
Abstract
Shiga toxin (Stx)-producing Escherichia coli (STEC) is the offending agent in post-diarrhea-associated hemolytic uremic syndrome (HUS), a disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney failure, with thrombi occluding the renal microvasculature. Endothelial dysfunction has been recognized as the trigger event in the development of microangiopathic processes. Glomerular endothelial cells are susceptible to the toxic effects of Stxs that, via nuclear factor kappa B (NF-κB) activation, induce the expression of genes encoding for adhesion molecules and chemokines, culminating in leukocyte adhesion and platelet thrombus formation on the activated endothelium. Complement activation via the alternative pathway has been seen in patients during the acute phase of STEC-associated HUS. Experimental evidence has highlighted the role of complement proteins in driving glomerular endothelium toward a thrombogenic phenotype. At the glomerular level, podocytes are also an important target of Stx-induced complement activation. Glomerular injury as a consequence of podocyte dysfunction and loss is thus a mechanism that might affect long-term renal outcomes in the disease. New approaches to targeting the complement system may be useful therapeutic options for patients with STEC-HUS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Shiga toxin (Stx, or verotoxin)-producing Escherichia coli (STEC) is the primary cause of the worldwide spread of hemorrhagic colitis complicated by diarrhea-associated hemolytic uremic syndrome (D + HUS), a disorder characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure, which develops principally in children under the age of 5 [1,2,3,4]. Thrombotic microangiopathy is the characteristic histological feature of HUS and consists of thickening of arterioles and capillaries, swelling and detachment of endothelial cells from the basement membrane, and fibrin- and platelet-rich thrombi that obstruct the microcirculation of different organs, mainly the kidney [5]. The overall incidence of STEC-HUS is about 2/100,000, with a peak of 6.1/100,000 in children <5 years of age [2]. In Latin America, where STEC infections are endemic, the incidence of the disease is markedly higher (10-17 cases per 100,000 children <5 years in Argentina) [6]. Concerning mortality, a review by Spinale et al. [7] indicates 1– 4% mortality rates in children during the acute phase of the disease. Accordingly, a study in STEC-HUS children hospitalized in the USA between 1997 and 2012 reported in-hospital mortality of 2.9% [8]. Evaluations of the percentage of patients who develop long-term sequelae (hypertension, impaired renal function, proteinuria, neurological symptoms) are variable among studies [7, 9, 10]. A systematic review by Garg et al. [9] estimated that 4 years after experiencing STEC-HUS, 25% of patients had renal sequelae and 3% developed end-stage renal disease (ESRD). In a study by Rosales et al. [10] in pediatric patients followed over a 6-year period, 70% of patients were fully recovered. The remaining 30% had renal impairment or neurological symptoms. Progression to ESRD within 2 months to 2 years was 1.4%. The frequency of neurological manifestations was 29% at the acute phase, decreased to 4% after 1 year, and remained stable at 5–years’ follow-up [10].
Specific therapies for STEC-HUS are lacking or still being investigated in preclinical studies, and patient care remains largely supportive [11,12,13]. The extreme clinical consequences of an outbreak in Germany in 2011 [14, 15] highlighted the continuing threat to public health that it poses and the need for effective treatments for STEC-HUS. Evidence that complement is dysregulated in STEC-HUS provided the rationale for using complement inhibitors as a therapeutic option. Results, however, were controversial [16,17,18,19,20,21,22].
In this paper, we will first review pathogenetic aspects of Stx-associated HUS. Then, we provide an update on complement system activation in this disease, highlighting the role of Stx-induced complement alternative pathway activation in glomerular microvascular injury and thrombosis. Finally, the impact of complement activation on glomerular podocyte damage and loss is discussed.
Pathogenetic aspects of STEC-HUS
The enterohemorrhagic E. coli (EHEC) O157:H7 serotype was the main offender in multiple outbreaks throughout the world until 2010 [1, 3]. In Europe and North America, other non-O157:H7 serotypes, including O26, O103, O111, O104, and O80, are all now as frequent as O157:H7 [3, 13], while O157:H7 remains the predominant (>70%) serotype in Latin America [6]. In 2011, a large outbreak of gastroenteritis and HUS linked to the consumption of raw fenugreek sprouts [14] occurred in northern Germany, with a satellite outbreak in western France, which was caused by an unusual E. coli O104:H4 strain combining the virulence potentials of enteroaggregative E. coli and typical Stx-producing enterohemorrhagic E. coli [23,24,25]. The outbreak predominantly affected adults (89%), with exceptionally severe manifestations and high mortality rates [15].
The main reservoirs of STEC are the intestinal tracts of healthy cattle, and foods of bovine origin, particularly undercooked beef and unpasteurized milk, and cattle-manure-contaminated vegetables and fruit or water are major sources of STEC infection in humans [3, 6]. Person-to-person transmission and contact with infected animals after farm or petting zoo visits have also been described. The production of Stx is the main virulence feature of STEC associated with the development of HUS but is not solely responsible for full pathogenicity. The risk of developing HUS for only a fraction of individuals following STEC infection may depend on host factors, including demography, immunity, lifestyle, use of antimotility agents, and pathogen factors such as the initial bacterial inoculum, the E. coli serotype, horizontally acquired genetic elements known as pathogenicity islands, and Stx type [6, 26, 27].
STEC colonization and Shiga toxin transport to the kidney
STEC colonize the large intestinal mucosa through peculiar attaching/effacing (A/E) lesions in a process characterized by the destruction of microvilli, intimate attachment of the pathogen to the host enterocyte, and changes in cytoskeletal structure of the enterocyte, with formation of an actin-rich pedestal around the bacteria [28]. This causes diarrhea and intestinal inflammation. Crucial colonization properties are carried by the locus of the enterocyte effacement (LEE) pathogenicity island, which encodes a diverse set of effector molecules, including a type III secretion system, the adhesin intimin, and its translocated receptor, Tir, which—together with several host proteins—is involved in actin polymerization and pedestal formation (see reviews in [29, 30]). Following Stx release, blood vessels in the colon are damaged, and bloody diarrhea occurs [1]. Stx translocates across the gastrointestinal epithelium, enters the circulation, and moves to the kidney and other target organs, such as brain and lungs, that express the glycosphingolipid globotriaosylceramide (Gb3Cer) or globotriaosylceramide (Gb4Cer) receptors [31, 32].
