytic uraemic syndrome (STEC-HUS): diagnosis, surveillance and public-health management in England. J Med Microbiol. 2020;69:1034–6.
Coppo P, Cuker A, George JN. Thrombotic thrombocytopenic purpura: toward targeted therapy and precision medicine. Res Pract Thromb Haemost. 2019. https://doi.org/10.1002/rth2.12160.
Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017. https://doi.org/10.1111/jth.13571.
Constantinescu AR, Bitzan M, Weiss LS, Christen E, Kaplan BS, Cnaan A, Trachtman H. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004. https://doi.org/10.1053/j.ajkd.2004.02.010.
Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of atypical hemolytic uremic syndrome: a systematic literature review. Clin Epidemiol. 2020. https://doi.org/10.2147/CLEP.S245642.
Bayer G, von Tokarski F, Thoreau B, et al. Etiology and outcomes of thrombotic microangiopathies. Clin J Am Soc Nephrol. 2019. CJN.11470918
Ardissino G, Salardi S, Colombo E, et al. Epidemiology of haemolytic uremic syndrome in children. Data from the North Italian HUS network. Eur J Pediatr. 2016. https://doi.org/10.1007/s00431-015-2642-1.
Durkan AM, Kim S, Craig J, Elliott E. The long-term outcomes of atypical haemolytic uraemic syndrome: a national surveillance study. Arch Dis Child. 2016. https://doi.org/10.1136/archdischild-2015-309471.
Zimmerhackl LB, Besbas N, Jungraithmayr T, et al. Epidemiology, clinical presentation, and pathophysiology of atypical and recurrent hemolytic uremic syndrome. Semin Thromb Hemost. 2006. https://doi.org/10.1055/s-2006-939767.
Mallett A, Patel C, Salisbury A, Wang Z, Healy H, Hoy W. The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis. 2014. https://doi.org/10.1186/1750-1172-9-98.
Jenssen GR, Hovland E, Bjerre A, Bangstad HJ, Nygard K, Vold L. Incidence and etiology of hemolytic-uremic syndrome in children in Norway, 1999-2008—a retrospective study of hospital records to assess the sensitivity of surveillance. BMC Infect Dis. 2014. https://doi.org/10.1186/1471-2334-14-265.
Wühl E, van Stralen KJ, Wanner C, et al. Renal replacement therapy for rare diseases affecting the kidney: an analysis of the ERA-EDTA Registry. Nephrol Dial Transplant. 2014;29(Suppl:4):iv1-8.
Cameron JS, Vick R. Letter: plasma-C3 in haemolytic-uraemic syndrome and thrombotic thrombocytopenic purpura. Lancet. 1973;2:975.
Thompson RA, Winterborn MH. Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin Exp Immunol. 1981;46:110–9.
Pichette V, Quérin S, Schürch W, Brun G, Lehner-Netsch G, Delâge JM. Familial hemolytic-uremic syndrome and homozygous factor H deficiency. Am J Kidney Dis. 1994. https://doi.org/10.1016/S0272-6386(12)81065-1.
Rougier N, Kazatchkine MD, Rougier JP, et al. Human complement factor H deficiency associated with hemolytic uremic syndrome. J Am Soc Nephrol. 1998. https://doi.org/10.1681/asn.v9122318.
Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, Goodship JA. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53:836–44.
Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2003;100:12966–71.
Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, Loirat C, Rondeau E, Fridman WH. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41:e84.
Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, Weiss L, Fridman WH, Fremeaux-Bacchi V. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:555–63.
Fremeaux-Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017–25.
Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, Lopez-Trascasa M, Sanchez-Corral P, Morgan BP, Rodriguez de Cordoba S. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2007;104:240–5.
Frémeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948–52.
Delvaeye M, Noris M, de Vriese A, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–57.
Lemaire M, Fremeaux-Bacchi V, Schaefer F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet. 2013;45:531–6.
Brocklebank V, Kumar G, Howie AJ, et al. Long-term outcomes and response to treatment in diacylglycerol kinase epsilon nephropathy. Kidney Int. 2020. https://doi.org/10.1016/j.kint.2020.01.045.
Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–97.
Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. 2016. https://doi.org/10.1111/imr.12475.
Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. 2017;18:1288–98.
Killick J, Morisse G, Sieger D, Astier AL. Complement as a regulator of adaptive immunity. Semin Immunopathol. 2018. https://doi.org/10.1007/s00281-017-0644-y.
Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6:257.
Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I—molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262.
