
Abstract
Hepatocyte nuclear factor-1β (HNF-1β) is an essential transcription factor that regulates the development and function of epithelia in the kidney, liver, pancreas, and genitourinary tract. Humans who carry HNF1B mutations develop heterogeneous renal abnormalities, including multicystic dysplastic kidneys, glomerulocystic kidney disease, renal agenesis, renal hypoplasia, and renal interstitial fibrosis. In the embryonic kidney, HNF-1β is required for ureteric bud branching, initiation of nephrogenesis, and nephron segmentation. Ablation of mouse Hnf1b in nephron progenitors causes defective tubulogenesis, whereas later inactivation in elongating tubules leads to cyst formation due to downregulation of cystic disease genes, including Umod, Pkhd1, and Pkd2. In the adult kidney, HNF-1β controls the expression of genes required for intrarenal metabolism and solute transport by tubular epithelial cells. Tubular abnormalities observed in HNF-1β nephropathy include hyperuricemia with or without gout, hypokalemia, hypomagnesemia, and polyuria. Recent studies have identified novel post-transcriptional and post-translational regulatory mechanisms that control HNF-1β expression and activity, including the miRNA cluster miR17 ∼ 92 and the interacting proteins PCBD1 and zyxin. Further understanding of the molecular mechanisms upstream and downstream of HNF-1β may lead to the development of new therapeutic approaches in cystic kidney disease and other HNF1B-related renal diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocyte nuclear factor-1β (HNF-1β, vHNF1) is a developmentally regulated transcription factor that is required for tissue-specific gene expression in the epithelial cells of many organs, including kidney, pancreas, liver, and genitourinary tract [1, 2]. In the developing kidney, HNF-1β is expressed in developing nephrons and the branching ureteric bud that gives rise to the renal collecting system [3,4,5]. In the mature kidney, expression of HNF-1β persists in the epithelial cells of renal tubules but not in the glomeruli, blood vessels, or interstitial cells. The importance of HNF-1β in renal tubules is underscored by the finding that combined expression of HNF-1β with the transcription factors Emx-2, HNF-4α, and Pax-8 is sufficient to reprogram fibroblasts into functional renal tubular epithelial cells [6].
HNF-1β activates or represses transcription of target genes through binding of its POU (Pit-1, OCT1/2, UNC-86; POUS)-specific domain and atypical POU homeodomain (POUH) to the DNA consensus sequence 5′-GTTAATNATTAAC-3′. The dimerization domain located at the N-terminus of the protein mediates the formation of HNF-1β homodimers or heterodimers with the related protein HNF-1α. The protein complex also includes two molecules of the dimerization cofactor of HNF1 known as pterin-4 alpha-carbinolamine dehydratase (PCBD1) [7]. The C-terminal region of HNF-1β contains a transactivation domain(s) that is responsible for the recruitment of coactivators and regulation of transcription [8]. To date, the DNA-binding domain is the only HNF-1β motif whose crystal structure has been determined (PDB file: 2H8R) [9]. HNF-1β and HNF-1α recognize the same DNA consensus sequence but can regulate gene transcription with different outcomes depending on the cellular context [10, 11].
In humans, heterozygous mutations of the HNF1B gene cause a spectrum of inherited and sporadic malformations of the kidney and genitourinary tract. Molecular genetic defects include whole-gene deletions in about 50% of patients; point mutations are detected in the remaining cases [12]. De novo mutations of HNF1B are encountered in up to 30–50% of cases [12, 13]. The first HNF1B-related disorder that was described was Renal Cysts and Diabetes (RCAD; MIM:137920), a syndrome characterized by autosomal dominant inheritance, renal cystic abnormalities, and maturity-onset diabetes of the young type 5 (MODY5) [12, 14]. Additional extrarenal manifestations include exocrine pancreatic failure, abnormal liver function tests, and genital tract malformations. HNF1B mutations also cause congenital anomalies of the kidney and urinary tract (CAKUT) [15, 16]. HNF1B mutations are found in 20–30% of fetuses with renal abnormalities and represent the most common prenatal cause of hyperechogenic kidneys with or without cysts [17]. Studies on large cohorts of fetuses and children showed that genetic variants in HNF1B are the most common cause of isolated renal hypodysplasia in humans [13, 18]. Other renal manifestations include multicystic dysplastic kidneys, glomerulocystic kidney disease, oligomeganephronia, renal agenesis, renal hypoplasia, familial juvenile hyperuricemic nephropathy, and renal interstitial fibrosis associated with a highly variable rate of decline of estimated glomerular filtration rate (eGFR) [12, 19, 20]. Humans with HNF1B mutations may also suffer from electrolyte disturbances, such as hypomagnesemia and hypokalemia [21, 22]. In addition, heterozygous deletions of HNF1B have been associated with prune belly syndrome (PBS, MIM: 100100), characterized by deficiency or absence of abdominal wall musculature, dilatation of the urinary tract and bilateral undescended testes [23, 24]. The renal phenotypes observed in patients with HNF1B mutations are highly heterogeneous and may manifest during the antenatal period, in childhood, or in adulthood [21]. This review summarizes recent insights into the roles of HNF-1β in the developing and mature kidney and reflects on future perspectives.
HNF-1β in kidney development
Mouse kidney development begins approximately at embryonic day E10.5 with the outgrowth of the ureteric bud (UB) from the Wolffian duct (WD) into the adjacent metanephric mesenchyme (MM, Fig. 1) [25]. Subsequently, the UB undergoes branching morphogenesis to generate the entire collecting system and ureter, whereas signals from the UB tips will induce an epithelial transition of surrounding MM cells to form renal vesicles (RV) [25]. Renal vesicles differentiate into comma- and S-shaped bodies, and subsequently into Bowman’s capsule of the glomerulus and nephron tubules that connect to the collecting system [25]. Functional nephrons are present by E16.5 in mice, but elongation and remodeling of tubular segments continue for 2 weeks after birth [25, 26].