Lipopolysaccharide (LPS) developed in the gut by STEC may also enter the systemic circulation and promote an inflammatory response that contributes to renal injury. LPS from E. coli O157 bound to circulating platelets has been found in children with Stx-HUS [33]. LPS potentiated the cytotoxic effect of Stx on cultured endothelial cells by enhancing the inhibiting activity of Stx on protein synthesis [34]. In a baboon model of HUS, LPS increased the toxicity of Stx by upregulating renal Stx receptors [35], and both Stx and LPS were required to elicit a HUS-like response in mice [36,37,38].
Free Stxs have been detected in the sera of HUS patients in only negligible amounts [39]; the toxin circulates preferentially in the cell-bound form. Human blood cells, such as erythrocytes, platelets, and monocytes, have been suggested as possible Stx carriers via the Gb3 receptor (for review, see [40]). Recently, the toll-like receptor 4 (TLR4) has been identified as the receptor that specifically recognizes and binds Stxs in human neutrophils [41]. This finding is particularly relevant because TLR4 polymorphisms, which have been described as influencing the frequency and course of infectious diseases, could contribute to explaining the individual susceptibility of humans to STEC-infections, resulting in mild or more severe clinical manifestations [41]. Microvesicles derived from Stx-infected blood cells have been proposed as a novel mechanism of Stx transfer to glomerular cells in vitro and in patients with HUS [4, 42].
Shiga toxin structure and cytotoxic effects
STEC may generate two major types of Stx: Stx1, a family consisting of Stx1, 1c, and 1d, and the more heterogeneous Stx2 family, comprising the variants Stx2c, 2c2, 2d, 2dactivatable, 2e, and 2f [43]. The type of Stx produced by E. coli greatly influences the clinical outcome of the infection. While Stx2, 2c, and 2activatable can cause hemorrhagic colitis and HUS, the other Stx types are responsible for asymptomatic infections and uncomplicated diarrhea. All Stxs share a common structure consisting of one biologically active A subunit of 32 kDa, associated with five identical B subunits, each of 7.7 kDa [44], which allow toxin binding to the Gb3Cer/CD77 receptor [45], except for Stx2e, which preferentially binds to globotetraosylceramide (Gb4Cer).
Following binding to Gb3-expressing cells, Stx is endocytosed, mainly via a clathrin-dependent mechanism, and undergoes retrograde transport through the Golgi apparatus to the endoplasmic reticulum [46]. Here, the A subunit is proteolytically processed to form a 27-kDa A1-fragment, which is translocated into the cytosol, where it triggers molecular damage within infected cells. The A1 fragment possesses ribosomal RNA (rRNA)-N-glycosidase activity and inactivates the host ribosomes by removing a single adenosine residue from the 28S rRNA of the 60S subunit, thereby inhibiting protein synthesis [47]. On the other hand Stx, at concentrations causing ribosomal lesions, also activates multiple stress-induced signaling pathways, with a mechanism culminating in the induction of inflammatory gene expression and apoptosis. There is abundant in vitro and in vivo evidence that Stx, with negligible effects on global protein synthesis, led to changes in gene expression that activate the vascular endothelium toward the apoptotic program and cell death [29, 48, 49].
Shiga toxin induces endothelial damage
It has been recognized that endothelial damage plays a central role in the events that lead to the microangiopathic process of HUS [29]. Glomerular endothelial cells are the primary target of the toxic effects of Stx, which triggers a cascade of signaling events resulting in loss of endothelial antiadhesive, anti-inflammatory, and thromboresistant properties. Studies conducted by us show that Stx, via activation of nuclear factor kappa B (NF-κB), induced adhesion of leukocytes to cultured human endothelial cells under flow conditions through upregulation of adhesive molecules (E-selectin, ICAM-1, and VCAM-1) and chemokines (MCP-1, IL-8, fractalkine) [38, 50, 51]. Consistently, gene-expression profiling in Stx-treated human endothelial cells demonstrated the upregulation of cytokines, cell adhesion molecules, and transcription factors belonging to the NF-κB and tumor necrosis factor (TNF)/stress-related signaling pathways [52].
The prothrombotic state in Stx-associated HUS has been principally ascribed to endothelial cell damage. In response to Stx, endothelial cells are activated and lose their thromboresistant phenotype. Thus, Stx promoted platelet adhesion and thrombus formation on cultured human microvascular endothelial cells under high shear stress, which mimics that encountered in the microcirculation [53]. Von Willebrand factor (VWF), which undergoes conformational changes under conditions of high shear stress, was found to mediate platelet adhesion on activated endothelial cells in response to Stx. Functional blockade of adhesive proteins, including P-selectin, was associated with a reduction of platelet thrombi on endothelial cells [53]. Data showing Stx-dependent upregulation of endothelial chemokine receptor 4 (CXCR4), and increased stromal-cell-derived factor-1 (SDF-1) levels have revealed that CXCR4 and its ligand SDF-1 play an important role. Inhibition of CXCR4/SDF-1 interaction in vitro prevented Stx-mediated platelet adhesion to the endothelium under flow conditions and restored platelets to basal levels in mice injected with Stx [54].
The complement system
The complement cascade, a component of the immune system, acts as an important effector of humoral defense against invading pathogens, but at the same time, it preserves normal “self” cells. The complement system orchestrates various responses during immune and inflammatory reactions. In addition to protecting against bacterial infections, it is involved in the disposal of immune complexes, chemotaxis for recruiting inflammatory cells, opsonization, phagocytosis of foreign particles, and cell lysis [55].
The complement system comprises >30 components, including serum proteins and cell-surface molecules. Activation of complement occurs through: (1) the classical pathway, which involves antibody-mediated activation via the C1 complex; (2) the lectin pathway, induced by mannose-binding lectin (MBL) bound to glycan-associated mannose on the cell surface; and (3) the alternative pathway, involving the direct activation of C3 through surface binding or tick-over. All three routes converge in the generation of C3b by C3 convertases and terminate with the formation of the membrane attack complex (MAC, C5b-9).