Bresin E, Rurali E, Caprioli J, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24:475–86.
Osborne A. Complement mutations database. https://www.complement-db.org/home.php.
Saunders RE, Abarrategui-Garrido C, Fremeaux-Bacchi V, et al. The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat. 2007;28:222–34.
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18:2392–400.
Besbas N, Gulhan B, Soylemezoglu O, Ozcakar ZB, Korkmaz E, Hayran M, Ozaltin F. Turkish pediatric atypical hemolytic uremic syndrome registry: initial analysis of 146 patients. BMC Nephrol. 2017;18:6.
Westland R, Bodria M, Carrea A, et al. Phenotypic expansion of DGKE-associated diseases. J Am Soc Nephrol. 2014;25:1408–14.
Chinchilla DS, Pinto S, Hoppe B, Adragna M, Lopez L, Roldan MLJ, Peña A, Trascasa ML, Sánchez-Corral P, de Córdoba SR. Complement mutations in diacylglycerol kinase-ɛ–associated atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2014. https://doi.org/10.2215/CJN.01640214.
Azukaitis K, Simkova E, Abdul Majid M, et al. The phenotypic spectrum of nephropathies associated with mutations in diacylglycerol Kinase ϵ. J Am Soc Nephrol. 2017. https://doi.org/10.1681/ASN.2017010031.
Gholizad-Kolveiri S, Hooman N, Alizadeh R, Hoseini R, Otukesh H, Talebi S, Akouchekian M. Whole exome sequencing revealed a novel homozygous variant in the DGKE catalytic domain: a case report of familial hemolytic uremic syndrome. BMC Med Genet. 2020; https://doi.org/10.1186/s12881-020-01097-9.
Dragon-Durey MA, Sethi SK, Bagga A, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–7.
Hofer J, Janecke AR, Zimmerhackl LB, et al. Complement factor H-related protein 1 deficiency and factor H antibodies in pediatric patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2013;8:407–15.
Sinha A, Gulati A, Saini S, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85:1151–60.
Hofer J, Giner T, Józsi M. Complement factor h-antibody-associated hemolytic uremic syndrome: pathogenesis, clinical presentation, and treatment. Semin Thromb Hemost. 2014. https://doi.org/10.1055/s-0034-1375297.
Puraswani M, Khandelwal P, Saini H, et al. Clinical and immunological profile of anti-factor H antibody associated atypical hemolytic uremic syndrome: a nationwide database. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.01282.
Štolbová Š, Bezdíčka M, Prohászka Z, Csuka D, Hrachovinová I, Burkert J, Šimánková N, Průhová Š, Zieg J. Molecular basis and outcomes of atypical haemolytic uraemic syndrome in Czech children. Eur J Pediatr. 2020. https://doi.org/10.1007/s00431-020-03666-9.
Schaefer F, Licht C, Ardissino G, Ariceta G, Kupelian V, Gasteyger C, Greenbaum LA, Ogawa M, van de Walle J, Fremeaux-Bacchi V. Manifestations of atypical haemolytic uraemic syndrome (AHUS) in children and adults: data from the global AHUS registry. Pediatr Nephrol. 2015;30:1544.
Condom P, Mansuy JM, Decramer S, Izopet J, Mengelle C. Atypical hemolytic uremic syndrome triggered by varicella infection. IDCases. 2017. https://doi.org/10.1016/j.idcr.2017.04.004.
Kobbe R, Schild R, Christner M, Oh J, Loos S, Kemper MJ. Case report—atypical hemolytic uremic syndrome triggered by influenza B. BMC Nephrol. 2017. https://doi.org/10.1186/s12882-017-0512-y.
van Hoeve K, Vandermeulen C, van Ranst M, Levtchenko E, van den Heuvel L, Mekahli D. Occurrence of atypical HUS associated with influenza B. Eur J Pediatr. 2017. https://doi.org/10.1007/s00431-017-2856-5.
Madden I, Roumenina LT, Langlois-Meurinne H, Guichoux J, Llanas B, Frémeaux-Bacchi V, Harambat J, Godron-Dubrasquet A. Hemolytic uremic syndrome associated with Bordetella pertussis infection in a 2-month-old infant carrying a pathogenic variant in complement factor H. Pediatr Nephrol. 2019. https://doi.org/10.1007/s00467-018-4174-1.
el Sissy C, Saldman A, Zanetta G, et al. COVID-19 as a potential trigger of complement-mediated atypical HUS. Blood. 2021. https://doi.org/10.1182/blood.2021012752.
