
Abstract
Mutations in HNF1B, the gene encoding hepatocyte nuclear factor 1β are the most commonly identified genetic cause of renal malformations. HNF1B was first identified as a disease gene for diabetes (MODY5) in 1997, and its involvement in renal disease was subsequently noted through clinical observations in pedigrees affected by MODY5. Since then, a whole spectrum of associated phenotypes have been reported, including genital malformations, autism, epilepsy, gout, hypomagnesaemia, primary hyperparathyroidism, liver and intestinal abnormalities and a rare form of kidney cancer. The most commonly identified mutation, in approximately 50 % of patients, is an entire gene deletion occurring in the context of a 17q12 chromosomal microdeletion that also includes several other genes. Some of the associated phenotypes, especially the neurologic ones, appear to occur only in the context of this microdeletion and thus may not be directly linked to HNF1B. Here we review the spectrum of associated phenotypes and discuss potential implications for clinical management.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
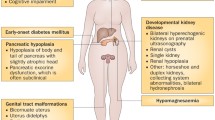
Hepatocyte nuclear factor 1β (HNF1B) is a member of the homeodomain-containing family of transcription factors and is encoded by the HNF1B gene located on chromosome 17. Heterozygous mutations in the encoding gene are the most commonly identified genetic cause of renal malformations [1–3]. Since the gene’s discovery, an ever-expanding spectrum of associated phenotypes has been reported, as well as an enormous variability of disease severity even within the same family. Moreover, there is an increasing body of knowledge about the role of HNF1B in organ development and maintenance. In this review, we briefly discuss the biological roles of HNF1B but concentrate on the associated phenotypic spectrum and discuss potential implications for managing patients with HNF1B mutations. An overview of renal and extrarenal HNF1B-associated features is as follows:
Renal malformations
-
Bilateral hyperechogenic kidneys on antenatal ultrasound
-
Renal cysts
-
Multicystic dysplastic kidney
-
Solitary kidney
-
Horseshoe kidney
-
Familial hypoplastic glomerulocystic kidney disease
-
Oligomeganephronia
-
Hydronephrosis and hydroureter
-
Tubular dysfunction
-
Hyperuricaemic nephropathy with gout
-
Renal magnesium wasting
-
-
Chromophobe renal carcinoma
Extrarenal features
-
Pancreas
-
Early-onset diabetes
-
New-onset diabetes after transplantation
-
Exocrine dysfunction
-
Agenesis of pancreatic body and tail
-
-
Female genitalia
-
Bicornate uterus
-
Uterus didelphys
-
Double vagina
-
Vaginal hypoplasia
-
Absent uterus
-
-
Male genitalia
-
Cryptorchidism
-
Hypospadia
-
Epididymal cyst
-
Agenesis of the vas deferens
-
-
Liver
-
Elevated liver enzymes
-
Neonatal cholestasis
-
-
Other
-
Primary hyperparathyroidism
-
Gout
-
Sensorineural deafness (reported only once)
-
-
Associated with 17q12 rearrangement
-
Mayer-Rokitansky-Küster-Hauser syndrome
-
Epilepsy
-
Lipodystrophy (reported only once)
-
Autism spectrum disorder
-
Developmental delay
-
Oesophageal malformations
-
History
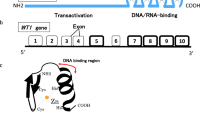
An important transcription factor in the liver was first identified in the 1980s by various groups and named HNF1 [4]. Subsequently, it was noted that this transcription factor was a homo- or heterodimer of two closely related proteins, then called HNF-1α and -1β, now HNF1A and HNF1B in new nomenclature [5]. Confusingly, HNF1B is sometimes also referred to as TCF2, although this term has been abandoned (http://www.genenames.org/cgi-bin/gene_symbol_report?hgnc_id=11630). The protein contains three functional domains important for dimerisation, DNA binding and transactivation [6].
After the discovery of HNF1A as a disease gene in maturity-onset diabetes in the young type 3 (MODY3) [7], HNF1B was an obvious candidate gene, and indeed, HNF1B-associated disease was shortly thereafter described in a Japanese family as MODY5 [8]. Interestingly, all affected family members had nondiabetic renal disease as part of their phenotype, and subsequently, other families were identified with MODY5 and renal cysts, leading to the term renal cysts and diabetes syndrome [9–12].
