
Abstract
Mycophenolic acid (MPA) was introduced into clinical practice as immunosuppressive drug therapy to prevent allograft rejection. Since then, its clinical application has widened. Our goal was to review the lessons learned from experimental nontransplant glomerular disease models on the mechanisms of MPA therapy. T and B lymphocytes are preferentially dependent on de novo purine synthesis. By inhibiting the rate-limiting enzyme of de novo purine synthesis, MPA depletes the pool of deoxyguanosine triphosphate (dGTP) and inhibits proliferation of these immune cells. Furthermore, MPA can also induce apoptosis of immune cells and is known to inhibit synthesis of fucose- and mannose-containing membrane glycoproteins altering the surface expression and binding ability of adhesion molecules. However, MPA exerts a direct effect also on nonimmune cells. Mesangial cells are partially dependent on de novo purine biosynthesis and are thus susceptible to MPA treatment. Additionally, MPA can inhibit apoptosis in podocytes and seems to be beneficial in preserving the expression of nephrin and podocin, and by attenuation of urokinase receptor expression leads to decreased foot-process effacement. In summary, our manuscript sheds light on the molecular mechanisms underlying the antiproteinuric effect of MPA. Overall, MPA is an excellent treatment option in many immunologic glomerulopathies because it possesses immunosuppressive properties, has a remarkable effect on nonimmune cells and counteracts the proliferation of mesangial cells, expansion of mesangial matrix, and foot-process effacement of podocytes combined with a low systemic toxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mycophenolate mofetil (MMF) was the product of a program of drug development led by Nelson, Allison, and Eugui of Roche Laboratories [1, 2]. This orally administered pro-drug is hydrolyzed by esterases in the intestine and blood to release mycophenolic acid (MPA), a potent, selective, noncompetitive inhibitor of inosine monophosphate dehydrogenase (IMPDH).
MMF was introduced into clinical practice in 1993 as a complementary immunosuppressive drug to glucocorticoids, calcineurin inhibitors, and cell cycle inhibitors to prevent allograft rejection (Fig. 1). Since 1993, the clinical application of MMF has significantly widened and has become part of first-line therapy in lupus nephritis [3, 4]. MMF has proven not to be inferior to cyclophosphamide for both induction and maintenance therapy of class III, IV, or V lupus nephritis. Moreover, African American and Hispanic patients had a better response rate to MMF than to cyclophosphamide [5–7]. There is a vast body of literature in favor of MMF being an inherent part of treatment in complicated cases of pediatric idiopathic nephrotic syndrome. Kidney Disease: Improving Global Outcomes (KDIGO) guidelines suggests MMF as second-line medication in frequently relapsing nephrotic syndrome (FRNS) [8]. Gellerman et al. carried out a randomized, multicenter, open-label, crossover trial comparing MMF and cyclosporin A (CsA) therapy in 60 children with FRNS over a 2-year period [9]. MMF was associated with significantly more relapses per patient in the first year, but this effect was no longer seen in the second year of the trial, potentially due to a carryover effect from the first year. At the same time, the mean glomerular filtration rate was lower in patients receiving CsA and a post hoc analysis demonstrated exposure-dependent comparable efficacy of MMF and CsA. In line with that, other investigators have shown the efficacy of MMF not to be inferior compared with CsA [10, 11]. These encouraging results have been obtained mostly in minimal-change nephropathy in which MMF may help to reduce the use of steroids in patients who are often very young and are therefore more susceptible to steroid-associated side effects. The results obtained in patients with focal segmental glomerulonephritis (FSGN) and idiopathic membranous nephropathy were less promising [12–16]. The effectiveness of MMF in immunolobulin (Ig)A nephritis as well as Henoch–Schönlein purpura nephritis (HSPN) again is auspicious. In HSPN, MMF can be considered as an option for maintenance therapy [17, 18]. In terms of IgA nephritis, conflicting results have been published, but in combination with renin-angiotensin system (RAAS) inhibition, MMF seems to be superior to monotherapy with an RAAS inhibitor [19, 20]. Albeit its general effectiveness in primary glomerulonephritis is still under debate [7, 12], MMF shows promising results especially in proteinuric renal diseases with an underlying immunological etiology via its active compound, MPA.