HNF1-β is required for UB branching, initiation of nephrogenesis, and nephron segmentation. Schematic showing the major stages of kidney development. HNF1-β-expressing cells are depicted in light pink. At E10.5, the UB emerges from the WD upon signaling from the MM. Starting from E12.5, HNF1-β is essential for UB branching. In developing nephrons, HNF1-β is expressed in the distal region of RVs, in the distal CSB, and in the distal to proximal SSB, but it is absent in the most proximal region that will form the glomerulus. By E17.5, HNF1-β is present in all tubular epithelial cells of the mature nephrons but not in the glomeruli. MM, metanephric mesenchyme; UB, ureteric bud; WD, Wolffian duct; RV, renal vesicle; CSB, comma-shaped bodies; SSB, S-shaped bodies; D, distal; I, intermediate; P, proximal; DCT, distal convoluted tubule; PT, proximal tubule; TAL, thick ascending limb of Henle; CD, collecting duct
HNF-1β is an essential transcriptional regulator of renal epithelial organization and differentiation [3,4,5, 27]. HNF-1β is expressed in the WD, UB, RV, comma-shaped bodies, S-shaped bodies, and developing renal tubules but not in MM (Fig. 1). Within comma- and S-shaped bodies, HNF-1β is expressed in the proximal and distal segments that are the anlagen of the renal tubules, but not in the most-proximal regions that will give rise to glomeruli [28]. Using constitutive inactivation of Hnf1b in the epiblast by tetraploid aggregation, Lokmane et al. found that HNF-1β is involved in the timing of UB outgrowth and its early branching. It is also required for maintenance of the WD epithelium and for early nephrogenesis [3]. HNF-1β exerts these functions, at least in part, through the direct control of several key regulatory genes [27]. HNF-1β acts directly upstream of Wnt9b, an essential factor that acts as a paracrine signal to orchestrate the mesenchymal-epithelial transitions underlying the initiation of nephrogenesis [29, 30]. HNF-1β also maintains the expression of Pax2 and Lim1, which have essential functions in multiple steps of renal epithelial tubular morphogenesis and are required for WD formation [3]. Recently, Desgrange et al. conditionally inactivated Hnf1b in the WD and UB using a HoxB7-Cre line [27]. They found that HNF-1β is required for the formation of a specialized UB tip domain through the maintenance of cell-cell adhesion, Pax2 expression, and Ret signaling. HNF-1β is subsequently required in the UB trunks for the establishment of normal apico-basal polarity and differentiation of the collecting system [27].
Inactivation of Hnf1b in MM using a Wnt4-Cre line or a Six2-Cre line leads to defective S-shaped bodies lacking an intermediate limb that gives rise to the loop of Henle loop and proximal tubule [4, 5]. HNF-1β regulates the acquisition of a proximal-intermediate nephron segment fate by controlling the expression of Irx1, Osr2, Pou3f3, and Notch signaling components [4, 5]. The importance of HNF-1β in nephron segmentation has also been demonstrated in zebrafish embryos [31].
Deletion of HNF-1β in elongating renal tubules using the Ksp-Cre line results in renal cyst formation [32]. The mitotic spindles of dividing tubular cells from HNF-1β-mutant kidneys are significantly misoriented compared with controls, a finding that suggests dysregulation of planar cell polarity (PCP) [33]. In elongating renal tubules, PCP signaling coordinates tubular narrowing and lengthening via convergent extension (CE) and the spatial distribution of daughter cells via oriented cell division (OCD) [34]. These processes control tubule lumen diameter and ensure that cell proliferation results in tubular elongation rather than dilation [34]. In HNF-1β-mutant kidneys, abnormalities in OCD are observed in precystic tubules, suggesting that loss of PCP may be responsible, at least in part, for cyst formation [35].
Role of HNF-1β in polycystic kidney disease
In experimental mice, kidney-specific deletion of Hnf1b by Cre/loxP recombination or transgenic expression of dominant-negative mutant HNF-1β leads to kidney cysts and renal failure, similar to the phenotype of humans with HNF1B mutations [32, 36]. Molecular characterization of the cystic kidneys has revealed that HNF-1β regulates the transcription of Pkhd1, the gene mutated in autosomal recessive polycystic kidney disease (ARPKD; MIM:263200); Pkd2, the gene mutated in autosomal dominant polycystic kidney disease (ADPKD; MIM:613295); Umod, associated with medullary cystic kidney disease (MCKD; MIM:174000); and Glis2, associated with nephronophthisis, and the cpk modifier gene Kif12 [32, 37, 38]. These findings suggest that mutations of HNF-1β produce kidney cysts by downregulating the expression of multiple cystic disease genes.
Most of the proteins encoded by cystic disease genes are located in the primary cilium, a sensory organelle on the cell surface, or the basal body, which anchors the cilium in the cell body. Primary cilia play essential roles during development and tissue homeostasis by regulating cell proliferation, migration, apoptosis, PCP, and differentiation through the activation of multiple signaling pathways, including canonical and non-canonical Wnt signaling and hedgehog, cAMP, and mTOR signaling [39,40,41]. Ciliary dysfunction or cilia loss in the kidney has been associated with renal cyst formation and renal failure [42]; however, the precise mechanism whereby the loss of renal cilia produces kidney cysts remains poorly understood.
Although HNF-1β is not directly involved in primary cilium function, it regulates the expression of proteins that localize to the cilium or that belong to biological processes mediated by cilia [43]. Polycystin-2 (PC2), encoded by the Pkd2 gene, is a Ca2+-permeable cation channel that interacts with polycystin-1 (PC1) in the primary cilium. Deletion of PC2 or mutations in HNF-1β are associated with decreased Ca2+ entry, activation of the Ca2+-inhibitable adenylyl cyclases AC5 and AC6 and elevated cAMP levels [40]. cAMP in turn promotes cyst growth by stimulating cell proliferation and fluid secretion. In addition, HNF-1β regulates the expression of phosphodiesterase 4C (PDE4C), which catabolizes cAMP in the primary cilium. In Hnf1b mutant kidney cells and mice, PDE4C is downregulated and cAMP levels are increased [40]. Inhibition of cAMP synthesis may be beneficial in the treatment of PKD as knockout of AC5 reduces renal cAMP levels and reduces cyst burden in Pkd2 mutant mice [44, 45].
The protein encoded by Pkhd1, named polyductin or fibrocystin, is an integral membrane protein that localizes to the basal body of primary cilia as well as the centrosomes and mitotic spindles of dividing cells [46]. Pkhd1 knockout mice develop kidney cysts, pancreatic cysts, and biliary ductal plate malformations, which confirms that downregulation of Pkhd1 is sufficient to produce renal cystic disease [47]. Expression of dominant-negative mutant HNF-1β or kidney-specific knockout of Hnf1b in mice reduces the levels of Pkhd1 mRNA transcripts in the kidney via direct downregulation of Pkhd1 promoter activity in renal collecting ducts (CDs) [32, 36, 48, 49].