Edey MM, Mead PA, Saunders RE, Strain L, Perkins SJ, Goodship THJ, Kanagasundaram NS. Association of a factor H mutation with hemolytic uremic syndrome following a diarrheal illness. Am J Kidney Dis. 2008. https://doi.org/10.1053/j.ajkd.2007.08.030.
Alberti M, Valoti E, Piras R, Bresin E, Galbusera M, Tripodo C, Thaiss F, Remuzzi G, Noris M. Two patients with history of STEC-HUS, posttransplant recurrence and complement gene mutations. Am J Transplant. 2013. https://doi.org/10.1111/ajt.12297.
Dragon-Durey MA, Blanc C, Garnier A, Hofer J, Sethi SK, Zimmerhackl LB. Anti-factor H autoantibody-associated hemolytic uremic syndrome: review of literature of the autoimmune form of HUS. Semin Thromb Hemost. 2010;36:633–40.
Durey MAD, Sinha A, Togarsimalemath SK, Bagga A. Anti-complement-factor H-associated glomerulopathies. Nat Rev Nephrol. 2016;12:563–78.
Togarsimalemath SK, Si-Mohammed A, Puraswani M, Gupta A, Vabret A, Liguori S, Mariani-Kurkdjian P, Bagga A, Dragon-Durey MA. Gastrointestinal pathogens in anti-FH antibody positive and negative Hemolytic Uremic Syndrome. Pediatr Res. 2018;84:118–24.
Fakhouri F, Roumenina L, Provot F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21:859–67.
Sethi S, Fervenza FC. Pathology of renal diseases associated with dysfunction of the alternative pathway of complement: C3 glomerulopathy and atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost. 2014. https://doi.org/10.1055/s-0034-1375701.
Benz K, Amann K. Pathological aspects of membranoproliferative glomerulonephritis (MPGN) and haemolytic uraemic syndrome (HUS)/thrombocyte thrombopenic purpura (TTP). Thromb Haemost. 2009. https://doi.org/10.1160/TH07-12-0761.
Goodship THJ, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017. https://doi.org/10.1016/j.kint.2016.10.005.
Formeck C, Swiatecka-Urban A. Extra-renal manifestations of atypical hemolytic uremic syndrome. Pediatr Nephrol. 2018. https://doi.org/10.1007/s00467-018-4039-7.
Hofer J, Rosales A, Fischer C, Giner T. Extra-renal manifestations of complement-mediated thrombotic microangiopathies. Front Pediatr. 2014. https://doi.org/10.3389/fped.2014.00097.
Gulleroglu K, Fidan K, Hancer VS, Bayrakci U, Baskin E, Soylemezoglu O. Neurologic involvement in atypical hemolytic uremic syndrome and successful treatment with eculizumab. Pediatr Nephrol. 2013;28:827–30.
Hu H, Nagra A, Haq MR, Gilbert RD. Eculizumab in atypical haemolytic uraemic syndrome with severe cardiac and neurological involvement. Pediatr Nephrol. 2014. https://doi.org/10.1007/s00467-013-2709-z.
Vilalta R, Lara E, Madrid A, Chocron S, Muñoz M, Casquero A, Nieto J. Long-term eculizumab improves clinical outcomes in atypical hemolytic uremic syndrome. Pediatr Nephrol. 2012. https://doi.org/10.1007/s00467-012-2276-8.
Diamante Chiodini B, Davin JC, Corazza F, Khaldi K, Dahan K, Ismaili K, Adams B. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics. 2014. https://doi.org/10.1542/peds.2013-1594.
Michaux K, Bacchetta J, Javouhey E, Cochat P, Frémaux-Bacchi V, Sellier-Leclerc A-L. Eculizumab in neonatal hemolytic uremic syndrome with homozygous factor H deficiency. Pediatr Nephrol. 2014;29:2415–9.
Malina M, Gulati A, Bagga A, Majid MA, Simkova E, Schaefer F. Peripheral gangrene in children with atypical hemolytic uremic syndrome. Pediatrics. 2013;131:e331–5.
Ariceta G, Arrizabalaga B, Aguirre M, Morteruel E, Lopez-Trascasa M. Eculizumab in the treatment of atypical hemolytic uremic syndrome in infants. Am J Kidney Dis. 2012. https://doi.org/10.1053/j.ajkd.2011.11.027.