Mechanism of action of different immunosuppressive drugs demonstrated in a T cell. In the treatment of glomerular diseases, the following immunosuppressive drugs can be administered (red): (1) Glucocorticoids act via nongenomic and genomic effects, e.g. by inhibiting transcription of genes that encode cytokines such as IL-2. (2) MPA is a noncompetitive inhibitor of the inosine monophosphate dehydrogenase, suppressing nucleotide synthesis. (3) Cyclophosphamide is an alkylating agent forming DNA crosslinks both between and within DNA strands, which leads to cell apoptosis. (4) Calcineurin inhibitors (cyclosporin A and tacrolimus) bind to cyclophilin and in this complex inhibit the phosphatase calcineurin and the transcription of IL-2, accordingly. (5) The monoclonal anti-CD20 antibody (rituximab) influences the function of B cells, leading to inactivation and apoptosis of these cells (not shown). The other immunosuppressive drugs shown are applied in transplant immunology (grey). MHC major histocompatibility complex, IL-2 interleukin-2, IL-2 R interleukin-2 receptor, TCR T-cell receptor, GCR glucocorticoid receptor, NFAT nuclear factor of activated T cells, mTOR mechanistic target of rapamycin, MPA mycophenolic acid
In the last two decades, a vast body of evidence has shown that this compound possesses effects not only on immune cells but also on different nonimmune cells, including glomerular cells, which makes MPA an attractive therapeutic choice in case of immune- as well as nonimmune-mediated glomerular diseases. The rational for use of MPA in glomerular diseases was thoroughly summarized years ago by Badid et al. in 2001 and by Zatz et al. in 2002 [21, 22]. However, since then, substantial new publications have emerged. Therefore, our goal was to review the growing evidence for direct effects of MPA on glomerular cells, shedding light on the potential underlying mechanisms of the clinically established MPA therapy in glomerular diseases.
Immune-mediated effects of mycophenolic acid
Inhibited proliferation of immune cells
MPA has a selective antiproliferative effect on lymphocytes. By inhibiting IMPDH, the rate-limiting enzyme of the de novo purine synthesis, MPA depletes the pool of deoxyguanosine triphosphate (dGTP) required for DNA synthesis. IMPDH occurs in two isoforms: types I and II. The type II isoform is nearly five times more susceptible to inhibition by MPA than the type I isoform. T and B lymphocytes possess only the type II isoform and are preferentially dependent on the de novo pathway of purine synthesis, making them an ideal target of the principal mechanism by which the immunosuppressive activity of MPA is exerted (Fig. 2). It is less well known, but important to note, that MPA significantly depletes GTP levels in monocytes also. This effect has been comprehensively summarized by Allison and Eugui [1, 2] and further confirmed by others in the last 20 years [23–25].

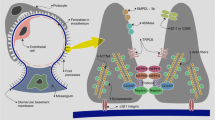
Potential mechanisms of action of mycophenolic acid (MPA) in glomerulopathies. MPA has several mechanisms of action, all of which may contribute in different degrees to treat glomerular diseases. MM mesangial matrix, MCP-1 monocyte chemoattractant protein 1, uPAR urokinase receptor
Facilitated apoptosis of immune cells
Although the principal mode of action of MPA on lymphocytes is cytostatic, it can also induce apoptosis of immune cells. Activated peripheral blood leukocytes showed 24 % apoptotic cells, while in the presence of 0.5 mM MPA, this was increased up to 67 %. A human T-lymphocytic cell line incubated with 1 mM MPA showed 82–98 % apoptotic cells as compared with 12 % in the absence of MPA [26]. Cohn et al. further examined the effects of MPA not only in a human T-lymphocytic cell line but also in two human monocytic precursor cell lines and observed MPA-induced apoptosis in each cell line. However, the incidence of apoptotic cells was lower in monocytic cell lines than in the lymphocytic line [27]. In concordance with these data, Andrikos et al. proved that therapeutic levels of MPA led to increased apoptosis rates of monocytic cells [28]. Moreover, the combination of CsA, steroid, and MMF increased the rate of apoptosis to as high as 95 %. Thus, immunosuppressive therapy can contribute to the high rate of apoptosis observed among mononuclear cells of transplanted patients. Takahashi et al. demonstrated that MPA is more effective in potentiating T-cell apoptosis compared with rapamycin and CsA [29]. Additionally, the Fas ligand, which belongs to the tumor necrosis factor family and induces apoptosis, accumulated during MPA therapy [30]. Thus, MPA action may not have a “guanine starvation” mechanism only but may also function via an active feedback control mechanism of cell proliferation.
Decreased recruitment of immune cells
It is accepted that MPA depletes pools of dGTP in human lymphocytes and monocytes, thereby inhibiting not only cell proliferation but also synthesis of fucose- and mannose-containing saccharide components of membrane glycoproteins. These are recognized by the family of adhesion molecules, which are involved in the interaction between lymphocytes and either endothelial or antigen-presenting cells or target cells. However, other studies concluded that MPA has no effect on the surface expression of adhesion molecules but alters the binding ability of these molecules. Nevertheless, MPA leads to impairment of lymphocyte and monocyte recruitment and adhesion to and penetration via endothelial cell surface. Thus, activation of these cells is highly disturbed and cytokine production reduced.