The complement cascade is finely regulated to guarantee activation focused on pathogen surfaces and to limit the deposition of complement proteins on normal cells [55]. To avoid self-destructive activation, host cells are protected by a battery of regulatory inhibitor proteins, present both in serum and on cell membranes. These regulatory proteins favor cleavage of C3b to the inactive form iC3b, dissociate C3/C5 convertases, and prevent C9 assembly into the C5b-9 complex [56].
Activation of the complement system in STEC-HUS
Clinical evidence
The first pieces of evidence of complement activation in patients with Stx-HUS date back to >30 years ago to anecdotes about reduced C3 and augmented serum levels of the C3 fragments C3b, c, and d during the active phase of the disease [57,58,59]. Later on, high plasma levels of factor Bb and C5b-9 were measured in 17 children during disease onset, indicating complement activation via the alternative pathway [60]. Consistently, factor Bb and soluble C5b-9 plasma levels were increased in active Stx-HUS patients and correlated with oliguria [61]. Plasma levels of C3a, generated by the cleavage of C3 in C3b, were significantly increased compared with controls in ten patients during HUS [62]. More recently, high levels of circulating microparticles, arising from platelets, monocytes, and red blood cells, with surface-bound C3 and C9, were found in patients with Stx-HUS [62, 62].
In addition to the systemic activation of complements, C3 and C5b-9 deposits associated with fibrin deposition were detected in the glomeruli of a Stx-HUS child [64], which supports the hypothesis that there is a functional link between complement activation and microvascular thrombosis. Intense glomerular C3 and C5b-9 deposits were also present in the kidney of a patient with a mutation of complement factor I who developed ESRD after STEC-HUS [65].
Experimental evidence
That Stx contributes directly to dysregulation and activation of the complement system is confirmed by several reports. Stx2 activates complement in the fluid phase, as described by Orth et al. [66], who detected the formation of soluble C5b-9 in normal human serum exposed to Stx2. Stimulation of whole blood with Stx2 increased formation of platelet–monocyte and platelet–neutrophil complexes and the release of blood-cell-derived microparticles, with surface-bound C3 and C9 [62]. Stx2 also caused the release of red-blood-cell-derived microvesicles coated with C5b-9, indicating that complement plays a role in the hemolytic process that occurs during STEC-HUS [63]. In vitro evidence indicates that Stx can activate complement through the alternative pathway. Stx2 binds factor H, the major soluble inhibitor of the alternative pathway, and this translates into a delay/reduction in factor H cofactor activity on the cell surface, with a consequent increase in complement activation and C3b deposition [66]. Stx2 is also a ligand for the factor H family proteins FHR-1 and FHL-1, which display amino acid sequence and regulatory function similarities with factor H [67]. In vitro studies have shown that FHR-1 competes with factor H for Stx2 binding [67]. FHR-1 exists in two allotypes: FHR-1*A and B, with FHR-1*B having lower binding capacity to Stx2 than allotype A. It has been proposed that in homozygous or heterozygous patients for FHR-1*B, because of the weaker binding capacity of FHR-1*B, Stx2 may preferentially bind to factor H, with consequent delay and/or reduction of cofactor activity of St x2-bound factor H [67]. Thus, interindividual variability of FHR-1 allotype might contribute to different degrees of disease susceptibility or severity in STEC-HUS patients.
Stx activates complement by acting at different levels of the cascade. A reduction in both messenger RNA (mRNA) and the surface expression of CD59, the membrane-bound complement inhibitor that prevents MAC formation, was found on human glomerular endothelial cells exposed to Stx2 [68]. On the other hand, high levels of CD59 were found on erythrocytes of HUS patients during the outbreak in Germany in 2011, suggesting that CD59 has a role in counteracting the hemolytic anemia caused by STEC infection [69].
Shiga-toxin-activated complement promotes glomerular endothelial cell damage
Animal models have provided valuable information about the importance of complement activation, via the alternative pathway, in the development of the thrombotic process, leading to kidney dysfunction in HUS. Mice with HUS induced by coinjection of Stx2/LPS exhibited thrombocytopenia and renal function impairment associated with glomerular deposition of C3 and fibrin(ogen) [38, 70, 71]. In this setting, factor-B-deficient mice were protected against platelet loss and renal dysfunction, indicating that the alternative complement pathway is a key mediator for microvascular thrombosis [70, 71]. Glomerular deposition of C3 and C5b-9 was found in mice infected with STEC. Renal disease progression was prevented when mice lacking C6 were used [64].
It is known that activation of the alternative pathway can be amplified by the lectin pathway through direct activation of factors B and D by MBL/ficolin-associated serine proteases (MASPs) [72]. The inhibition of MBL2 in a mouse model of Stx-HUS significantly attenuated renal C3d deposition and limited renal injury, with new implications for the lectin pathway in disease onset [73].
Several lines of evidence point to thrombomodulin, a membrane-bound complement and coagulation regulator, as having a role in the pathogenesis of Stx-associated HUS [74]. Thrombomodulin is an endothelial transmembrane receptor for thrombin, the best-known cofactor in the protein C anticoagulant pathway [75], which possesses different properties that impact on fibrinolysis, complement activation, inflammation, and cell proliferation [76]. Our group has demonstrated that mutations that impair thrombomodulin function and cause defective complement regulation increase the human risk of developing atypical HUS, a form that is not Stx-associated [76]. In vitro Stx2 reduced thrombomodulin expression in human glomerular endothelial cells [77]. Stx-induced thrombomodulin loss was due to the ability of the toxin to increase thrombomodulin shedding from the endothelial cell surface through release of serine proteases from endothelial-cell-specific intracellular storage vesicles [70]. In a mouse model of Stx-HUS, the glomerular expression of thrombomodulin was reduced in association with C3, fibrin (ogen), and platelet deposition [70, 74]. Mice lacking the lectin-like domain of thrombomodulin were more susceptible to developing thrombocytopenia and renal dysfunction than were wild-type mice exhibiting a stronger inflammatory reaction and earlier intraglomerular fibrin (ogen) deposits [74]. These data suggested that genetic or acquired functional defects in thrombomodulin may contribute to an adverse outcome during the course of Stx-HUS.