Ardissino G, Tel F, Testa S, Marzano AV, Lazzari R, Salardi S, Edefonti A. Skin involvement in atypical hemolytic uremic syndrome. Am J Kidney Dis. 2014. https://doi.org/10.1053/j.ajkd.2013.09.020.
Santos C, Lopes D, Gomes A, Ventura A, Tente D, Seabra J. Cutaneous involvement in haemolytic uraemic syndrome. Clin Kidney J. 2013. https://doi.org/10.1093/ckj/sft114.
Larakeb A, Leroy S, Frémeaux-Bacchi V, Montchilova M, Pelosse B, Dunand O, Deschênes G, Bensman A, Ulinski T. Ocular involvement in hemolytic uremic syndrome due to factor H deficiency—are there therapeutic consequences? Pediatr Nephrol. 2007. https://doi.org/10.1007/s00467-007-0540-0.
Davin JC, Majoie C, Groothoff J, Gracchi V, Bouts A, Goodship TH, Loirat C. Prevention of large-vessel stenoses in atypical hemolytic uremic syndrome associated with complement dysregulation. Pediatr Nephrol. 2011;26:155–7.
Loirat C, Macher MA, Elmaleh-Berges M, et al. Non-atheromatous arterial stenoses in atypical haemolytic uraemic syndrome associated with complement dysregulation. Nephrol Dial Transplant. 2010;25:3421–5.
Békássy ZD, Kristoffersson AC, Cronqvist M, Roumenina LT, Rybkine T, Vergoz L, Hue C, Fremeaux-Bacchi V, Karpman D. Eculizumab in an anephric patient with atypical haemolytic uraemic syndrome and advanced vascular lesions. Nephrol Dial Transplant. 2013. https://doi.org/10.1093/ndt/gft340.
Ažukaitis K, Loirat C, Malina M, Adomaitienė I, Jankauskienė A. Macrovascular involvement in a child with atypical hemolytic uremic syndrome. Pediatr Nephrol. 2014;29:1273–7.
Noris M, Remuzzi G. Cardiovascular complications in atypical haemolytic uraemic syndrome. NRN. 2014:10–5.
Ohanian M, Cable C, Halka K. Eculizumab safely reverses neurologic impairment and eliminates need for dialysis in severe atypical hemolytic uremic syndrome. Clin Pharmacol. 2011;3:5–12.
Salem G, Flynn JM, Cataland SR. Profound neurological injury in a patient with atypical hemolytic uremic syndrome. Ann Hematol. 2013. https://doi.org/10.1007/s00277-012-1615-y.
McFarlane PA, Bitzan M, Broome C, et al. Making the correct diagnosis in thrombotic microangiopathy: a narrative review. Can J Kidney Health Dis. 2021. https://doi.org/10.1177/20543581211008707.
Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016. https://doi.org/10.1007/s00467-015-3076-8.
le Quintrec M, Zuber J, Moulin B, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. 2013. https://doi.org/10.1111/ajt.12077.
Mele C, Remuzzi G, Noris M. Hemolytic uremic syndrome. Semin Immunopathol. 2014;36:399–420.
Espié E, Grimont F, Mariani-Kurkdjian P, et al. Surveillance of hemolytic uremic syndrome in children less than 15 years of age, a system to monitor o157 and non-o157 shiga toxin-producing escherichia coli infections in France, 1996-2006. Pediatr Infect Dis J. 2008. https://doi.org/10.1097/INF.0b013e31816a062f.
Loirat C, Girma JP, Desconclois C, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura related to severe ADAMTS13 deficiency in children. Pediatr Nephrol. 2009. https://doi.org/10.1007/s00467-008-0863-5.
Loirat C, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura in children. Curr Opin Pediatr. 2013. https://doi.org/10.1097/MOP.0b013e32835e7888.
Hassenpflug WA, Budde U, Schneppenheim S, Schneppenheim R. Inherited thrombotic thrombocytopenic purpura in children. Semin Thromb Hemost. 2014. https://doi.org/10.1055/s-0034-1376152.
Sharma AP, Greenberg CR, Prasad AN, Prasad C. Hemolytic uremic syndrome (HUS) secondary to cobalamin C (cblC) disorder. Pediatr Nephrol. 2007;22:2097–103.
Cornec-Le Gall E, Delmas Y, de Parscau L, Doucet L, Ogier H, Benoist JF, Fremeaux-Bacchi V, le Meur Y. Adult-onset eculizumab-resistant hemolytic uremic syndrome associated with cobalamin C deficiency. Am J Kidney Dis. 2014. https://doi.org/10.1053/j.ajkd.2013.08.031.