Allison et al. first published data using flow cytometry and showed that MPA depletes the terminal mannose content of different membrane glycoproteins, thus altering cell adhesion in activated human peripheral blood lymphocytes [1]. In accordance with that, MPA was shown to reduce adhesion of peripheral blood lymphocytes to human umbilical vein endothelial cells (HUVEC)-cultured monlayers by up to 70–80 % and T-lymphocyte penetration up to 50–60 % [31]. In another study, MPA prevented not only the accumulation of T-cell adhesion ligands, very late antigen 4 (VLA-4), and P-selectin glycoprotein ligand 1 (PSGL-1) receptor on the membranes of T-cell pseudopodia, but reduced the binding capacity of PSGL-1, VLA-4, and leukocyte-function-associated antigen 1 by 70 %, 50 %, and 30 %, respectively [32]. Beyond adhesion molecules, cytokines were also found to play a crucial role in the mechanism of action of MPA. Some authors demonstrated that the monocyte chemoattractant protein 1 (MCP-1), an important chemotactic factor for macrophages and lymphocytes, was strongly reduced by MPA treatment [33, 34]. Another molecule, osteopontin, which is involved in the accumulation of intrarenal macrophages, was also described as being highly down-regulated by MPA treatment [35]. Finally, because of suppression of macrophage activation, the production of various macrophage cytokines is also highly reduced [21, 24]. Accordingly, a great body of evidence shows that the infiltration of immune cells is prevented by MPA treatment in diabetic glomerulopathy [33, 36, 37], in the 5/6 ablation model [38–40], in spontaneously hypertensive rats [41], in a model of adriamycin-induced nephropathy followed by unilateral nephrectomy [42], in Heymann nephritis [43], and in rapidly progressive glomerulonephritis [35]. It can be concluded that MPA possesses potent infiltration-blocking properties.
Non-immune-mediated effects of mycophenolic acid
All results detailed above show that a major part of the clinically observed glomeruloprotective action of MPA is mediated through immune cells. However, there is data showing that MPA also exerts a direct effect on nonimmune cells, such as mesangial cells, endothelial cells, and podocytes.
Inhibited proliferation of mesangial cells
Activation and proliferation of mesangial cells is a key feature of many glomerular diseases caused by immunological or other mechanism of injury. It is also known that in mesangial cells, the salvage pathway for guanosine synthesis is less active than in other cell types in glomeruli such as, for example, endothelial cells. Nevertheless, mesangial cells are partially dependent on de novo purine biosynthesis and susceptible to MPA.
Cell proliferation is a highly regulated process that includes enhancing factors such as cyclins, cyclin-dependent kinases, and inhibitory proteins. Chiara et al. applied a model of anti-thy1-antigen nephritis and showed that MPA treatment during the early phase of mesangial cell proliferation causes a decrease in cyclin gene expression (in both B and D groups) and a parallel increase in levels of cell-cycle inhibitor proteins, such as p27 kip1 [44]. A direct effect of MPA on mesangial cell proliferation was further supported by experimental results. Ziswiler et al. and Hauser et al. demonstrated that in vitro, MPA can dose-dependently inhibit growth of cultured mesangial cells, leading to almost total arrest of cell proliferation. This effect seemed to be reversible, since administration of guanosine was able to prevent it [45, 46]. Furthermore, in an in vivo model of anti-thy1.1 nephritis, MPA decreased the bromodeoxyuridine-positive proliferating cells in the mesangium [45]. In summary, in vitro and in vivo results identify mesangial cells as being sensitive targets of MPA’s antiproliferative action, which is mediated by guanosine depletion.
Inhibition of mesangial matrix expansion
The response of mesangial cells to different challenges includes an excessive accumulation of matrix proteins, leading to fibrosis. Some studies clearly show that MPA has intrinsic antifibrotic properties. Namely, MPA exerts a direct inhibitory effect on the secretion of osteopontin in a platelet-derived growth factor (PDGF)-treated rat mesangial cell line in vitro [47]. Furthermore, MPA was found to counteract the stimulatory effect of fetal calf serum and transforming growth factor (TGF)-β on the production of fibronectin and collagen type I, which may be mediated at least in part by dGTP depletion [48]. In agreement with these results, collagen type III deposition in the 5/6 ablation model was also significantly reduced by MPA treatment [49]. In addition, Ziswiler et al. demonstrated a decrease in the expression of α-smooth muscle actin and an attenuated accumulation of extracellular matrix (ECM) in a nephritic animal model in case of MPA treatment [45]. However, the authors found that in cultured mesangial cells, MPA did not affect the expression of three major matrix proteins: fibronectin, and collagen types I and IV. Therefore, the authors concluded that the attenuation of matrix deposition in vivo appeared to be most likely the consequence of the reduced cell number rather than of a specific effect of MPA on ECM protein synthesis. In summary \in vivo data clearly show an antifibrotic effect of MPA, but to date, whether this reflects a direct inhibitory effect on production of cytokines and ECM proteins or rather an indirect response of the mesangium due to prevention of cell proliferation in the glomeruli has not been fully determined.