Umod encodes uromodulin, a protein produced in the thick ascending limb of the loop of Henle (TAL) [50]. Uromodulin is found in the mitotic spindles of dividing cells. In addition, the decreased number of uromodulin-positive cilia in the kidney biopsies of patients with known UMOD mutations suggests a specific function of the protein in cilia [50].
Genome-wide chromatin immunoprecipitation combined with DNA microarray (ChIP-chip) and transcription profiling identified the PKD modifier gene Kif12 and the suppressor of cytokine signaling Socs3 as novel HNF-1β target genes [37, 51]. KIF12 participates in mitotic spindle formation, and variation in KIF12 copy number has been associated with CAKUT in humans [52]. HNF-1β promotes tubulogenesis of renal cells by repressing the transcription of Socs3, which enables activation of hepatocyte growth factor (HGF) signaling [51]. Conversely, upregulation of Socs3 in dominant-negative HNF-1β-expressing cells inhibits HGF signaling and impairs tubule formation. Consistent with these findings, HNF1B is downregulated and SOCS3 is highly upregulated in human polycystic kidneys [53]. Moreover, humans with heterozygous mutations of both PKD1 and HNF1B have a more severe phenotype, suggesting that HNF1B functions as a modifier gene in PKD [54].
Recently, it was shown that HNF-1β regulates the expression of a long noncoding RNA (lncRNA) containing the miR-200 cluster of microRNAs [55]. Kidney tubule-specific deletion of Hnf1b results in decreased expression of miR-200, and in turn, knockdown of miR-200 in cultured renal epithelial cells inhibits tubulogenesis and produces cyst-like structures [55]. The molecular mechanism whereby miR-200 regulates cyst pathogenesis may involve modulation of PKD1 expression via binding to the 3′UTR of the PKD1 transcript. miR-200 also regulates epithelial-to-mesenchymal transition (EMT), a process in which epithelial cells acquire mesenchymal properties, such as increased migratory capacity [56, 57]. EMT is integral to development and is reactivated during wound healing and tissue fibrosis. miR-200 and its transcriptional regulator HNF-1β inhibit EMT [58, 59]. Downregulation of HNF-1β is associated with EMT [58], consistent with the observed decrease in miR-200 levels, and inactivation of HNF-1β in CDs using a Pkhd1-Cre line causes interstitial fibrosis [55, 60]. Clinically, some patients with HNF1B mutations present primarily with renal fibrosis, a syndrome called autosomal dominant tubulointerstitial kidney disease (ADTKD) [22]. The role of EMT in the pathogenesis of ADTKD requires further investigation.
HNF-1β regulates ion transport in kidney
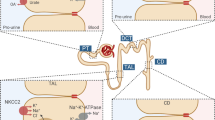
Almost 50% of patients with HNF-1β mutations develop hypomagnesemia due to renal Mg2+ wasting [21]. In the kidney, the major sites of Mg2+ reabsorption are the thick ascending limb of the loop of Henle (TAL) and distal convoluted tubule (DCT). Whereas defects in Mg2+ handling in the TAL lead to concomitant renal wasting of Ca2+, defects in the DCT cause hypermagnesuria associated with normo- or hypocalciuria [61]. In patients with HNF1B mutations, hypomagnesemia is usually accompanied by hypocalciuria, although a few affected adults have normocalciuria suggesting impaired reabsorption of Mg2+ in DCT [21, 62]. Hypomagnesemia is attributed to decreased expression of FXYD2, which encodes the γ subunit of Na+-K+-ATPase. Expression of FXYD2 is directly regulated by HNF-1β (Fig. 2) [62, 63], and mutations of FXYD2 cause autosomal dominant renal hypomagnesemia with hypocalciuria (MIM 601814) [64]. Recently, it was demonstrated that HNF-1β is a transcriptional activator of CaSR, encoding the calcium-sensing receptor, in the TAL [65]. Consequently, patients with HNF1B mutations may have reduced CaSR activity in the kidney, which could contribute to the hypocalciuria.

HNF-1β regulates the expression of solute transporters along the nephron. Defects in ion homeostasis reported in patients with HNF1B mutations include hypomagnesemia, hypokalemia, hyperuricemia, and rarely Fanconi syndrome. Whereas HNF-1α is restricted to the PT, HNF-1β is expressed in all epithelial cells of the nephron. HNF-1β target genes in the mature kidney encode OAT1/3/4, URAT1, NPT1, collectrin, uromodulin, NKCC2, the γ-subunit of Na+-K+-ATPase, and FXR. Refer to the text for detailed information. PT, proximal tubule; TAL, thick ascending limb of Henle; DCT, distal convoluted tubule; CD, collecting duct; HNF-1α/β, hepatocyte nuclear factor-1α/β; OAT1/3/4, organic anion transporter 1/3/4; NPT1, Na+-Pi cotransporter 1; URAT1, urate anion exchanger 1; OA−, organic anion; UA, uric acid/urate; ROMK, renal outer medullary K+ channel; NKCC2, Na+-K+-Cl− transporter; CaSR, calcium-sensing receptor; TRPM6, transient receptor potential melastatin 6; FXR, farnesoid X nuclear receptor
Faguer et al. found that 46% of adults with HNF1B mutations had hypokalemia [21]. Since hypokalemia is frequently associated with Mg2+ deficiency [66], it was hypothesized that a decrease in intracellular Mg2+ concentrations leads to the release of inhibition of renal outer medullary K+ channel (ROMK) causing urinary K+ wasting [67]. In addition, HNF-1β can directly regulate the transcription of genes involved in renal K+ handling, such as UMOD and SLC12A1 encoding respectively uromodulin and the Na+-K+-Cl− transporter (NKCC2) in the TAL (Fig. 2). Under physiological conditions, NKCC2 mediates Na+ and Cl− transport across the apical membrane, which is maintained by K+ recycling through ROMK. Uromodulin regulates both NKCC2 and ROMK [68, 69]. Mutations in UMOD result in urinary salt wasting, gout, and hypertension [70], whereas mutations in SLC12A1 and in KCNJ1, encoding ROMK, are associated with antenatal Bartter’s syndrome (MIM 601668, MIM 241200), a severe salt wasting syndrome that leads to a pronounced urinary concentrating defect and metabolic alkalosis [71, 72]. HNF-1β also directly regulates the transcription of KCNJ10 that encodes the K+ channel Kir5.1 [73]. Kir5.1 is a major contributor to the basolateral K+ conductance in the DCT, and mutations in KCNJ10 cause a salt-wasting syndrome that includes hypomagnesemia and hypokalemia (MIM 612780) [73]. Hence, dysregulation of multiple transporters may contribute to the K+ and Mg2+ wasting observed in patients with HNF1B mutations.