Lemoine M, François A, Grangé S, et al. Cobalamin C deficiency induces a typical histopathological pattern of renal arteriolar and glomerular thrombotic microangiopathy. Kidney Int Rep. 2018. https://doi.org/10.1016/j.ekir.2018.05.015.
Menni F, Testa S, Guez S, Chiarelli G, Alberti L, Esposito S. Neonatal atypical hemolytic uremic syndrome due to methylmalonic aciduria and homocystinuria. Pediatr Nephrol. 2012. https://doi.org/10.1007/s00467-012-2152-6.
Lawrence J, Gwee A, Quinlan C. Pneumococcal haemolytic uraemic syndrome in the postvaccine era. Arch Dis Child. 2018;103:957–61.
Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005. https://doi.org/10.1111/j.1365-2141.2005.05420.x.
Mackie I, Mancini I, Muia J, Kremer Hovinga J, Nair S, Machin S, Baker R. International Council for Standardization in Haematology (ICSH) recommendations for laboratory measurement of ADAMTS13. Int J Lab Hematol. 2020. https://doi.org/10.1111/ijlh.13295.
Meyer SC, Sulzer I, Lämmle B, Kremer Hovinga JA. Hyperbilirubinemia interferes with ADAMTS-13 activity measurement by FRETS-VWF73 assay: diagnostic relevance in patients suffering from acute thrombotic microangiopathies [4]. J Thromb Haemost. 2007. https://doi.org/10.1111/j.1538-7836.2007.02438.x.
Mackie I, Langley K, Chitolie A, Liesner R, Scully M, Machin S, Peyvandi F. Discrepancies between ADAMTS13 activity assays in patients with thrombotic microangiopathies. Thromb Haemost. 2013. https://doi.org/10.1160/TH12-08-0565.
Thouzeau S, Capdenat S, Stepanian A, Coppo P, Veyradier A. Evaluation of a commercial assay for ADAMTS13 activity measurement. Thromb Haemost. 2013. https://doi.org/10.1160/TH13-05-0393.
Ardissino G, Perrone M, Tel F, Testa S, Morrone A, Possenti I, Tagliaferri F, Dilena R, Menni F. Late onset cobalamin disorder and hemolytic uremic syndrome: a rare cause of nephrotic syndrome. Case Rep Pediatr. 2017. https://doi.org/10.1155/2017/2794060.
Topaloglu R, Inözü M, Gülhan B, Gürbüz B, Talim B, Coşkun T. Do not miss rare and treatable cause of early-onset hemolytic uremic syndrome: cobalamin C deficiency. Nephron. 2019. https://doi.org/10.1159/000497822.
Sloan JL, Carrillo N, Adams D. Disorders of intracellular cobalamin metabolism. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®; 2008.
Watson R, Lindner S, Bordereau P, Hunze EM, Tak F, Ngo S, Zipfel PF, Skerka C, Dragon-Durey MA, Marchbank KJ. Standardisation of the factor H autoantibody assay. Immunobiology. 2014;219:9–16.
Cataland SR, Holers VM, Geyer S, Yang S, Wu HM. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood. 2014. https://doi.org/10.1182/blood-2013-12-547067.
Noris M, Galbusera M, Gastoldi S, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. 2014;124:1715–26.
Volokhina EB, Westra D, van der Velden TJAM, van de Kar NCAJ, Mollnes TE, van den Heuvel LP. Complement activation patterns in atypical haemolytic uraemic syndrome during acute phase and in remission. Clin Exp Immunol. 2015. https://doi.org/10.1111/cei.12426.
Barnum SR, Bubeck D, Schein TN. Soluble membrane attack complex: biochemistry and immunobiology. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.585108.
Moore I, Strain L, Pappworth I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115:379–87.
Venables JP, Strain L, Routledge D, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3:e431.
Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ. A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant. 2011;26:739–41.
Francis NJ, McNicholas B, Awan A, et al. A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood. 2012:119:591–601.
Heinen S, Wiehl U, Lauer N, et al. Disease associated protein complement factor H related protein 1 (CFHR1) is a regulator of the human alternative complement pathway. Mol Immunol. 2008. https://doi.org/10.1016/j.molimm.2008.08.027.
Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119:4182–91.
Marinozzi MC, Vergoz L, Rybkine T, et al. Complement factor b mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol. 2014. https://doi.org/10.1681/ASN.2013070796.