Maintenance of podocyte function
It is well known that nephrin, the podocyte transmembrane slit-diaphragm protein and podocin, the membrane-associated protein of band-7-stromatin family, are necessary for the proper function of the renal filtration barrier. Nakhoul et al. investigated an adriamycin-induced nephrotic model accompanied by severe disruption of slit diaphragm structure and consequent rapid and profound loss of nephrin and podocin. MPA treatment seemed to be beneficial and preserved the expression of nephrin and podocin. However, this favorable response did not reach a significant level [50]. In line with that, in a diabetic model, it was revealed that MPA is able to protect against the loss of nephrin [51]. Furthermore, Wu et al. demonstrated that glomeruli of diabetic rat express significantly less nephrin and podocin, which can be restored by MPA treatment. Furthermore, they described that macrophage recruitment and expression of interleukin (IL)-1 and tumor necrosis factor (TNF)-α was also ameliorated [36]. To note, Takano et al. found that the macrophage-derived cytokines IL-1 and TNF-α markedly suppressed activity of the nephrin gene promoter [52], thus one can postulate that in this experimental setting, less infiltration of macrophages prevented the phenotypic changes of podocytes and the consequent loss of nephrin. As a further aspect, MPA was found to inhibit podocyte depletion in the early stage of streptozotocin-induced diabetes mellitus, partly by inhibiting apoptosis via decreasing cleaved caspase-3 and bax expression and increasing bcl-2 expression [51, 53]. Furthermore, urokinase receptor (uPAR) has been recently identified to have a down-stream signal pathway in podocytes, leading to foot-process effacement and urinary protein loss via a mechanism that includes lipid-dependent activation of alphavbeta3 integrin [54]. MPA appeared to decrease uPAR messenger RNA (mRNA) expression and protein levels, leading to decreased foot-process effacement and proteinuria in lupus nephritis [55].
Effects on endothelial cells
Data on how expression of endothelial cell adhesion molecules is influenced by MPA are rather contradictory. Olejarz et al. reported that MPA significantly inhibits TNF-α-induced intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) mRNA, and protein expression in human aortic endothelial cells [56]. Additionally, Huang et al. proved that not only did inhibition of TNF-α induce expression of ICAM-1 but also a multifactorial effect of MPA on endothelial cells showing decreased angiogenesis of endothelial cells in a three-dimensional collagen gel culture system, reduced migration of endothelial cells, and proliferation inhibition of these cells [57]. While others could not confirm the reduction of ICAM-1 by MPA treatment, they found a diminished E- and P-selectin expression on endothelial cell membranes in the presence of MPA [31, 32, 58, 59]. Nonetheless, there are reports showing no effect of MPA on adhesion molecule expression [60–62]. So, further studies are warranted to delineate the effect of MPA on endothelial cells.
Effects on tubular epithelial cells
The effect of MPA on tubular epithelial cells deserves attention in order to understand the complexity of MPA mechanism of action. In the pathomechanism of ischemia–reperfusion (I/R) renal failure, which is mainly characterized by tubular injury, endothelin-1 (ET-1) plays a pivotal deteriorating role. When cultured rabbit proximal tubular epithelial cells (PTEC) were treated with different immunosuppressives, a slight to moderate decrease in ET-1 was observed following administration of MPA, while treatment with CsA or tacrolimus (Tac) resulted in a significant increase in ET-1 [63]. Another mechanism of tubular injury is exposure to high levels of proteinuria, which increases MCP-1 expression in tubular epithelial cells, thus facilitating the infiltration of immune cells. Shui et al. showed that this effect was prevented by MPA, which was partly dependent on the p38—mitogen activated protein kinase (MAPK) pathway [64]. Furthermore, Copland et al. demonstrated that MPA even influences epithelial mesenchymal transformation (EMT). MPA not only prevented a transforming-growth-factor-induced transition of human PTEC into myofibroblasts, but it markedly inhibited proliferation of PTEC and returned myofibroblasts to a PTEC morphology [65].
The beneficial effect of MPA on tubular epithelial cells was also described in vivo. In diabetic nephropathy, MPA has a protective function via suppression of the EMT [66]. Additionally, Iturbe et al. showed that angiotensin-II-induced tubulointerstitial injury, including tubular dilation and sloughing, was significantly reduced by MPA treatment, as were tubulointerstitial proliferative activity and T-cell infiltration and activation [67]. Finally, several publications emphasize the capability of MPA to inhibit expression of adhesion molecules on the surface of inflammatory cells and ultimately prevent their infiltration into the kidney. Accordingly, a significantly better tubulointerstitial score was seen in Fisher 344→Lewis rat transplant model, in anti-thy1-induced chronic–progressive glomerulosclerosis, in subtotal ablation, in long-term I/R injury, and in diabetic nephropathy [33, 68–71].