Overactivation of the complement system on renal and blood cells contributes significantly to the abnormal vascular function and prothrombotic processes that typically occur in Stx-HUS. Stx directly induces complement activation, via the alternative pathway, on the surface of endothelial cells, as revealed by increased C3 deposition and thrombus formation on microvascular endothelial cells after perfusion with human serum [70]. Proof that C3 deposition is functionally linked to thrombus growth derives from data that confirms that the complement inhibitor soluble complement receptor 1 (sCR1) completely blocks thrombus formation on the endothelial surface. Loss of endothelial thromboresistance due to Stx depends on upregulation of P-selectin—a membrane adhesion molecule acting as a specific ligand of C3 [70, 78]—and on the reduction of thrombomodulin expression. This evidence was confirmed in the mouse model of Stx-HUS in which both treatment with anti-P-selectin antibody and factor-B deficiency protected mice from glomerular endothelial damage and thrombosis [70].
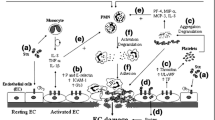
Complement proteins such as C3a, C5a, and C5b-9 cause profound perturbation of the thromboresistance phenotype of endothelial cells. The binding of C5a to its receptor and the deposition of a sublytic amount of C5b-9 on the endothelial surface induce proinflammatory and procoagulant activities [79, 80]. C3a promotes cytokine production and increases endothelial cell permeability [81, 82]. We demonstrated that C3a increased P-selectin expression and thrombomodulin release from cultured endothelial cells, all of which culminated in thrombus growth on the cell surface [70] (Fig. 1). The role of C3a in potentiating microvascular thrombosis in Stx-HUS has been highlighted by data showing a marked reduction in fibrin(ogen) and limited thrombomodulin loss in glomeruli of mice with Stx-HUS after treatment with a C3a receptor antagonist [70].

Glomerular complement activation in Shiga toxin (Stx)-associated hemolytic uremic syndrome (HUS). Possible sequence of events through which Stx promotes complement activation on glomerular endothelial cells and podocytes. Stx, by binding to its specific endothelial receptor Gb3, favors the surface expression of P-selectin, which is responsible for thrombus formation on endothelial cells. Excessive glomerular C3 activation in response to Stx generates C3a, which interacts with the specific receptor C3aR, thereby potentiating P-selectin expression and thrombomodulin shedding, with final thrombus formation and reduced protection from complement activation. In parallel, C3a binds to the podocyte surface via C3aR, causing a decrease in α-actinin-4 expression and promoting integrin-linked kinase (ILK)-dependent nuclear translocation of Snail, with consequent nephrin downregulation and podocyte dysfunction
Shiga-toxin-activated complement induces podocyte injury
Long-term renal sequelae have been observed in about 25–30% of STEC-HUS patients who undergo a critical reduction in nephron numbers, with consequent hyperfiltration, proteinuria, glomerular injury, and chronic kidney disease [7, 9]. Glomerular podocyte injury and loss have been recognized as the inciting event in the complex process that ends in glomerulosclerosis, whatever the primary kidney disease [83,84,85,86]. Podocytes are postmitotic cells that are incapable of proliferating and replenishing their numbers following migration and detachment from the basement membrane in disease [87, 88]. In STEC-HUS, little information is available regarding the role of Stx in the development of proteinuria and glomerular podocyte damage due to the fact that kidney biopsies are rarely performed in these patients. The structural hallmarks of Stx-associated HUS, such as collapse and retraction of the glomerular microvascular endothelium, are often associated with swelling of podocytes and the effacement of their foot processes [58, 89]. The presence of nephrin and synaptopodin mRNA in the urine of 15 patients during active Stx-HUS means that podocyte injury and loss may occur during the acute phase of the disease [90]. Consistently, in a primate model of HUS, glomerular endothelial injury was linked to structural podocyte alterations [91]. Microalbuminuria occurred in rats injected with Stx2, being an early sign of podocyte injury as evidenced by alteration of the slit-diaphragm proteins nephrin and podocalyxin. These glomerular changes were also functionally linked to proximal tubular alteration of megalin-dependent endocytosis [92].
The proof that podocytes represent a relevant target for Stx rests on in vitro studies showing that podocytes are susceptible to the direct cytotoxic effects of Stx1 and 2 isoforms because they express discrete levels of Gb3 receptor [93,94,95,96]. Upon binding to the receptor, Stxs activate podocytes to release cytokines such as interleukin-1 (IL-1) and TNF, which remarkably increase Gb3 expression, thus enhancing cell sensitivity toward the toxins [94, 97] and favoring apoptosis [96]. That Stxs elicit inflammatory responses is also suggested by findings that in cultured mouse podocytes, Stx2 activates p38 and p42/44 mitogen-activated protein kinases (MAPKs) and upregulates transcription factors NF-κB and activator protein (AP-1) [93], known regulators of cytokine and chemokine gene expression. In this setting, the increased podocyte expression and production of the vasoactive peptide endothelin-1 (ET-1) in response to the toxin suggests that Stx may also deleteriously impact on glomerular microcirculation via an autocrine and paracrine action of podocyte-derived ET1, thus contributing to glomerular dysfunction.
In search of important determinants of glomerular damage to predict the cause of long-term renal prognosis in HUS, we recently determined that complement activation, via the alternative pathway, is a functional player of podocyte dysregulation and loss [71]. The robust presence of C3 deposits was observed in the proximity of podocytes, as shown by the costaining of complement with nephrin in glomeruli of mice with Stx/LPS-induced HUS. Moreover, glomerular complement deposition was associated with increased expression of activated integrin-linked kinase (ILK), a signal responsible for podocyte adhesion/motility on the glomerular basement membrane (GBM). The early engagement of ILK preceded the activation of Snail, a transcription factor known to regulate nephrin expression [71]. In parallel, a marked reduction of alpha-actinin 4, an actin cross-linking protein, involved both in the stabilization of podocyte attachment and regulation of cell motility through its interaction with integrins, was also described [71]. The instrumental role of the complement alternative pathway activation was demonstrated by applying the Stx2/LPS model to factor-B-deficient mice and the pharmacological inhibition of factor B with a functional blocking antibody. Both strategies successfully normalized complement-dependent glomerular structural and functional changes in response to Stx2 [71]. Our data also substantiated the novel concept that C3a, generated during C3 deposition, could be a key driver of podocyte dysfunction as well as endothelial injury. The proof-of-concept was based on data confirming that treatment with a C3a receptor antagonist reduced podocyte ultrastructural changes and loss by limiting ILK and Snail activation and preserving alpha-actinin 4 in mice with HUS. Thrombocytopenia and renal function were also improved. In vitro data with human cultured podocytes showed that C3a affected podocyte phenotype and motility via activated ILK [71] (Fig. 1). In this context, the possible contribution of C5a and MAC in processes leading to glomerular damage cannot be excluded.