Roumenina LT, Jablonski M, Hue C, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood. 2009;114:2837–45.
Fremeaux-Bacchi V, Kemp EJ, Goodship JA, Dragon-Durey MA, Strain L, Loirat C, Deng HW, Goodship TH. The development of atypical haemolytic-uraemic syndrome is influenced by susceptibility factors in factor H and membrane cofactor protein: evidence from two independent cohorts. J Med Genet. 2005;42:852–6.
Abarrategui-Garrido C, Martínez-Barricarte R, López-Trascasa M, Rodríguez De Córdoba S, Sánchez-Corral P. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. 2009. https://doi.org/10.1182/blood-2009-05-223834.
Meri S. Complement activation in diseases presenting with thrombotic microangiopathy. Eur J Intern Med. 2013;24:496–502.
Riedl M, Fakhouri F, le Quintrec M, Noone DG, Jungraithmayr TC, Fremeaux-Bacchi V, Licht C. Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost. 2014;40:444–64.
Turner NA, Moake J. Assembly and activation of alternative complement components on endothelial cell-anchored ultra-large von willebrand factor links complement and hemostasis-thrombosis. PLoS One. 2013; https://doi.org/10.1371/journal.pone.0059372.
Rayes J, Roumenina LT, Dimitrov JD, et al. The interaction between factor H and VWF increases factor H cofactor activity and regulates VWF prothrombotic status. Blood. 2014. https://doi.org/10.1182/blood-2013-04-495853.
Feng S, Liang X, Cruz MA, Vu H, Zhou Z, Pemmaraju N, Dong JF, Kroll MH, Afshar-Kharghan V. The interaction between factor H and Von Willebrand factor. PLoS One. 2013. https://doi.org/10.1371/journal.pone.0073715.
Ståhl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008. https://doi.org/10.1182/blood-2007-08-106153.
Pickering MC, Goicoechea De Jorge E, Martinez-Barricarte R, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007. https://doi.org/10.1084/jem.20070301.
Martin Merinero H, Zhang Y, Arjona E, del Angel G, Goodfellow R, Gomez-Rubio E, Ji R-R, Michelena M, Smith R, Rodríguez de Córdoba S. Functional characterization of 105 Factor H variants associated with atypical HUS: lessons for variant classification. Blood. 2021. https://doi.org/10.1182/blood.2021012037.
Fakhouri F, Frémeaux-Bacchi V. Thrombotic microangiopathy in aHUS and beyond: clinical clues from complement genetics. Nat Rev Nephrol. 2021. https://doi.org/10.1038/s41581-021-00424-4.
Quaggin SE. DGKE and atypical HUS. Nat Genet. 2013;45:475–6.
Bu F, Zhang Y, Wang K, et al. Genetic analysis of 400 patients refines understanding and implicates a new gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2018; ASN.2018070759.
Thergaonkar RW, Narang A, Gurjar BS, et al. Targeted exome sequencing in anti-factor H antibody negative HUS reveals multiple variations. Clin Exp Nephrol. 2018. https://doi.org/10.1007/s10157-017-1478-6.
Challis RC, Ring T, Xu Y, et al. Thrombotic microangiopathy in inverted formin 2-mediated renal disease. J Am Soc Nephrol. 2017. https://doi.org/10.1681/ASN.2015101189.
Ståhl A-L, Kristoffersson AC, Olin AI, Olsson ML, Roodhooft AM, Proesmans W, Karpman D. A novel mutation in the complement regulator clusterin in recurrent hemolytic uremic syndrome. Mol Immunol. 2009. https://doi.org/10.1016/j.molimm.2009.04.012.
Brocklebank V, Johnson S, Sheerin TP, et al. Factor H autoantibody is associated with atypical hemolytic uremic syndrome in children in the United Kingdom and Ireland. Kidney Int. 2017;92:1261–71.
Blanc C, Roumenina LT, Ashraf Y, et al. Overall neutralization of complement factor H by autoantibodies in the acute phase of the autoimmune form of atypical hemolytic uremic syndrome. J Immunol. 2012;189:3528–37.
Holmes LV, Strain L, Staniforth SJ, Moore I, Marchbank K, Kavanagh D, Goodship JA, Cordell HJ, Goodship THJ. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS One. 2013. https://doi.org/10.1371/journal.pone.0060352.