Taken together, the beneficial effects of MPA therapy extend beyond glomeruli to preventing accompanying tubular injury via suppressing the proteinuria-induced infiltration of immune cells in the tubulointerstitium and via inhibiting EMT.
Conclusion
Overall, MPA seems to be an excellent treatment option in many immunologic glomerular diseases. First MPA possesses widely accepted immunosuppressive properties. Moreover, it has a remarkable effect on nonimmune cells and ability to counteract with proliferation of mesangial cells, expansion of mesangial matrix, and foot-process effacement of podocytes, as discussed in this review (Fig. 2). In addition, it has a relatively low systemic toxicity. Therefore, future prospective randomized studies will need to examine the antiproteinuric value of MPA in glomerular diseases beyond established use in immunological renal disease, such as lupus nephritis.
References
Allison AC, Kowalski WJ, Muller CD, Eugui EM (1993) Mechanisms of action of mycophenolic acid. Ann N Y Acad Sci 696:63–87
Allison AC, Eugui EM (2000) Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 47:85–118
Rovin BH (2012) Lupus nephritis: guidelines for lupus nephritis–more recommendations than data? Nat Rev Nephrol 8:620–621
Conti F, Ceccarelli F, Perricone C, Massaro L, Cipriano E, Pacucci VA, Truglia S, Miranda F, Morello F, Alessandri C, Spinelli FR, Valesini G (2014) Mycophenolate mofetil in systemic lupus erythematosus: results from a retrospective study in a large monocentric cohort and review of the literature. Immunol Res 60:270–276
Punaro MG (2013) The treatment of systemic lupus proliferative nephritis. Pediatr Nephrol 28:2069–2078
Ortega LM, Schultz DR, Lenz O, Pardo V, Contreras GN (2010) Review: Lupus nephritis: pathologic features, epidemiology and a guide to therapeutic decisions. Lupus 19:557–574
Ostalska-Nowicka D, Malinska A, Silska M, Perek B, Zachwieja J, Nowicki M (2011) Mycophenolate mofetil (MMF) treatment efficacy in children with primary and secondary glomerulonephritis. Arch Med Sci D 7:1042–1048
Lombel RM, Gipson DS, Hodson EM (2013) Kidney Disease: Improving Global outcomes. Treatment of steroid-sensitive nephrotic syndrome: new guidelines from KDIGO. Pediatr Nephrol 28:415–426
Gellermann J, Weber L, Pape L, Tönshoff B, Hoyer P, Querfeld U, Gesellschaft für Pädiatrische Nephrologie (GPN) (2013) Mycophenolate mofetil versus cyclosporin A in children with frequently relapsing nephrotic syndrome. J Am Soc Nephrol 24:1689–1697
Larkins N, Kim S, Craig J, Hodson E (2016) Steroid-sensitive nephrotic syndrome: an evidence-based update of immunosuppressive treatment in children. Arch Dis Child 101:404–408
Hackl Á, Cseprekál O, Gessner M, Liebau MC, Habbig S, Ehren R, Müller C, Taylan C, Dötsch J, Weber LT (2016) Mycophenolate Mofetil Therapy in Children With Idiopathic Nephrotic Syndrome: Does Therapeutic Drug Monitoring Make a Difference? Ther Drug Monit 38:274–279
Sepe V, Libetta C, Giuliano MG, Adamo G, Dal Canton A (2008) Mycophenolate mofetil in primary glomerulopathies. Kidney Int 73:154–162
Paydas S, Kurt C, Taskapan H, Balal M, Sertdemir Y, Pembegul I (2009) The effect of mycophenolate mofetil on primary and secondary treatment of primary glomerulonephritis and lupus nephritis. Int Urol Nephrol 41:145–152
Chen Y, Schieppati A, Cai G, Chen X, Zamora J, Giuliano GA, Braun N, Perna A (2013) Immunosuppression for membranous nephropathy: a systematic review and meta-analysis of 36 clinical trials. Clin J Am Soc Nephrol 8:787–796
Chen Y, Schieppati A, Chen X, Cai G, Zamora J, Giuliano GA, Braun N, Perna A (2014) Immunosuppressive treatment for idiopathic membranous nephropathy in adults with nephrotic syndrome. Cochrane Database Syst Rev 10:CD004293
Tran TH, J Hughes G, Greenfeld C, Pham JT (2015) Overview of current and alternative therapies for idiopathic membranous nephropathy. Pharmacotherapy 35:396–411
Hahn D, Hodson EM, Willis NS, Craig JC (2015) Interventions for preventing and treating kidney disease in Henoch-Schönlein Purpura (HSP). Cochrane Database Syst Rev 8:CD005128