The aberrant role of C3/C3a in podocyte dysregulation and glomerular injury has been highlighted in a model of proteinuric progressive nephropathy [98]. In mice with protein overload proteinuria, marked glomerular accumulation of C3 and local generation of C3a was an indispensable factor in podocyte dysfunction and loss that subsequently led to parietal epithelial cell activation and development of glomerular lesions.
Conclusions
In recent years, a substantial amount of evidence has shown that complement activation has a role in Stx-HUS. Glomerular endothelial cells have long been considered the main target of Stx-induced renal toxicity. However, it was recently recognized that another cell population in proximity to injured glomerular endothelium—the podocytes—is involved in the disease pathogenesis. Abnormal glomerular activation of the complement cascade via the alternative pathway, with local C3 deposition and C3a generation in response to Stx, causes a series of events beginning with loss of endothelial thromboresistance and followed by the development of microvascular thrombi. Complement-dependent endothelial injury may be accompanied by dysfunction and loss of podocytes. This is possibly the cause of the long-term renal sequelae in 25–30% of Stx-HUS patients. Indeed, podocytes—the critical regulators of the glomerular filtration barrier—change their phenotype in response to activated complement proteins, which translates into abnormal cell–cell interaction and cell matrix adhesion to the GBM, ending in podocyte detachment. The recent findings obtained in patients and in experimental models of HUS suggest complement inhibition at C3 and C3a levels is a possible therapeutic tool for counteracting Stx-induced glomerular damage. Using C3a receptor antagonist, currently available in preclinical studies [70, 71, 99], could offer the advantage of avoiding the complete shutdown of C3—which could increase susceptibility to infections—and provide a promising opportunity for long-term systemic intervention. So far, the only complement inhibitor used in clinical studies has been the anti-C5 antibody (eculizumab), which exhibited beneficial effects in paroxysmal nocturnal hemoglobinuria, myocardial infarction, age-related maculopathy, atypical HUS, and C3 glomerulopathy [100]. Eculizumab showed encouraging results in three children with severe Stx-associated HUS [16], but clear effects could not be demonstrated during the large O104:H4 STEC-HUS outbreak in 2011 [17,18,19, 22]. Subsequent smaller studies reported a rapid improvement after eculizumab in severe STEC-HUS patients [20, 21]. The controversial results regarding the use of eculizumab in STEC-HUS may reflect statistical bias in retrospective or uncontrolled studies, which emphasizes the need for prospective studies. The ongoing phase III prospective randomized controlled trial (NCT02205541) in pediatric patients affected by STEC-HUS should help clarify the benefit of eculizumab in this devastating disease.
References
Tarr PI, Gordon CA, Chandler WL (2005) Shiga-toxin-producing Escherichia Coli and haemolytic uraemic syndrome. Lancet 365:1073–1086
Noris M, Remuzzi G (2005) Hemolytic uremic syndrome. J Am Soc Nephrol 16:1035–1050
Karmali MA, Gannon V, Sargeant JM (2010) Verocytotoxin-producing Escherichia Coli (VTEC). Vet Microbiol 140:360–370
Karpman D, Loos S, Tati R, Arvidsson I (2017) Haemolytic uraemic syndrome. J Intern Med 281:123–148
Ruggenenti P, Noris M, Remuzzi G (2001) Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int 60:831–846
Rivas M, Chinen I, Miliwebsky E, Masana M (2014) Risk factors for Shiga toxin-producing Escherichia Coli-associated human diseases. Microbiol Spectr 2. https://doi.org/10.1128/microbiolspec.EHEC-0002-2013
Spinale JM, Ruebner RL, Copelovitch L, Kaplan BS (2013) Long-term outcomes of Shiga toxin hemolytic uremic syndrome. Pediatr Nephrol 28:2097–2105
Mody RK, Gu W, Griffin PM, Jones TF, Rounds J, Shiferaw B, Tobin-D'Angelo M, Smith G, Spina N, Hurd S, Lathrop S, Palmer A, Boothe E, Luna-Gierke RE, Hoekstra RM (2015) Postdiarrheal hemolytic uremic syndrome in United States children: clinical spectrum and predictors of in-hospital death. J Pediatr 166:1022–1029
Garg AX, Suri RS, Barrowman N, Rehman F, Matsell D, Rosas-Arellano MP, Salvadori M, Haynes RB, Clark WF (2003) Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. JAMA 290:1360–1370
Rosales A, Hofer J, Zimmerhackl LB, Jungraithmayr TC, Riedl M, Giner T, Strasak A, Orth-Holler D, Wurzner R, Karch H (2012) Need for long-term follow-up in enterohemorrhagic Escherichia Coli-associated hemolytic uremic syndrome due to late-emerging sequelae. Clin Infect Dis 54:1413–1421
Bitzan M (2009) Treatment options for HUS secondary to Escherichia Coli O157:H7. Kidney Int Suppl:S62–S66
Petruzziello-Pellegrini TN, Marsden PA (2012) Shiga toxin-associated hemolytic uremic syndrome: advances in pathogenesis and therapeutics. Curr Opin Nephrol Hypertens 21:433–440
Fakhouri F, Zuber J, Fremeaux-Bacchi V, Loirat C (2017) Haemolytic uraemic syndrome. Lancet 390:681–696
Buchholz U, Bernard H, Werber D, Bohmer MM, Remschmidt C, Wilking H, Delere Y, an der Heiden M, Adlhoch C, Dreesman J, Ehlers J, Ethelberg S, Faber M, Frank C, Fricke G, Greiner M, Hohle M, Ivarsson S, Jark U, Kirchner M, Koch J, Krause G, Luber P, Rosner B, Stark K, Kuhne M (2011) German outbreak of Escherichia Coli O104:H4 associated with sprouts. N Engl J Med 365:1763–1770
Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Muller L, King LA, Rosner B, Buchholz U, Stark K, Krause G (2011) Epidemic profile of Shiga-toxin-producing Escherichia Coli O104:H4 outbreak in Germany. N Engl J Med 365:1771–1780
Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, Schaefer F (2011) Eculizumab in severe Shiga-Toxin-Associated HUS. N Engl J Med 364:2561–2563
Menne J, Nitschke M, Stingele R, Abu-Tair M, Beneke J, Bramstedt J, Bremer JP, Brunkhorst R, Busch V, Dengler R, Deuschl G, Fellermann K, Fickenscher H, Gerigk C, Goettsche A, Greeve J, Hafer C, Hagenmuller F, Haller H, Herget-Rosenthal S, Hertenstein B, Hofmann C, Lang M, Kielstein JT, Klostermeier UC, Knobloch J, Kuehbacher M, Kunzendorf U, Lehnert H, Manns MP, Menne TF, Meyer TN, Michael C, Munte T, Neumann-Grutzeck C, Nuernberger J, Pavenstaedt H, Ramazan L, Renders L, Repenthin J, Ries W, Rohr A, Rump LC, Samuelsson O, Sayk F, Schmidt BM, Schnatter S, Schocklmann H, Schreiber S, von Seydewitz CU, Steinhoff J, Stracke S, Suerbaum S, van de Loo A, Vischedyk M, Weissenborn K, Wellhoner P, Wiesner M, Zeissig S, Buning J, Schiffer M, Kuehbacher T (2012) Validation of treatment strategies for enterohaemorrhagic Escherichia Coli O104:H4 induced haemolytic uraemic syndrome: case-control study. BMJ 345:e4565
Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, Kuhlmann U, Bramstedt J, Panzer U, Vischedyk M, Busch V, Ries W, Mitzner S, Mees S, Stracke S, Nurnberger J, Gerke P, Wiesner M, Sucke B, Abu-Tair M, Kribben A, Klause N, Schindler R, Merkel F, Schnatter S, Dorresteijn EM, Samuelsson O, Brunkhorst R (2012) Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-Toxin-Producing E. Coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant 27:3807–3815
Ruggenenti P, Remuzzi G (2012) Thrombotic microangiopathy: E. Coli O104:H4 German outbreak: a missed opportunity. Nat Rev Nephrol 8:558–560
Saini A, Emke AR, Silva MC, Perlman SJ (2015) Response to Eculizumab in Escherichia Coli O157: H7-induced hemolytic uremic syndrome with severe neurological manifestations. Clin Pediatr (Phila) 54:387–389
Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T (2015) Eculizumab in typical Hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore) 94:e1000
Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, Helou S, Bordes C, Reffet A, Llanas B, Skopinski S, Rolland P, Gruson D, Combe C (2014) Outbreak of Escherichia Coli O104:H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant 29:565–572
Bielaszewska M, Mellmann A, Zhang W, Kock R, Fruth A, Bauwens A, Peters G, Karch H (2011) Characterisation of the Escherichia Coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect Dis 11:671–676
Ruggenenti P, Remuzzi G (2011) A German outbreak of haemolytic uraemic syndrome. Lancet 378:1057–1058
Hauswaldt S, Nitschke M, Sayk F, Solbach W, Knobloch JK (2013) Lessons learned from outbreaks of Shiga toxin producing Escherichia Coli. Curr Infect Dis Rep 15:4–9
Karmali MA (2009) Host and pathogen determinants of verocytotoxin-producing Escherichia Coli-associated hemolytic uremic syndrome. Kidney Int Suppl:S4–S7
Karmali MA (2017) Emerging public health challenges of Shiga toxin-producing Escherichia Coli related to changes in the pathogen, the population, and the environment. Clin Infect Dis 64:371–376
Frankel G, Phillips AD (2008) Attaching effacing Escherichia Coli and paradigms of Tir-triggered actin polymerization: getting off the pedestal. Cell Microbiol 10:549–556
Zoja C, Buelli S, Morigi M (2010) Shiga toxin-associated hemolytic uremic syndrome: pathophysiology of endothelial dysfunction. Pediatr Nephrol 25:2231–2240
Campellone KG (2010) Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia Coli: Tir, EspFU and actin pedestal assembly. FEBS J 277:2390–2402
Bauwens A, Betz J, Meisen I, Kemper B, Karch H, Muthing J (2013) Facing glycosphingolipid-Shiga toxin interaction: dire straits for endothelial cells of the human vasculature. Cell Mol Life Sci 70:425–457
Rutjes NW, Binnington BA, Smith CR, Maloney MD, Lingwood CA (2002) Differential tissue targeting and pathogenesis of verotoxins 1 and 2 in the mouse animal model. Kidney Int 62:832–845
Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, Watkins SL, Johnson R, Karpman D (2006) Lipopolysaccharide from enterohemorrhagic Escherichia Coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 108:167–176
Louise CB, Obrig TG (1992) Shiga toxin-associated hemolytic uremic syndrome: combined cytotoxic effects of shiga toxin and lipopolysaccharide (endotoxin) on human vascular endothelial cells in vitro. Infect Immun 60:1536–1543
Clayton F, Pysher TJ, Lou R, Kohan DE, Denkers ND, Tesh VL, Taylor FB Jr, Siegler RL (2005) Lipopolysaccharide upregulates renal shiga toxin receptors in a primate model of hemolytic uremic syndrome. Am J Nephrol 25:536–540
Ikeda M, Ito S, Honda M (2004) Hemolytic uremic syndrome induced by lipopolysaccharide and Shiga-like toxin. Pediatr Nephrol 19:485–489
Keepers TR, Psotka MA, Gross LK, Obrig TG (2006) A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol 17:3404–3414
Zanchi C, Zoja C, Morigi M, Valsecchi F, Liu XY, Rottoli D, Locatelli M, Buelli S, Pezzotta A, Mapelli P, Geelen J, Remuzzi G, Hawiger J (2008) Fractalkine and CX3CR1 mediate leukocyte capture by endothelium in response to Shiga toxin. J Immunol 181:1460–1469
Brigotti M, Tazzari PL, Ravanelli E, Carnicelli D, Rocchi L, Arfilli V, Scavia G, Minelli F, Ricci F, Pagliaro P, Ferretti AV, Pecoraro C, Paglialonga F, Edefonti A, Procaccino MA, Tozzi AE, Caprioli A (2011) Clinical relevance of shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr Infect Dis J 30:486–490
Obrig TG, Karpman D (2012) Shiga toxin pathogenesis: kidney complications and renal failure. Curr Top Microbiol Immunol 357:105–136
Brigotti M, Carnicelli D, Arfilli V, Tamassia N, Borsetti F, Fabbri E, Tazzari PL, Ricci F, Pagliaro P, Spisni E, Cassatella MA (2013) Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J Immunol 191:4748–4758