In the study by Faguer et al., two HNF1B mutant patients had PT dysfunction presenting as Fanconi syndrome, which is more typically associated with HNF1A mutations. This finding is in line with the evidence that HNF-1α and HNF-1β are co-expressed in the PT and co-regulate expression of renal organic anion transporters such as OAT1 (Slc22A6), OAT3 (Slc22A83), and OAT4 (Slc22A11), the renal urate transporter (URAT1/Slc22A12) and renal Na+-phosphate (Pi) transporter 1 (NPT1/Slc17A1; Fig. 2) [10, 74,75,76]. Another HNF1-1β target gene involved in PT transport is Tmem27 encoding collectrin [77, 78]. Collectrin increases the surface expression of amino acid transporters, and disruption of Tmem27 in mice results in a severe defect in renal amino acid uptake [79, 80]. In general, PT dysfunction in humans with HNF1B mutations is rare, probably because HNF-1α is able to compensate for the loss of HNF-1β. Hyperuricemia with reduced fractional excretion of uric acid is the only widespread finding in HNF1B mutant patients that can be ascribed to an effect in the PT [81]. This phenotype is probably due to inhibition of the OAT1/3/4 transporters and/or uromodulin. Mutation of the latter also causes hyperuricemia [70], attributed to inhibition of NKCC2 in the TAL with a consequent increase in proximal Na+ reabsorption, which drives uric acid reabsorption (Fig. 2) [81].
Finally, hydronephrosis and polyuria have been observed in humans with mutations of HNF1B [62]. Proposed etiologies of polyuria include glycosuria due to early-onset diabetes mellitus and renal Na+ and K+ wasting causing intrauterine polyuria and polyhydramnios. Another possible mechanism was recently suggested by Aboudehen et al. [60] who found that inactivation of Hnf1b in mouse CDs produces polyuria and defects in urinary concentration by: (i) direct inhibition of osmosensitive genes, such as FXR; (ii) indirect downregulation of the urea transporter UT-A1; and (iii) impaired apical trafficking of aquaporin 2 (AQP2) [60].
HNF-1β and intrarenal metabolism
A combination of chromatin immunoprecipitation/next generation sequencing (Chip-seq) and gene expression profiling in renal epithelial cells uncovered a novel role of HNF-1β in a transcriptional network that regulates intrarenal cholesterol metabolism [82]. Studies in Hnf1b mutant kidney cells showed that HNF-1β stimulates cholesterol synthesis and inhibits cholesterol uptake, a mechanism that is reminiscent of the function of SREBP-2, which also activates transcription of cholesterol biosynthetic genes [82]. Indeed, ChIP-seq and reporter gene assays demonstrated that HNF-1β directly regulates Srebf2 (SREBP-2) transcription as well as the expression of Pcsk9, which encodes a serine protease that plays a crucial role in regulating cholesterol influx by internalizing the LDL receptor [83]. The physiological consequences of the regulation of intrarenal cholesterol metabolism, without affecting circulating cholesterol levels, are unknown. It is possible that HNF-1β may regulate metabolic changes in response to renal injury. Renal expression of HNF-1β shows an early downregulation followed by transient over-expression after ischemic acute kidney injury (AKI) [84, 85], a condition that is also associated with stimulation of renal cholesterol synthesis [86]. The increase in renal cholesterol, which seems to have a cytoprotective effect, may be mediated by the transient upregulation of HNF-1β expression. In sepsis-induced AKI, HNF-1β levels decrease within 24 h after injury and quickly normalize by 36 h [87]. HNF-1β downregulation during sepsis in vivo is associated with a decrease in the expression of PPARGC1A, which is involved in mitochondrial biogenesis and function. In vitro experiments have shown that HNF-1β directly regulates PPARGC1A and mitochondrial respiration, whereas ablation of Hnf1b in proximal tubule cells leads to a shift from oxidative phosphorylation to glycolysis [87]. Kang et al. reported that favoring glycolysis in renal tubular cells worsens the development of fibrosis [88], a common finding in HNF1B mutant humans. The contribution of the metabolic events affected by HNF-1β to the pathogenesis of cystic kidney disease and other mutant phenotypes remains to be further defined.
Mechanisms of HNF-1β regulation
Many intracellular events that occur at multiple levels regulate the abundance and activity of HNF-1β (Fig. 3). Whereas the upstream effectors that are involved in HNF1B gene transcription are largely unknown, with the exception of SNAIL and C/EBP, microRNAs (miRNAs) have been found to control Hnf1b mRNA abundance in kidney. Patel et al. reported that the miR-17 ∼ 92 miRNA cluster regulates the posttranscriptional expression of PKD genes, including Pkd1, Pkd2, and Hnf1b [89]. Within the cluster, miR-92a repressed the 3′ UTR of Hnf1b, whereas mutations of the miR-92a binding site abrogated this repression. Since miR-17 ∼ 92 is upregulated in PKD, miR-17 ∼ 92-dependent post-transcriptional silencing of PKD genes may represent a mechanism underlying cyst growth.