Pohl M, Dittrich K, Ehrich JHH, Hoppe B, Kemper MJ, Klaus G, Schmitt CP, Hoyer PF (2013) Behandlung der Purpura-Schönlein-Henoch-Nephritis bei Kindern und Jugendlichen Therapieempfehlungen der Gesellschaft für Pädiatrische Nephrologie (GPN). Monatsschr Kinderheilkd 161:543–553
Vecchio M, Bonerba B, Palmer SC, Craig JC, Ruospo M, Samuels JA, Molony DA, Schena FP, Strippoli GF (2015) Immunosuppressive agents for treating IgA nephropathy. Cochrane Database Syst Rev 8:CD003965
Chen Y, Li Y, Yang S, Li Y, Liang M (2014) Efficacy and safety of mycophenolate mofetil treatment in IgA nephropathy: a systematic review. BMC Nephrol 15:193
Badid C, Desmouliere A, Laville M (2001) Mycophenolate mofetil: implications for the treatment of glomerular disease. Nephrol Dial Transplant 16:1752–1756
Zatz R, Noronha IL, Fujihara CK (2002) Experimental and clinical rationale for use of MMF in nontransplant progressive nephropathies. Am J Physiol Renal Physiol 283:1167–1175
Azuma H, Binder J, Heemann U, Schmid C, Tullius SG, Tilney NL (1995) Effects of RS61443 on functional and morphological changes in chronically rejecting rat kidney allografts. Transplantation 59:460–466
Srinivas TR, Kaplan B, Meier-Kriesche HU (2003) Mycophenolate mofetil in solid-organ transplantation. Expert Opin Pharmacother 4:2325–2345
Staatz CE, Tett SE (2014) Pharmacology and toxicology of mycophenolate in organ transplant recipients: an update. Arch Toxicol 88:1351–1389
Cohn RG, Mirkovich A, Caulfield J, Eugui EM (1999) Apoptosis of human activated peripheral T-cells and Tlymphocytic and promonocytic cell lines induced by mycophe nolic acid, the active metabolite of CellCept. In: The Sixth Basic Sciences Symposium of the Transplantation Society, Monterey, CA. p 173
Cohn RG, Mirkovich A, Dunlap B, Burton F, Chiu SH, Dugui E, Caulfield JP (1996) Mycophenolic acid increases apoptosis, lysosomes and lipid droplets in human lymphoid and monocytic cell lines. Transplantation 68:411–418
Andrikos E, Yavuz A, Bordoni V, Ratanarat R, De Cal M, Bonello M, Salvatori G, Levin N, Yakupoglu G, Pappas M, Ronco C (2005) Effect of cyclosporine, mycophenolate mofetil, and their combination with steroids on apoptosis in a human cultured monocytic U937 cell line. Transplant Proc 37:3226–3229
Takahashi K, Reynolds M, Ogawa N, Longo DL, Burdick J (2004) Augmentation of T-cell apoptosis by immunosuppressive agents. Clin Transplant 12:72–75
Nakamura M, Ogawa N, Shalabi A, Maley WR, Longo D, Burdick JF (2001) Positive effect on T-cell regulatory apoptosis by mycophenolate mofetil. Clin Transplant 6:36–40
Blaheta RA, Leckel K, Wittig B, Zenker D, Oppermann E, Harder S, Scholz M, Weber S, Encke A, Markus BH (1999) Mycophenolate mofetil impairs transendothelial migration of allogeneic CD4 and CD8 T-cells. Transplant Proc 31:1250–1252
Blaheta RA, Leckel K, Wittig B, Zenker D, Oppermann E, Harder S, Scholz M, Weber S, Schuldes H, Encke A, Markus BH (1998) Inhibition of endothelial receptor expression and of T-cell ligand activity by mycophenolate mofetil. Transpl Immunol 6:251–259
Wu YG, Lin H, Qian H, Zhao M, Qi XM, Wu GZ, Lin ST (2006) Renoprotective effects of combination of angiotensin converting enzyme inhibitor with mycophenolate mofetil in diabetic rats. Inflamm Res 55:192–199
Chen FQ, Wang QY, Wei GZ, Ma XY, Ma DW, Deng WW, Sun WB (2014) Effects of mycophenolate mofetil on the expression of monocyte chemoattractant protein-1 and fibronectin in high glucose cultured human mesangial cells. Genet Mol Res 13:3154–3161
Takeda S, Takahashi M, Sado Y, Takeuchi K, Hakamata Y, Shimizu H, Kaneko T, Yamamoto H, Ito C, Ookawara S, Asano Y, Kusano E, Kobayashi E (2004) Prevention of glomerular crescent formation in glomerulonephritis by mycophenolate mofetil in rats. Nephrol Dial Transplant 19:2228–2236
Wu Y, Dong J, Yuan L, Liang C, Ren K, Zhang W, Fang F, Shen J (2008) Nephrin and podocin loss is prevented by mycophenolate mofetil in early experimental diabetic nephropathy. Cytokine 44:85–91