Bhattacharjee A, Reuter S, Trojnár E, et al. The major autoantibody epitope on factor H in atypical hemolytic uremic syndrome is structurally different from its homologous site in factor H-related protein 1, supporting a novel model for induction of autoimmunity in this disease. J Biol Chem. 2015;290:9500–10.
Kavanagh D, Pappworth IY, Anderson H, et al. Factor i autoantibodies in patients with atypical hemolytic uremic syndrome: disease-associated or an epiphenomenon? Clin J Am Soc Nephrol. 2012;7:417–26.
Bagga A, Khandelwal P, Mishra K, et al. Hemolytic uremic syndrome in a developing country: consensus guidelines. Pediatr Nephrol. 2019;34:1465–82.
Zuber J, Quintrec ML, Krid S, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012. https://doi.org/10.1111/j.1600-6143.2012.04252.x.
Sinibaldi S, Guzzo I, Piras R, Bresin E, Emma F, dello Strologo L. Post-transplant recurrence of atypical hemolytic uremic syndrome in a patient with thrombomodulin mutation. Pediatr Transplant. 2013. https://doi.org/10.1111/petr.12151.
Saland JM, Ruggenenti P, Remuzzi G. Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2009;20:940–9.
Zuber J, le Quintrec M, Morris H, Frémeaux-Bacchi V, Loirat C, Legendre C. Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation. Transplant Rev. 2013. https://doi.org/10.1016/j.trre.2013.07.003.
Kellum JA, Lameire N, Aspelin P, et al. Kidney disease: improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012. https://doi.org/10.1038/kisup.2012.1.
Hillmen P, Hall C, Marsh JC, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350:552–9.
Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:544–6.
Nurnberger J, Philipp T, Witzke O, et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:542–4.
Christmann M, Hansen M, Bergmann C, Schwabe D, Brand J, Schneider W. Eculizumab as first-line therapy for atypical hemolytic uremic syndrome. Pediatrics. 2014. https://doi.org/10.1542/peds.2013-1787.
Vaisbich MH, Henriques LDS, Watanabe A, et al. [Eculizumab for the treatment of atypical hemolytic uremic syndrome: case report and revision of the literature]. J Bras Nefrol. 2013. https://doi.org/10.5935/0101-2800.20130037.
Besbas N, Gulhan B, Karpman D, Topaloglu R, Duzova A, Korkmaz E, Ozaltin F. Neonatal onset atypical hemolytic uremic syndrome successfully treated with eculizumab. Pediatr Nephrol. 2013. https://doi.org/10.1007/s00467-012-2296-4.
Giordano M, Castellano G, Messina G, Divella C, Bellantuono R, Puteo F, Colella V, Depalo T, Gesualdo L. Preservation of renal function in atypical hemolytic uremic syndrome by eculizumab: a case report. Pediatrics. 2012. https://doi.org/10.1542/peds.2011-1685.
Cayci FS, Cakar N, Hancer VS, Uncu N, Acar B, Gur G. Eculizumab therapy in a child with hemolytic uremic syndrome and CFI mutation. Pediatr Nephrol. 2012. https://doi.org/10.1007/s00467-012-2283-9.
Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015. https://doi.org/10.1038/ki.2014.423.
Greenbaum LA, Fila M, Ardissino G, et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016;89:701–11.
Sheridan D, Yu ZX, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti-C5 antibody with extended duration of action. PLoS One. 2018. https://doi.org/10.1371/journal.pone.0195909.
Rondeau E, Scully M, Ariceta G, et al. The long-acting C5 inhibitor, ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97:1287–96.
Barbour T, Scully M, Ariceta G, et al. Long-term efficacy and safety of the long-acting complement C5 inhibitor ravulizumab for the treatment of atypical hemolytic uremic syndrome in adults. Kidney Int Rep. 2021. https://doi.org/10.1016/j.ekir.2021.03.884.
Tanaka K, Adams B, Aris AM, Fujita N, Ogawa M, Ortiz S, Vallee M, Greenbaum LA. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr Nephrol. 2021;36:889–98.
https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-002370-39/ES.
McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High risk for invasive meningococcal disease among patients receiving eculizumab (soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66:734–7.
Hillmen P, Muus P, Röth A, et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013. https://doi.org/10.1111/bjh.12347.
Struijk GH, Bouts AHM, Rijkers GT, Kuin EAC, ten Berge IJM, Bemelman FJ. Meningococcal sepsis complicating eculizumab treatment despite prior vaccination. Am J Transplant. 2013. https://doi.org/10.1111/ajt.12032.