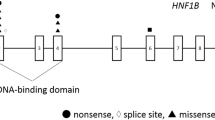
With increased use of genetic testing for HNF1B mutations, the associated phenotypic spectrum has increasingly expanded to include pancreatic, renal, genital, liver, intestinal and neurological abnormalities, some of which can be isolated [13] [14] or occur in the context of multiorgan involvement [15]. In 2005, entire gene deletions were identified as the most common mutation involving HNF1B [16], which occur in the context of a chromosomal microdeletion at 17q12, involving a further 14 genes [17, 18].
Functional studies in animals
Homozygous deletion of Hnf1b in mice is embryonically lethal due to visceral endoderm defects, whereas heterozygous deletion, unlike in humans, is associated with no phenotype [19, 20]. Consequently, studies in mice rely on sophisticated transgenic models that either rescue visceral expression during development or that allow conditional knockout in specific organs and/or developmental stages [21]. These have demonstrated a critical role for Hnf1b in pancreas development, providing an explanation for the diabetes and occasional exocrine impairment observed in humans [22]. During early kidney development, the absence of Hnf1b leads to impaired cross-talk between the ureteric bud and the metanephric mesenchyme, leading to defective ureteric-bud branching and the absence of mesenchymal to epithelial transition [23]. In contrast, if Hnf1b is deleted at a later time point, when tubules have already formed but are still elongating, a polycystic phenotype is observed [24]. Specific deletion of Hnf1b in the metanephric mesenchyme alone leads to complete impairment in tubular expansion and differentiation during nephron tubular development. However, nephron precursors are able to form glomerular structures, resulting in the formation of aberrant nephrons characterised by glomeruli with dilated Bowman’s (urinary) space without their normal tubular segments, which may reflect glomerular cysts commonly observed in human patients [25].
Clinical phenotype
Mutations in HNF1B are inherited in an autosomal dominant pattern, although up to 50 % of mutations occur de novo [26, 27], explaining the absence of a family history in many patients. The spectrum of severity can vary enormously, ranging from isolated MODY [13] or kidney involvement [14] to multiorgan disease [15, 28]. Since HNF1B is expressed in many organs, predominantly liver, intestine, pancreas, kidney and the urogenital tract, the occurrence of phenotypic abnormalities in these organs are perhaps not surprising with HNF1B mutations.
Kidneys
The first manifestation of HNF1B-related disease typically occurs in the kidneys. Mutations in this gene are found in up to 30 % of children with renal abnormalities, depending on cohort selection [14, 26, 29–32]. Some of these features can be identified by antenatal ultrasound, and the most frequent antenatal presentation is bilateral hyperechogenic kidneys with normal or increased size. This hyperechogenicity is diagnosed after 17 weeks of pregnancy when the kidney appears more echogenic than the liver or spleen. This can be secondary to multiple microscopic cysts, renal dysplasia or tubular dilation. Other causes of fetal hyperechogenic kidneys include autosomal recessive polycystic kidney disease (ARPKD), autosomal dominant polycystic kidney disease (ADPKD) and cystic dysplasia [33]. Recognising HNF1B-associated disease as a cause of echogenic foetal kidneys is important, as the common assumption of ARPKD as the typical cause may lead to misinformation during antenatal counselling [14]. Family history and the presence of associated extrarenal abnormalities can improve diagnostic accuracy and direct genetic testing.
The majority of neonates with HNF1B-associated kidney disease exhibits renal cysts with normal or small kidneys. Cysts can derive from all nephron segments, frequently include glomeruli and are usually small. Other renal manifestations include multicystic kidney dysplasia, familial hypoplastic glomerulocystic kidney disease, solitary kidney [34], oligomeganephronia [10], hyperuricaemic nephropathy with gout [28, 35], renal magnesium wasting [14], horseshoe kidney [27, 31] and hydronephrosis and hydroureter [14] (see also the subheading "Renal malformations").
End-stage kidney disease
Whilst renal malformations appear to be the most common manifestation of HNF1B mutations, progression to end-stage kidney disease (ESKD) seems rare, at least in childhood. In our own cohort of now 38 HNF1B mutation carriers, only two progressed to chronic kidney disease (CKD) stage 5 during childhood, with renal replacement therapy (RRT) commenced at the age of 3 and 13 years, respectively. In another larger cohort of 71 live births, only one was reported to have reached ESKD (age 3 months) [29].
Tubular dysfunction
The identification of tubular abnormalities associated with HNF1B mutations was surprising, as it implied that this transcription factor is not only important for renal development but also for the maintenance of functional tubules [14].