Utimura R, Fujihara CK, Mattar AL, Malheiros DM, Noronha IL, Zatz R (2003) Mycophenolate mofetil prevents the development of glomerular injury in experimental diabetes. Kidney Int 63:209–216
Fujihara CK, Malheiros DM, Zatz R, Noronha IL (1998) Mycophenolate mofetil attenuates renal injury in the rat remnant kidney. Kidney Int 54:1510–1519
Romero F, Rodríguez-Iturbe B, Parra G, González L, Herrera-Acosta J, Tapia E (1999) Mycophenolate mofetil prevents the progressive renal failure induced by 5/6 renal ablation in rats. Kidney Int 55:945–955
Remuzzi G, Zoja C, Gagliardini E, Corna D, Abbate M, Benigni A (1999) Combining an antiproteinuric approach with mycophenolate mofetil fully suppresses progressive nephropathy of experimental animals. J Am Soc Nephrol 10:1542–1549
Rodríguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chávez M, Herrera-Acosta J, Johnson RJ, Pons HA (2002) Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol 282:191–201
Van den Branden C, Ceyssens B, Pauwels M, Van Wichelen G, Heirman I, Jie N, Verbeelen D (2003) Effect of mycophenolate mofetil on glomerulosclerosis and renal oxidative stress in rats. Nephron Exp Nephrol 95:93–99
Penny MJ, Boyd RA, Hall BM (1998) Mycophenolate mofetil prevents the induction of active Heymann nephritis: association with Th2 cytokine inhibition. J Am Soc Nephrol 9:2272–2282
Chiara M, Menegatti E, Di Simone D, Davit A, Bellis D, Sferch D, De Rosa G, Giachino O, Sena LM, Roccatello D (2005) Mycophenolate mofetil and roscovitine decrease cyclin expression and increase p27(kip1) expression in anti Thy1 mesangial proliferative nephritis. Clin Exp Immunol 139:225–235
Ziswiler R, Steinmann-Niggli K, Kappeler A, Daniel C, Marti HP (1998) Mycophenolic acid: a new approach to the therapy of experimental mesangial proliferative glomerulonephritis. J Am Soc Nephrol 9:2055–2066
Hauser IA, Renders L, Radeke HH, Sterzel RB, Goppelt-Struebe M (1999) Mycophenolate mofetil inhibits rat and human mesangial cell proliferation by guanosine depletion. Nephrol Dial Transplant 14:58–63
Wang W, Mo S, Chan L (1999) Mycophenolic acid inhibits PDGF-induced osteopontin expression in rat mesangial cells. Transplant Proc 31:1176–1177
Dubus I, Vendrely B, Christophe I, Labouyrie JP, Delmas Y, Bonnet J, Combe C (2002) Mycophenolic acid antagonizes the activation of cultured human mesangial cells. Kidney Int 62:857–867
Badid C, Vincent M, McGregor B, Melin M, Hadj-Aissa A, Veysseyre C, Hartmann DJ, Desmouliere A, Laville M (2000) Mycophenolate mofetil reduces myofibroblast infiltration and collagen III deposition in rat remnant kidney. Kidney Int 58:51–61
Nakhoul F, Ramadan R, Khankin E, Yaccob A, Kositch Z, Lewin M, Assady S, Abassi Z (2005) Glomerular abundance of nephrin and podocin in experimental nephrotic syndrome: different effects of antiproteinuric therapies. Am J Physiol Renal Physiol 289:880–890
Lv W, Lou J, Zhang Y, Lian P, Qi D, Wang J (2015) Mycophenolate mofetil inhibits hypertrophy and apoptosis of podocyte in vivo and in vitro. Int J Clin Exp Med 8:19781–19790
Takano Y, Yamauchi K, Hayakawa K, Hiramatsu N, Kasai A, Okamura M, Yokouchi M, Shitamura A, Yao J, Kitamura M (2007) Transcriptional suppression of nephrin in podocytes by macrophages: roles of inflammatory cytokines and involvement of the PI3K/Akt pathway. FEBS Lett 581:421–426
Lv W, Zhang Y, Guan G, Li P, Wang J, Qi D (2013) Mycophenolate mofetil and valsartan inhibit podocyte apoptosis in streptozotocin-induced diabetic rats. Pharmacology 92:227–234
Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, Cowan P, Kretzler M, Parrilla R, Bendayan M, Gupta V, Nikolic B, Kalluri R, Carmeliet P, Mundel P, Reiser J (2008) Modification of kidney barrier function by the urokinase receptor. Nat Med 14:55–63
Cheng CC, Lee YF, Lan JL, Wu MJ, Hsieh TY, Lin NN, Wang JM, Chiu YT (2013) Mycophenolate mofetil alleviates lupus nephritis through urokinase receptor signaling in a mice model. Lupus 22:554–561