Stahl AL, Arvidsson I, Johansson KE, Chromek M, Rebetz J, Loos S, Kristoffersson AC, Bekassy ZD, Morgelin M, Karpman D (2015) A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog 11:e1004619
Muthing J, Schweppe CH, Karch H, Friedrich AW (2009) Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thromb Haemost 101:252–264
O'Brien AD, Tesh VL, Donohue-Rolfe A, Jackson MP, Olsnes S, Sandvig K, Lindberg AA, Keusch GT (1992) Shiga toxin: biochemistry, genetics, mode of action, and role in pathogenesis. Curr Top Microbiol Immunol 180:65–94
Lingwood CA (1996) Role of verotoxin receptors in pathogenesis. Trends Microbiol 4:147–153
Sandvig K, Garred O, Prydz K, Kozlov JV, Hansen SH, van Deurs B (1992) Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 358:510–512
Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K (1988) Site of action of a Vero toxin (VT2) from Escherichia Coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur J Biochem 171:45–50
Petruzziello-Pellegrini TN, Moslemi-Naeini M, Marsden PA (2013) New insights into Shiga toxin-mediated endothelial dysfunction in hemolytic uremic syndrome. Virulence 4:556–563
Tesh VL (2012) The induction of apoptosis by Shiga toxins and ricin. Curr Top Microbiol Immunol 357:137–178
Morigi M, Micheletti G, Figliuzzi M, Imberti B, Karmali MA, Remuzzi A, Remuzzi G, Zoja C (1995) Verotoxin-1 promotes leukocyte adhesion to cultured endothelial cells under physiologic flow conditions. Blood 86:4553–4558
Zoja C, Angioletti S, Donadelli R, Zanchi C, Tomasoni S, Binda E, Imberti B, te Loo M, Monnens L, Remuzzi G, Morigi M (2002) Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via NF-kappaB dependent up-regulation of IL-8 and MCP-1. Kidney Int 62:846–856
Matussek A, Lauber J, Bergau A, Hansen W, Rohde M, Dittmar KE, Gunzer M, Mengel M, Gatzlaff P, Hartmann M, Buer J, Gunzer F (2003) Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood 102:1323–1332
Morigi M, Galbusera M, Binda E, Imberti B, Gastoldi S, Remuzzi A, Zoja C, Remuzzi G (2001) Verotoxin-1-induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood 98:1828–1835
Petruzziello-Pellegrini TN, Yuen DA, Page AV, Patel S, Soltyk AM, Matouk CC, Wong DK, Turgeon PJ, Fish JE, Ho JJ, Steer BM, Khajoee V, Tigdi J, Lee WL, Motto DG, Advani A, Gilbert RE, Karumanchi SA, Robinson LA, Tarr PI, Liles WC, Brunton JL, Marsden PA (2012) The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome in humans and mice. J Clin Invest 122:759–776
Walport MJ (2001) Complement. First of two parts. N Engl J Med 344:1058–1066
Zipfel PF, Skerka C (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9:729–740
Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P (1980) The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol 13:168–171
Koster FT, Boonpucknavig V, Sujaho S, Gilman RH, Rahaman MM (1984) Renal histopathology in the hemolytic-uremic syndrome following shigellosis. Clin Nephrol 21:126–133
Robson WL, Leung AK, Fick GH, McKenna AI (1992) Hypocomplementemia and leukocytosis in diarrhea-associated hemolytic uremic syndrome. Nephron 62:296–299
Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, Holers VM, Lesser M, Kline M, Hoffman C, Christen E, Trachtman H (2009) Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol 4:1920–1924
Ferraris JR, Ferraris V, Acquier AB, Sorroche PB, Saez MS, Ginaca A, Mendez CF (2015) Activation of the alternative pathway of complement during the acute phase of typical haemolytic uraemic syndrome. Clin Exp Immunol 181:118–125
Stahl AL, Sartz L, Karpman D (2011) Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia Coli-induced hemolytic uremic syndrome. Blood 117:5503–5513
Arvidsson I, Stahl AL, Hedstrom MM, Kristoffersson AC, Rylander C, Westman JS, Storry JR, Olsson ML, Karpman D (2015) Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J Immunol 194:2309–2318
Arvidsson I, Rebetz J, Loos S, Herthelius M, Kristoffersson AC, Englund E, Chromek M, Karpman D (2016) Early terminal complement blockade and C6 deficiency are protective in Enterohemorrhagic Escherichia Coli-infected mice. J Immunol 197:1276–1286
Alberti M, Valoti E, Piras R, Bresin E, Galbusera M, Tripodo C, Thaiss F, Remuzzi G, Noris M (2013) Two patients with history of STEC-HUS, posttransplant recurrence and complement gene mutations. Am J Transplant 13:2201–2206
Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, Stoiber H, Dierich MP, Zimmerhackl LB, Wurzner R (2009) Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol 182:6394–6400
Poolpol K, Orth-Holler D, Speth C, Zipfel PF, Skerka C, de Cordoba SR, Brockmeyer J, Bielaszewska M, Wurzner R (2014) Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol Immunol 58:77–84
Ehrlenbach S, Rosales A, Posch W, Wilflingseder D, Hermann M, Brockmeyer J, Karch H, Satchell SC, Wurzner R, Orth-Holler D (2013) Shiga toxin 2 reduces complement inhibitor CD59 expression on human renal tubular epithelial and glomerular endothelial cells. Infect Immun 81:2678–2685
Dammermann W, Schipper P, Ullrich S, Fraedrich K, Schulze Zur Wiesch J, Frundt T, Tiegs G, Lohse A, Luth S (2013) Increased expression of complement regulators CD55 and CD59 on peripheral blood cells in patients with EAHEC O104:H4 infection. PLoS One 8:e74880
Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, Pagani C, Noris M, Gobbi M, Stravalaci M, Rottoli D, Tedesco F, Remuzzi G, Zoja C (2011) Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 187:172–180
Locatelli M, Buelli S, Pezzotta A, Corna D, Perico L, Tomasoni S, Rottoli D, Rizzo P, Conti D, Thurman JM, Remuzzi G, Zoja C, Morigi M (2014) Shiga toxin promotes podocyte injury in experimental Hemolytic uremic syndrome via activation of the alternative pathway of complement. J Am Soc Nephrol 25:1786–1798