Schematic model of the molecular mechanisms involved in the regulation of HNF-1β expression and activity. Control of HNF1-β expression and activity in the cell nucleus can occur at multiple levels: (1) HNF1B gene transcription; (2) HNF1B mRNA stability; and (3) HNF1-β dimerization and interaction with regulatory proteins. EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; PCBD1, pterin-4 alpha-carbinolamine dehydratase; P, phosphorylation site
After translation in the cytosol, HNF-1β is targeted to the nucleus by a nuclear localization signal. Within the nucleus, HNF-1β binds DNA target sites as homodimers or heterodimers with its homolog HNF-1α (Fig. 3). This complex is able to recruit a number of cofactors necessary for transcription. The dimerization domain of HNF-1β interacts with the transcriptional coactivator PCBD1, a 12-kD protein that shuttles between cytosol and nucleus (Fig. 3) [7]. A few disease mutations have been mapped to the dimerization domain of HNF-1β in patients [8, 90]. These mutations are primarily premature termination codons that render the HNF1B mRNA susceptible to nonsense-mediated decay. Therefore, the phenotype in these patients is driven by HNF-1β haploinsufficiency rather than lack of interaction with PCBD1. Interestingly, homozygous or compound heterozygous PCBD1 mutations in humans are associated with MODY diabetes and renal Mg2+ wasting, although with variable penetrance [91]. These manifestations are in line with a defective function of PCBD1 as a dimerization cofactor for HNF-1β.
The transactivation domain at the C-terminus of HNF-1β interacts with CBP, P/CAF, and HDAC1 [8, 49, 92]. Disease-causing mutations in HNF-1β can lead to defective transactivation function through impaired CBP and P/CAF recruitment in vitro [8]. Accordingly, expression of an HNF-1β C-terminal deletion mutant in transgenic mice causes downregulation of Pkhd1 transcriptions and development of renal cysts, possibly due to a defect in recruiting CBP and P/CAF [49].
Zyxin is a novel HNF-1β interacting protein that binds to the POU domain and the C-terminal transactivation domain of HNF-1β, forming a transcriptional complex that also includes CBP [93]. Zyxin localizes in focal adhesions at sites of cell-matrix interaction but can translocate to the nucleus to enhance HNF-1β-mediated transcription upon stimulation by epidermal growth factor (EGF) (Fig. 3) [93]. Since signaling from the extracellular matrix is important for epithelial polarity and tubular morphogenesis in kidney tubules [94], the regulation of HNF-1β by zyxin may play an important role in kidney development and cystic kidney diseases [93].
In conclusion, lack of interaction between mutated HNF-1β and its partners may contribute to the onset of the HNF-1β-related disease in vivo [8, 49, 92]. In addition, HNF-1β-like renal abnormalities may be attributable to mutations in HNF-1β partners.
Conclusion and future perspectives
During the last decade, the identification of novel HNF-1β target genes in kidney by use of genome-wide techniques has provided new insights into the roles of HNF-1β in the developing and adult kidney [3, 32]. HNF-1β coordinates transcriptional networks involved in nephrogenesis, epithelial differentiation, tubular transport, and intrarenal metabolism. Therefore, patients who carry heterozygous mutations in HNF1B may present with complex renal phenotypes. To date, genotype-phenotype studies have not found a correlation between the type of mutations and the type and/or severity of renal disease, and patients who harbor the same HNF1B mutation show inter- and intrafamilial variability [12]. The multiple steps that influence HNF-1β-mediated gene transcription could explain this diversity. First, genetic variations in HNF-1β interacting proteins may affect the expression of target genes. Depending on the developmental stage, the ability of a regulatory protein to bind HNF-1β could give rise to either hypoplasia or cysts. Second, variations in non-coding regions may affect HNF1B gene expression and/or HNF1B mRNA stability. Finally, additional germline or de novo mutations in other genes that belong to the same biological pathways as HNF-1β may contribute to the phenotypic variability observed in HNF1B mutant patients. To test the hypothesis of modifier effects, a screen of HNF-1β interacting genes in patients should be performed. Future studies should also address the upstream signaling pathways that regulate HNF1B transcription and HNF-1β activity. Dissecting the molecular basis of HNF-1β regulation may offer new targets for the treatment of renal anomalies associated with HNF1B mutations.
References
Ott MO, Rey-Campos J, Cereghini S, Yaniv M (1991) vHNF1 is expressed in epithelial cells of distinct embryonic origin during development and precedes HNF1 expression. Mech Dev 36:47–58
Coffinier C, Barra J, Babinet C, Yaniv M (1999) Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev 89:211–213
Lokmane L, Heliot C, Garcia-Villalba P, Fabre M, Cereghini S (2010) vHNF1 functions in distinct regulatory circuits to control ureteric bud branching and early nephrogenesis. Development 137:347–357
Massa F, Garbay S, Bouvier R, Sugitani Y, Noda T, Gubler MC, Heidet L, Pontoglio M, Fischer E (2013) Hepatocyte nuclear factor 1beta controls nephron tubular development. Development 140:886–896
Heliot C, Desgrange A, Buisson I, Prunskaite-Hyyrylainen R, Shan J, Vainio S, Umbhauer M, Cereghini S (2013) HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signalling components and Irx1/2. Development 140:873–885
Kaminski MM, Tosic J, Kresbach C, Engel H, Klockenbusch J, Muller AL, Pichler R, Grahammer F, Kretz O, Huber TB, Walz G, Arnold SJ, Lienkamp SS (2016) Direct reprogramming of fibroblasts into renal tubular epithelial cells by defined transcription factors. Nat Cell Biol 18:1269–1280
Mendel DB, Khavari PA, Conley PB, Graves MK, Hansen LP, Admon A, Crabtree GR (1991) Characterization of a cofactor that regulates dimerization of a mammalian homeodomain protein. Science 254:1762–1767