Olejarz W, Bryk D, Zapolska-Downar D, Małecki M, Stachurska A, Sitkiewicz D (2014) Mycophenolic acid attenuates the tumour necrosis factor-α-mediated proinflammatory response in endothelial cells by blocking the MAPK/NF-kB and ROS pathways. Eur J Clin Invest 44:54–64
Huang Y, Liu Z, Huang H, Liu H, Li L (2005) Effects of mycophenolic acid on endothelial cells. Int Immunopharmacol 5:1029–1039
Bertalanffy P, Dubsky P, Wolner E, Weigel G (1999) Alterations of endothelial nucleotide levels by mycophenolic acid result in changes of membrane glycosylation and E-selectin expression. Clin Chem Lab Med 37:259–264
Raab M, Daxecker H, Karimi A, Markovic S, Cichna M, Markl P, Müller MM (2001) In vitro effects of mycophenolic acid on the nucleotide pool and on the expression of adhesion molecules of human umbilical vein endothelial cells. Clin Chim Acta 310:89–98
Hauser IA, Johnson DR, Thévenod F, Goppelt-Strübe M (1997) Effect of mycophenolic acid on TNF alpha-induced expression of cell adhesion molecules in human venous endothelial cells in vitro. Br J Pharmacol 122:1315–1322
Weigel G, Bertalanffy P, Dubsky P, Griesmacher A, Wolner E (1999) Mycophenolic acid influences T helper 2 (Th2) cytokine induced expression of intercellular cell adhesion molecule-1 (ICAM-1) on human endothelial cells. Clin Chem Lab Med 37:253–257
Weigel G, Bertalanffy P, Wolner E (2002) Depletion of intracellular GTP results in nuclear factor-kappaB activation and intercellular adhesion molecule-1 expression in human endothelial cells. Mol Pharmacol 62:453–462
Haug C, Schmid-Kotsas A, Linder T, Jehle PM, Bachem MG, Gruenert A, Rozdzinski E (2002) The immunosuppressive drug mycophenolic acid reduces endothelin-1 synthesis in endothelial cells and renal epithelial cells. Clin Sci 48:76S–80S
Shui H, Gao P, Si X, Ding G (2010) Mycophenolic acid inhibits albumin-induced MCP-1 expression in renal tubular epithelial cells through the p38 MAPK pathway. Mol Biol Rep 37:1749–1754
Copeland JW, Beaumont BW, Merrilees MJ, Pilmore HL (2007) Epithelial-to-mesenchymal transition of human proximal tubular epithelial cells: effects of rapamycin, mycophenolate, cyclosporin, azathioprine, and methylprednisolone. Transplantation 83:809–814
Xiao X, Wang J, Chang X, Zhen J, Zhou G, Hu Z (2015) Mycophenolate mofetil ameliorates diabetic nephropathy through epithelial mesenchymal transition in rats. Mol Med Rep 12:4043–4050
Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gómez-Garre D, Largo R, Egido J, Johnson RJ (2001) Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59:2222–2232
Noris M, Azzollini N, Pezzotta A, Mister M, Benigni A, Marchetti G, Gagliardini E, Perico N, Remuzzi G (2001) Combined treatment with mycophenolate mofetil and an angiotensin II receptor antagonist fully protects from chronic rejection in a rat model of renal allograft. J Am Soc Nephrol 12:1937–1946
Krämer S, Loof T, Martini S, Rückert M, Wang Y, Böhler T, Shimizu F, Kawachi H, Neumayer HH, Peters H (2005) Mycophenolate mofetil slows progression in anti-thy1-induced chronic renal fibrosis but is not additive to a high dose of enalapril. Am J Physiol Renal Physiol 289:359–368
Tapia E, Franco M, Sánchez-Lozada LG, Soto V, Avila-Casado C, Santamaría J, Quiroz Y, Rodríguez-Iturbe B, Herrera-Acosta J (2003) Mycophenolate mofetil prevents arteriolopathy and renal injury in subtotal ablation despite persistent hypertension. Kidney Int 63:994–1002
Yang B, Jain S, Pawluczyk IZ, Imtiaz S, Bowley L, Ashra SY, Nicholson L (2005) Inflammation and caspase activation in long-term renal ischemia/reperfusion injury and immunosuppression in rats. Kidney Int 68:2050–2067
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
LTW has received unrestricted scientific grants from Novartis Pharma GmbH and Roche Pharma AG.
AH and RE declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Hackl, A., Ehren, R. & Weber, L.T. Effect of mycophenolic acid in experimental, nontransplant glomerular diseases: new mechanisms beyond immune cells. Pediatr Nephrol 32, 1315–1322 (2017). https://doi.org/10.1007/s00467-016-3437-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-016-3437-y