Iwaki D, Kanno K, Takahashi M, Endo Y, Matsushita M, Fujita T (2011) The role of mannose-binding lectin-associated serine protease-3 in activation of the alternative complement pathway. J Immunol 187:3751–3758
Ozaki M, Kang Y, Tan YS, Pavlov VI, Liu B, Boyle DC, Kushak RI, Skjoedt MO, Grabowski EF, Taira Y, Stahl GL (2016) Human mannose-binding lectin inhibitor prevents Shiga toxin-induced renal injury. Kidney Int 90:774–782
Zoja C, Locatelli M, Pagani C, Corna D, Zanchi C, Isermann B, Remuzzi G, Conway EM, Noris M (2012) Lack of the lectin-like domain of thrombomodulin worsens Shiga toxin-associated hemolytic uremic syndrome in mice. J Immunol 189:3661–3668
Esmon CT (1995) Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J 9:946–955
Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, Zoja C, Remuzzi G, Conway EM (2009) Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med 361:345–357
Fernandez GC, Te Loo MW, van der Velden TJ, van der Heuvel LP, Palermo MS, Monnens LL (2003) Decrease of thrombomodulin contributes to the procoagulant state of endothelium in hemolytic uremic syndrome. Pediatr Nephrol 18:1066–1068
Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V (2005) Platelet activation leads to activation and propagation of the complement system. J Exp Med 201:871–879
Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y (1997) C5a induces tissue factor activity on endothelial cells. Thromb Haemost 77:394–398
Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A (1997) The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med 185:1619–1627
Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC (2003) Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J 17:1003–1014
Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R (2002) Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol 169:2102–2110
Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R (2001) Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int 60:957–968
Macconi D, Bonomelli M, Benigni A, Plati T, Sangalli F, Longaretti L, Conti S, Kawachi H, Hill P, Remuzzi G, Remuzzi A (2006) Pathophysiologic implications of reduced podocyte number in a rat model of progressive glomerular injury. Am J Pathol 168:42–54
Wiggins RC (2007) The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71:1205–1214
Hodgin JB, Bitzer M, Wickman L, Afshinnia F, Wang SQ, O'Connor C, Yang Y, Meadowbrooke C, Chowdhury M, Kikuchi M, Wiggins JE, Wiggins RC (2015) Glomerular aging and focal global glomerulosclerosis: a podometric perspective. J Am Soc Nephrol 26:3162–3178
Marshall CB, Shankland SJ (2007) Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol 106:e51–e59
Liapis H, Romagnani P, Anders HJ (2013) New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol 183:1364–1374
Habib R (1992) Pathology of the Hemolytic uremic syndrome. In: Kaplan BS, Trompeter RS, Moake JL (eds) Hemolytic uremic syndrome and thrombotic thrombocytopenic Purpura. Marcel Dekker, Inc., New York, pp 315–353
De Petris L, Patrick J, Christen E, Trachtman H (2006) Urinary podocyte mRNA excretion in children with D+HUS: a potential marker of long-term outcome. Ren Fail 28:475–482
Taylor FB Jr, Tesh VL, DeBault L, Li A, Chang AC, Kosanke SD, Pysher TJ, Siegler RL (1999) Characterization of the baboon responses to Shiga-like toxin: descriptive study of a new primate model of toxic responses to Stx-1. Am J Pathol 154:1285–1299
Ochoa F, Oltra G, Gerhardt E, Hermes R, Cohen L, Damiano AE, Ibarra C, Lago NR, Zotta E (2012) Microalbuminuria and early renal response to lethal dose Shiga toxin type 2 in rats. Int J Nephrol Renov Dis 5:29–36
Morigi M, Buelli S, Zanchi C, Longaretti L, Macconi D, Benigni A, Moioli D, Remuzzi G, Zoja C (2006) Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol 169:1965–1975
Hughes AK, Stricklett PK, Schmid D, Kohan DE (2000) Cytotoxic effect of Shiga toxin-1 on human glomerular epithelial cells. Kidney Int 57:2350–2359
Ergonul Z, Clayton F, Fogo AB, Kohan DE (2003) Shigatoxin-1 binding and receptor expression in human kidneys do not change with age. Pediatr Nephrol 18:246–253
Dettmar AK, Binder E, Greiner FR, Liebau MC, Kurschat CE, Jungraithmayr TC, Saleem MA, Schmitt CP, Feifel E, Orth-Holler D, Kemper MJ, Pepys M, Wurzner R, Oh J (2014) Protection of human podocytes from shiga toxin 2-induced phosphorylation of mitogen-activated protein kinases and apoptosis by human serum amyloid P component. Infect Immun 82:1872–1879
Hughes AK, Stricklett PK, Kohan DE (2001) Shiga toxin-1 regulation of cytokine production by human glomerular epithelial cells. Nephron 88:14–23
Morigi M, Locatelli M, Rota C, Buelli S, Corna D, Rizzo P, Abbate M, Conti D, Perico L, Longaretti L, Benigni A, Zoja C, Remuzzi G (2016) A previously unrecognized role of C3a in proteinuric progressive nephropathy. Sci Rep 6:28445
Li L, Chen L, Zang J, Tang X, Liu Y, Zhang J, Bai L, Yin Q, Lu Y, Cheng J, Fu P, Liu F (2015) C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/beta-catenin signaling pathway in diabetic kidney disease. Metabolism 64:597–610
Morgan BP, Harris CL (2015) Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov 14:857–877
Acknowledgements
The authors thank Professor Giuseppe Remuzzi for reviewing the manuscript and Dr. Antonella Piccinelli for helping in figure preparation. Dr. Kerstin Mierke and Manuela Passera helped with preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Zoja, C., Buelli, S. & Morigi, M. Shiga toxin triggers endothelial and podocyte injury: the role of complement activation. Pediatr Nephrol 34, 379–388 (2019). https://doi.org/10.1007/s00467-017-3850-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-017-3850-x