Uric acid
Abnormalities in uric acid handling were first reported in 2003, when genetic analysis of a kindred diagnosed with familial juvenile hyperuricaemic nephropathy (FJHN) surprisingly showed that the disease cosegregated with a splice-site mutation in HNF1B [35]. Subsequent, analysis of serum uric acid levels in patients with HNF1B mutations, compared with those with kidney disease or MODY without HNF1B mutation, demonstrated significantly higher levels in HNF1B mutation carriers. The finding of elevated uric acid levels in HNF1B mutation carriers has not been systematically analysed in other cohorts but was often associated [14, 29]. The cause is not clear, but kidney-specific deletion of Hnf1b in mice is associated with reduced expression of UMOD, the gene first identified to underlie FJHN, and HNF1B has been shown to bind to the promoter of this gene [24].
Magnesium
We first reported the association of hypomagnesaemia with HNF1B mutations in 2009 after investigations performed in two siblings with renal malformation and marked hypomagnesaemia [14]. Further analysis of our entire cohort showed significantly reduced plasma magnesium values in HNF1B-mutation-positive patients compared with those with renal malformations from other causes. Increased urinary losses of magnesium suggested a renal leak, and subsequent in silico and in vitro analysis showed HNF1B binding to the promoter of FXYD2, a subunit of the Na+-K+-ATPase, previously associated with autosomal dominant hypomagnesaemia [36]. Consistent with this role of HNF1B in magnesium transport in the distal convoluted tubule, mutations in PCBD1, a dimerization cofactor for HNF1B, were recently also associated with renal magnesium wasting, as well as with diabetes [37].
Extrarenal manifestations
Diabetes
MODY is an autosomal dominant form of noninsulin diabetes mellitus secondary to a dysfunction in pancreatic β cells, with onset typically before the age of 25 years [38]. After the discovery of HNF1A mutation as common genetic cause [7], the highly homologous HNF1B was an obvious candidate, and mutations were soon identified [8]. Despite its initial identification as a diabetes disease gene, HNF1B is a rare cause of MODY, accounting for <2 % of cases, compared with ∼60 % attributed to HNF1A [39].
Consistent with an important role of HNF1B in pancreatic development, pancreatic malformations have been described in mutation carriers [40, 41], as well as in animal models [42]. HNF1B mutations are not usually associated with diabetes in childhood: although single cases with onset as early as the neonatal age have been described [27, 43], diabetes typically manifests in the third or fourth decade of life and is seen in approximately half of adults with HNF1B mutations [15]. In our own cohort, diabetes occurred in approximately a quarter of patients during childhood, with age at onset between 10 and 14 years [14]. In another larger cohort of 75 patients with HNF1B mutations, only one developed diabetes before the age of 18 years [29].
The report of new-onset diabetes after transplantation (NODAT) in a patient with an HNF1B mutation has raised the question of whether HNF1B mutation carriers are at higher risk for this complication and whether transplant management should be modified [44, 45].
Exocrine pancreas dysfunction
Consistent with the reported pancreatic malformations, not only endocrine dysfunction in the form of diabetes but also exocrine abnormalities can be observed. When checked carefully, subclinical dysfunction in the form of reduced bicarbonate and enzyme secretion can be found in mutation carriers compared with controls [46].
Female genitalia
HNF1B has a high expression in the Müllerian duct and thus in the developing female genitalia but also in the mature endometrium and fallopian tube. Consistent with this, abnormalities of female genitalia are a common finding in HNF1B mutation carriers. In a gynaecologic cohort of 108 females with congenital uterine abnormalities, mutations in HNF1B were identified in nine [47]. Interestingly, these were all associated with renal abnormalities and never occurred in isolation. Observed phenotypes include abnormalities from incomplete fusion of the Müllerian ducts, such as bicornate uterus, uterus didelphys and double vagina [34]. Other observed phenotypes are absent uterus and vaginal hypoplasia [48].
Male genitalia
In contrast to female genital abnormalities, abnormal male genitalia are rarely associated with HNF1B mutations. Observed phenotypes in single cases include cryptorchidism, hypospadia, epididymal cysts and agenesis of the vas deferens [34, 48, 49].
Liver abnormalities
Despite the prominent expression of HNF1B in the liver, mutations in HNF1B are not associated with severe liver abnormalities. Mild elevation of liver enzymes has been reported recurrently [48, 49], and there are isolated case reports of neonatal cholestasis with reduced number of bile ducts on biopsy [50, 51]. No further liver abnormalities have been reported.