Barbacci E, Chalkiadaki A, Masdeu C, Haumaitre C, Lokmane L, Loirat C, Cloarec S, Talianidis I, Bellanne-Chantelot C, Cereghini S (2004) HNF1beta/TCF2 mutations impair transactivation potential through altered co-regulator recruitment. Hum Mol Genet 13:3139–3149
Lu P, Rha GB, Chi YI (2007) Structural basis of disease-causing mutations in hepatocyte nuclear factor 1beta. Biochemistry 46:12071–12080
Saji T, Kikuchi R, Kusuhara H, Kim I, Gonzalez FJ, Sugiyama Y (2008) Transcriptional regulation of human and mouse organic anion transporter 1 by hepatocyte nuclear factor 1 alpha/beta. J Pharmacol Exp Ther 324:784–790
Liu H, Ren H, Spear BT (2011) The mouse alpha-albumin (afamin) promoter is differentially regulated by hepatocyte nuclear factor 1alpha and hepatocyte nuclear factor 1beta. DNA Cell Biol 30:137–147
Heidet L, Decramer S, Pawtowski A, Moriniere V, Bandin F, Knebelmann B, Lebre AS, Faguer S, Guigonis V, Antignac C, Salomon R (2010) Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol 5:1079–1090
Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, Ghiggeri GM, Jankauskiene A, Mir S, Montini G, Peco-Antic A, Wuhl E, Zurowska AM, Mehls O, Antignac C, Schaefer F, Salomon R (2006) Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 17:2864–2870
Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, Clauin S, Deschenes G, Bouissou F, Bensman A, Bellanne-Chantelot C (2006) Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol 17:497–503
Madariaga L, Moriniere V, Jeanpierre C, Bouvier R, Loget P, Martinovic J, Dechelotte P, Leporrier N, Thauvin-Robinet C, Jensen UB, Gaillard D, Mathieu M, Turlin B, Attie-Bitach T, Salomon R, Gubler MC, Antignac C, Heidet L (2013) Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol 8:1179–1187
Renkema KY, Winyard PJ, Skovorodkin IN, Levtchenko E, Hindryckx A, Jeanpierre C, Weber S, Salomon R, Antignac C, Vainio S, Schedl A, Schaefer F, Knoers NV, Bongers EM (2011) Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT). Nephrol Dial Transplant 26:3843–3851
Decramer S, Parant O, Beaufils S, Clauin S, Guillou C, Kessler S, Aziza J, Bandin F, Schanstra JP, Bellanne-Chantelot C (2007) Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol 18:923–933
Thomas R, Sanna-Cherchi S, Warady BA, Furth SL, Kaskel FJ, Gharavi AG (2011) HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 26:897–903
Bellanne-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Beaufils S, Wilhelm JM, Boitard C, Noel LH, Velho G, Timsit J (2004) Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med 140:510–517
Edghill EL, Bingham C, Ellard S, Hattersley AT (2006) Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet 43:84–90
Faguer S, Decramer S, Chassaing N, Bellanne-Chantelot C, Calvas P, Beaufils S, Bessenay L, Lengele JP, Dahan K, Ronco P, Devuyst O, Chauveau D (2011) Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 80:768–776
Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, Deltas C, Hosking A, Kmoch S, Rampoldi L, Wiesener M, Wolf MT, Devuyst O; Kidney Disease: Improving Global Outcomes (2015) Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management—a KDIGO consensus report. Kidney Int 88:676–683
Haeri S, Devers PL, Kaiser-Rogers KA, Moylan VJ Jr, Torchia BS, Horton AL, Wolfe HM, Aylsworth AS (2010) Deletion of hepatocyte nuclear factor-1-beta in an infant with prune belly syndrome. Am J Perinatol 27:559–563
Murray PJ, Thomas K, Mulgrew CJ, Ellard S, Edghill EL, Bingham C (2008) Whole gene deletion of the hepatocyte nuclear factor-1beta gene in a patient with the prune-belly syndrome. Nephrol Dial Transplant 23:2412–2415
Short KM, Combes AN, Lefevre J, Ju AL, Georgas KM, Lamberton T, Cairncross O, Rumballe BA, McMahon AP, Hamilton NA, Smyth IM, Little MH (2014) Global quantification of tissue dynamics in the developing mouse kidney. Dev Cell 29:188–202
Phua YL, Gilbert T, Combes A, Wilkinson L, Little MH (2016) Neonatal vascularization and oxygen tension regulate appropriate perinatal renal medulla/papilla maturation. J Pathol 238:665–676
Desgrange A, Heliot C, Skovorodkin I, Akram SU, Heikkila J, Ronkainen VP, Miinalainen I, Vainio SJ, Cereghini S (2017) HNF1B controls epithelial organization and cell polarity during ureteric bud branching and collecting duct morphogenesis. Development 144:4704–4719
Lindstrom NO, McMahon JA, Guo J, Tran T, Guo Q, Rutledge E, Parvez RK, Saribekyan G, Schuler RE, Liao C, Kim AD, Abdelhalim A, Ruffins SW, Thornton ME, Basking L, Grubbs B, Kesselman C, McMahon AP (2018) Conserved and divergent features of human and mouse kidney organogenesis. J Am Soc Nephrol 29:785–805
Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP (2005) Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell 9:283–292
Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ (2009) Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet 41:793–799
Naylor RW, Przepiorski A, Ren Q, Yu J, Davidson AJ (2013) HNF1beta is essential for nephron segmentation during nephrogenesis. J Am Soc Nephrol 24:77–87
Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, Fiette L, Igarashi P, Yaniv M, Pontoglio M (2004) A transcriptional network in polycystic kidney disease. EMBO J 23:1657–1668
Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M (2006) Defective planar cell polarity in polycystic kidney disease. Nat Genet 38:21–23
Simons M, Walz G (2006) Polycystic kidney disease: cell division without a c(l)ue? Kidney Int 70:854–864
Verdeguer F, Le Corre S, Fischer E, Callens C, Garbay S, Doyen A, Igarashi P, Terzi F, Pontoglio M (2010) A mitotic transcriptional switch in polycystic kidney disease. Nat Med 16:106–110
Hiesberger T, Bai Y, Shao X, McNally BT, Sinclair AM, Tian X, Somlo S, Igarashi P (2004) Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest 113:814–825
Gong Y, Ma Z, Patel V, Fischer E, Hiesberger T, Pontoglio M, Igarashi P (2009) HNF-1beta regulates transcription of the PKD modifier gene Kif12. J Am Soc Nephrol 20:41–47