Rondeau E, Cataland SR, Al-Dakkak I, Miller B, Webb NJA, Landau D. Eculizumab safety: five-year experience from the global atypical hemolytic uremic syndrome registry. Kidney Int Rep. 2019;4:1568.
Noone D, Waters A, Pluthero FG, Geary DF, Kirschfink M, Zipfel PF, Licht C. Successful treatment of DEAP-HUS with eculizumab. Pediatr Nephrol. 2014. https://doi.org/10.1007/s00467-013-2654-x.
Brocklebank V, Johnson S, Sheerin TP, et al. Factor H autoantibody associated atypical haemolytic uraemic syndrome in children in the United Kingdom and Ireland. Kidney Int. 2017;92(5):1261–71.
2021. https://www.ema.europa.eu/en/medicines/human/EPAR/soliris.
Nishimura J, Yamamoto M, Hayashi S, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014. https://doi.org/10.1056/nejmoa1311084.
Cugno M, Gualtierotti R, Possenti I, et al. Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Thromb Haemost. 2014;12:1440–8.
Gilbert RD, Fowler DJ, Angus E, Hardy SA, Stanley L, Goodship TH. Eculizumab therapy for atypical haemolytic uraemic syndrome due to a gain-of-function mutation of complement factor B. Pediatr Nephrol. 2013;28:1315–8.
Willrich MAV, Ladwig PM, Martinez MA, Sridharan MR, Go RS, Murray DL. Monitoring ravulizumab effect on complement assays. J Immunol Methods. 2021;490:112944.
Ardissino G, Testa S, Possenti I, Tel F, Paglialonga F, Salardi S, Tedeschi S, Belingheri M, Cugno M. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis. 2014;64:633–7.
Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–79.
Fakhouri F, Delmas Y, Provot F, et al. Insights from the use in clinical practice of eculizumab in adult patients with atypical hemolytic uremic syndrome affecting the native kidneys: an analysis of 19 cases. Am J Kidney Dis. 2014. https://doi.org/10.1053/j.ajkd.2013.07.011.
Fila M, Caillez M, Hogan J, Louillet F, Loirat C, Fremeaux Bacchi V, Fakhouri F. Outcome of 11 pediatric patients with atypical hemolytic and uremic syndrome after Eculizumab discontinuation. Pediatr Nephrol. 2016;31:233.
Wijnsma KL, Duineveld C, Volokhina EB, van den Heuvel LP, van de Kar NCAJ, Wetzels JFM. Safety and effectiveness of restrictive eculizumab treatment in atypical haemolytic uremic syndrome. Nephrol Dial Transplant. 2018;33:635–45.
Ariceta G, Fakhouri F, Sartz L, Miller B, Nikolaou V, Cohen D, Siedlecki AM, Ardissino G. Eculizumab discontinuation in atypical haemolytic uraemic syndrome: TMA recurrence risk and renal outcomes. Clin Kidney J. 2021. https://doi.org/10.1093/ckj/sfab005.
Neto ME, de Moraes SL, Vasconcelos HVG, et al. Eculizumab interruption in atypical hemolytic uremic syndrome due to shortage: analysis of a Brazilian cohort. J Nephrol. 2021. https://doi.org/10.1007/s40620-020-00920-z.
Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021. https://doi.org/10.1182/blood.2020009280.
ISRCTN17503205. Stopping Eculizumab Treatment Safely in atypical Haemolytic Uraemic Syndrome (SETS aHUS). 2018. http://www.who.int/trialsearch/Trial2.aspx?TrialID=ISRCTN17503205.
Saland J. Liver-kidney transplantation to cure atypical HUS: still an option post-eculizumab? Pediatr Nephrol. 2014. https://doi.org/10.1007/s00467-013-2722-2.
Park SH, Kim GS. Anesthetic management of living donor liver transplantation for complement factor h deficiency hemolytic uremic syndrome: a case report. Korean J Anesthesiol. 2014. https://doi.org/10.4097/kjae.2014.66.6.481.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Malina, M., Fremeaux-Bacchi, V., Johnson, S. (2023). Atypical Hemolytic Uremic Syndrome. In: Schaefer, F., Greenbaum, L.A. (eds) Pediatric Kidney Disease. Springer, Cham. https://doi.org/10.1007/978-3-031-11665-0_22
Download citation
DOI: https://doi.org/10.1007/978-3-031-11665-0_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-11664-3
Online ISBN: 978-3-031-11665-0
eBook Packages: MedicineMedicine (R0)