Attanasio M, Uhlenhaut NH, Sousa VH, O'Toole JF, Otto E, Anlag K, Klugmann C, Treier AC, Helou J, Sayer JA, Seelow D, Nurnberg G, Becker C, Chudley AE, Nurnberg P, Hildebrandt F, Treier M (2007) Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet 39:1018–1024
Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. N Engl J Med 364:1533–1543
Choi YH, Suzuki A, Hajarnis S, Ma Z, Chapin HC, Caplan MJ, Pontoglio M, Somlo S, Igarashi P (2011) Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc Natl Acad Sci U S A 108:10679–10684
Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK (2009) The primary cilium as a complex signaling center. Curr Biol 19:R526–R535
Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S (2013) Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45:1004–1012
Ong AC, Wheatley DN (2003) Polycystic kidney disease—the ciliary connection. Lancet 361:774–776
Calvet JP (2008) Strategies to inhibit cyst formation in ADPKD. Clin J Am Soc Nephrol 3:1205–1211
Wang Q, Cobo-Stark P, Patel V, Somlo S, Han PL, Igarashi P (2017) Adenylyl cyclase 5 deficiency reduces renal cyclic AMP and cyst growth in an orthologous mouse model of polycystic kidney disease. Kidney Int 93:403–415
Zhang J, Wu M, Wang S, Shah JV, Wilson PD, Zhou J (2010) Polycystic kidney disease protein fibrocystin localizes to the mitotic spindle and regulates spindle bipolarity. Hum Mol Genet 19:3306–3319
Williams SS, Cobo-Stark P, James LR, Somlo S, Igarashi P (2008) Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr Nephrol 23:733–741
Williams SS, Cobo-Stark P, Hajarnis S, Aboudehen K, Shao X, Richardson JA, Patel V, Igarashi P (2014) Tissue-specific regulation of the mouse Pkhd1 (ARPKD) gene promoter. Am J Physiol Ren Physiol 307:F356–F368
Hiesberger T, Shao X, Gourley E, Reimann A, Pontoglio M, Igarashi P (2005) Role of the hepatocyte nuclear factor-1beta (HNF-1beta) C-terminal domain in Pkhd1 (ARPKD) gene transcription and renal cystogenesis. J Biol Chem 280:10578–10586
Zaucke F, Boehnlein JM, Steffens S, Polishchuk RS, Rampoldi L, Fischer A, Pasch A, Boehm CW, Baasner A, Attanasio M, Hoppe B, Hopfer H, Beck BB, Sayer JA, Hildebrandt F, Wolf MT (2010) Uromodulin is expressed in renal primary cilia and UMOD mutations result in decreased ciliary uromodulin expression. Hum Mol Genet 19:1985–1997
Ma Z, Gong Y, Patel V, Karner CM, Fischer E, Hiesberger T, Carroll TJ, Pontoglio M, Igarashi P (2007) Mutations of HNF-1beta inhibit epithelial morphogenesis through dysregulation of SOCS-3. Proc Natl Acad Sci U S A 104:20386–20391
Westland R, Verbitsky M, Vukojevic K, Perry BJ, Fasel DA, Zwijnenburg PJ, Bokenkamp A, Gille JJ, Saraga-Babic M, Ghiggeri GM, D'Agati VD, Schreuder MF, Gharavi AG, van Wijk JA, Sanna-Cherchi S (2015) Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int 88:1402–1410
Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, Pei Y (2009) Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 18:2328–2343
Bergmann C, von Bothmer J, Ortiz Bruchle N, Venghaus A, Frank V, Fehrenbach H, Hampel T, Pape L, Buske A, Jonsson J, Sarioglu N, Santos A, Ferreira JC, Becker JU, Cremer R, Hoefele J, Benz MR, Weber LT, Buettner R, Zerres K (2011) Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol 22:2047–2056
Hajarnis SS, Patel V, Aboudehen K, Attanasio M, Cobo-Stark P, Pontoglio M, Igarashi P (2015) Transcription factor hepatocyte nuclear factor-1beta (HNF-1beta) regulates microRNA-200 expression through a long noncoding RNA. J Biol Chem 290:24793–24805
Park SM, Gaur AB, Lengyel E, Peter ME (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283:14910–14914
Boutet A, De Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA (2006) Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J 25:5603–5613
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Aboudehen K, Noureddine L, Cobo-Stark P, Avdulov S, Farahani S, Gearhart MD, Bichet DG, Pontoglio M, Patel V, Igarashi P (2017) Hepatocyte nuclear factor-1beta regulates urinary concentration and response to hypertonicity. J Am Soc Nephrol 28:2887–2900
Dimke H, Monnens L, Hoenderop JG, Bindels RJ (2013) Evaluation of hypomagnesemia: lessons from disorders of tubular transport. Am J Kidney Dis 62:377–383
Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van’t Hoff W, Marks SD, Trompeter RS, Tullus K, Winyard PJ, Cansick J, Mushtaq I, Dhillon HK, Bingham C, Edghill EL, Shroff R, Stanescu H, Ryffel GU, Ellard S, Bockenhauer D (2009) HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 20:1123–1131
Ferre S, Veenstra GJ, Bouwmeester R, Hoenderop JG, Bindels RJ (2011) HNF-1B specifically regulates the transcription of the gammaa-subunit of the Na+/K+-ATPase. Biochem Biophys Res Commun 404:284–290
Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV (2000) Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)-ATPase gamma-subunit. Nat Genet 26:265–266
Kompatscher A, de Baaij JHF, Aboudehen K, Farahani S, LHJ VS, Milatz S, Himmerkus N, Veenstra GJC, Bindels RJM, Hoenderop JGJ (2018) Transcription factor HNF1beta regulates expression of the calcium-sensing receptor in the thick ascending limb of the kidney. Am J Physiol Ren Physiol. https://doi.org/10.1152/ajprenal.00601.2017
Huang CL, Kuo E (2007) Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 18:2649–2652
Yang L, Frindt G, Palmer LG (2010) Magnesium modulates ROMK channel-mediated potassium secretion. J Am Soc Nephrol 21:2109–2116
Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille C, Dosche C, Kumar S, Castaneda-Bueno M, Gamba G, Bachmann S (2011) Activation of the bumetanide-sensitive Na+,K+,2Cl- cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J Biol Chem 286:30200–30210
Renigunta A, Renigunta V, Saritas T, Decher N, Mutig K, Waldegger S (2011) Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J Biol Chem 286:2224–2235
Trudu M, Janas S, Lanzani C, Debaix H, Schaeffer C, Ikehata M, Citterio L, Demaretz S, Trevisani F, Ristagno G, Glaudemans B, Laghmani K, Dell'antonio G, Bochud M, Burnier M, Devuyst O, Martin PY, Mohaupt M, Paccaud F, Pechere-Bertschi A, Vogt B, Ackermann D, Ehret G, Guessous I, Ponte B, Pruijm M, Loffing J, Rastaldi MP, Manunta P, Devuyst O, Rampoldi L (2013) Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 19:1655–1660
Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP (1996) Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13:183–188
Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP (1996) Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 14:152–156
Kompatscher A, de Baaij JHF, Aboudehen K, Hoefnagels A, Igarashi P, Bindels RJM, Veenstra GJC, Hoenderop JGJ (2017) Loss of transcriptional activation of the potassium channel Kir5.1 by HNF1beta drives autosomal dominant tubulointerstitial kidney disease. Kidney Int 92:1145–1156
Kikuchi R, Kusuhara H, Hattori N, Shiota K, Kim I, Gonzalez FJ, Sugiyama Y (2006) Regulation of the expression of human organic anion transporter 3 by hepatocyte nuclear factor 1alpha/beta and DNA methylation. Mol Pharmacol 70:887–896
Jin L, Kikuchi R, Saji T, Kusuhara H, Sugiyama Y (2012) Regulation of tissue-specific expression of renal organic anion transporters by hepatocyte nuclear factor 1 alpha/beta and DNA methylation. J Pharmacol Exp Ther 340:648–655
Cheret C, Doyen A, Yaniv M, Pontoglio M (2002) Hepatocyte nuclear factor 1 alpha controls renal expression of the Npt1-Npt4 anionic transporter locus. J Mol Biol 322:929–941
Akpinar P, Kuwajima S, Krutzfeldt J, Stoffel M (2005) Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab 2:385–397
Fukui K, Yang Q, Cao Y, Takahashi N, Hatakeyama H, Wang H, Wada J, Zhang Y, Marselli L, Nammo T, Yoneda K, Onishi M, Higashiyama S, Matsuzawa Y, Gonzalez FJ, Weir GC, Kasai H, Shimomura I, Miyagawa J, Wollheim CB, Yamagata K (2005) The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab 2:373–384
Danilczyk U, Sarao R, Remy C, Benabbas C, Stange G, Richter A, Arya S, Pospisilik JA, Singer D, Camargo SM, Makrides V, Ramadan T, Verrey F, Wagner CA, Penninger JM (2006) Essential role for collectrin in renal amino acid transport. Nature 444:1088–1091
Malakauskas SM, Quan H, Fields TA, McCall SJ, Yu MJ, Kourany WM, Frey CW, Le TH (2007) Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am J Physiol Ren Physiol 292:F533–F544
Bingham C, Ellard S, van’t Hoff WG, Simmonds HA, Marinaki AM, Badman MK, Winocour PH, Stride A, Lockwood CR, Nicholls AJ, Owen KR, Spyer G, Pearson ER, Hattersley AT (2003) Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int 63:1645–1651
Aboudehen K, Kim MS, Mitsche M, Garland K, Anderson N, Noureddine L, Pontoglio M, Patel V, Xie Y, DeBose-Boyd R, Igarashi P (2016) Transcription factor hepatocyte nuclear factor-1beta regulates renal cholesterol metabolism. J Am Soc Nephrol 27:2408–2421
Horton JD, Cohen JC, Hobbs HH (2009) PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res 50(Suppl):S172–S177
Faguer S, Mayeur N, Casemayou A, Pageaud AL, Courtellemont C, Cartery C, Fournie GJ, Schanstra JP, Tack I, Bascands JL, Chauveau D (2013) Hnf-1beta transcription factor is an early hif-1alpha-independent marker of epithelial hypoxia and controls renal repair. PLoS One 8:e63585
Ogata K, Shimamura Y, Hamada K, Hisa M, Bun M, Okada N, Inoue K, Taniguchi Y, Ishihara M, Kagawa T, Horino T, Fujimoto S, Terada Y (2012) Upregulation of HNF-1beta during experimental acute kidney injury plays a crucial role in renal tubule regeneration. Am J Physiol Ren Physiol 303:F689–F699
Naito M, Bomsztyk K, Zager RA (2009) Renal ischemia-induced cholesterol loading: transcription factor recruitment and chromatin remodeling along the HMG CoA reductase gene. Am J Pathol 174:54–62
Casemayou A, Fournel A, Bagattin A, Schanstra J, Belliere J, Decramer S, Marsal D, Gillet M, Chassaing N, Huart A, Pontoglio M, Knauf C, Bascands JL, Chauveau D, Faguer S (2017) Hepatocyte nuclear factor-1beta controls mitochondrial respiration in renal tubular cells. J Am Soc Nephrol 28:3205–3217
Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K (2015) Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21:37–46
Patel V, Williams D, Hajarnis S, Hunter R, Pontoglio M, Somlo S, Igarashi P (2013) miR-17~92 miRNA cluster promotes kidney cyst growth in polycystic kidney disease. Proc Natl Acad Sci U S A 110:10765–10770
Waller SC, Rees L, Woolf AS, Ellard S, Pearson ER, Hattersley AT, Bingham C (2002) Severe hyperglycemia after renal transplantation in a pediatric patient with a mutation of the hepatocyte nuclear factor-1beta gene. Am J Kidney Dis 40:1325–1330
Ferre S, de Baaij JH, Ferreira P, Germann R, de Klerk JB, Lavrijsen M, van Zeeland F, Venselaar H, Kluijtmans LA, Hoenderop JG, Bindels RJ (2013) Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 25:574–586
Soutoglou E, Viollet B, Vaxillaire M, Yaniv M, Pontoglio M, Talianidis I (2001) Transcription factor-dependent regulation of CBP and P/CAF histone acetyltransferase activity. EMBO J 20:1984–1992
Choi YH, McNally BT, Igarashi P (2013) Zyxin regulates migration of renal epithelial cells through activation of hepatocyte nuclear factor-1beta. Am J Physiol Ren Physiol 305:F100–F110
Wilson PD, Burrow CR (1999) Cystic diseases of the kidney: role of adhesion molecules in normal and abnormal tubulogenesis. Exp Nephrol 7:114–124
Funding
Work from the authors’ laboratory was supported by NIH grant R37DK042921 and the University of Texas Southwestern Medical Center O’Brien Kidney Research Core Center P30DK079328. Silvia Ferrè was supported by the Ben J. Lipps Research Fellowship Program of the American Society of Nephrology Foundation for Kidney Research and the Charles and Jane Pak Center for Mineral Metabolism and Clinical Research Innovative Research Support Award.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ferrè, S., Igarashi, P. New insights into the role of HNF-1β in kidney (patho)physiology. Pediatr Nephrol 34, 1325–1335 (2019). https://doi.org/10.1007/s00467-018-3990-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-018-3990-7