Primary hyperparathyroidism
In a recent report of a single-centre cohort of ten HNF1B mutation carriers with available parathyroid hormone (PTH) plasma levels, seven had CKD stage <3 [glomerular filtration rate (GFR) > 60 ml/min/1.73 m2 based on either estimated or measured creatinine clearances], yet elevated PTH levels (6.6–16.3 pmol/l, normal <6.5) [52]. One of these patients had a diagnosis of primary hyperparathyroidism with parathyroidectomy at age 23 years, several years before the HNF1B mutation was identified. The group subsequently identified an HNF1B-responsive element in the PTH promoter and demonstrated repression of PTH transcription by HNF1B [52]. The elevated PTH levels were made more remarkable by the fact that six of these seven patients had concomitant hypomagnesaemia (0.41–0.64 mmol/l): hypomagnesaemia is usually associated with hypoparathyroidism, as magnesium is an important cofactor for the calcium-sensing receptor to initiate PTH release [53].
Sensorineural deafness
We previously reported sensorineural deafness in two siblings with a splice-site mutation in HNF1B [28]. However, there are no further reports of this phenotype, and thus it may be coincidental. Careful screening of a larger number of mutation carriers is needed to further assess an association.
Chromophobe renal carcinoma
Exceptionally, the combination of monoallelic germline and somatic mutations has been described in a patient with chromophobe renal carcinoma [54]. This patient inherited a heterozygote nonsense mutation in HNF1B from her mother, and after tumor resection, a second somatic mutation was identified in form of a 17q12 deletion (see below). So far, this is the only report of this cancer occurring in an HNF1B mutation carrier.
Extrarenal manifestations associated with 17q12 rearrangements
In addition to the above phenotypes, some abnormalities are reported only in association with a 17q12 microdeletion. Initial reports assayed the HNF1B deletion only [16, 55], but a subsequent study suggests that all patients with an entire HNF1B deletion have, in fact, a common 17q12 microdeletion [18]. A closer investigation of this chromosomal region identified segmental duplications, which make it susceptible to chromosomal rearrangements such as deletion or duplication by nonallelic homologous recombination [17]. These duplications flank a region between 1.3 and 1.7 Mb, which—despite the size difference—contains the same 15 genes, one being HNF1B [18]. Therefore it is not clear to what degree HNF1B, rather than the other 14 genes, actually contributes to the associated phenotypes. However, given that deletions make up ∼50 % of identified HNF1B mutations, it is important to be aware of these potential complications.
Mayer-Rokitansky-Küster-Hauser syndrome
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome refers to the constellation of congenital aplasia of the uterus and upper part of the vagina due to anomalous development of Müllerian ducts, often associated with other congenital malformations (OMIM %277000). Given the above-mentioned complications from incomplete fusion of the Müllerian ducts observed in HNF1B mutation carriers, it is easily conceivable that the complete absence of uterus and upper vagina may also be related, especially when associated with renal malformations. However, genetic studies so far have only associated this syndrome with chromosomal microdeletions, including the 17q12 deletion [56, 57].
Epilepsy
There are only isolated case reports of seizures associated with this deletion. In a detailed study of four patients with the deletion, two had early-onset seizures [58]. Interestingly, in one of them, there was a family history of seizures in the mother, who was found not to carry the deletion, suggesting a potential other cause for the seizures. In a separate report of a large cohort of 3812 patients with epilepsy investigated for genomic copy-number variations, only one patient was found to have the 17q12 deletion [59]. Overall, there is no strong association between epilepsy and this deletion. Interestingly, a duplication of this identical region at chromosome 17q12 appears to be enriched in patients with epilepsy and yet is not usually associated with renal disease [17, 58]. It has been speculated that LHX1, a gene in this region encoding a transcription factor important for brain development, could be responsible for central nervous system complications [58].
Lipodsytrophy
This phenotype has so far been reported in only one patient with mild renal disease (CKD stage 1) and epilepsy [59]. It was speculated that the gene ACACA, which is included in the deletion and encodes a key enzyme in hepatic fatty acid synthesis, might be responsible. In our own cohort, we failed to identify an association with body mass index (BMI) with the 17q12 deletion, but there are, of course, many confounders, including CKD.
Autism and developmental delay
Autism and developmental delay have been recurrently reported with the 17q12 deletion but mostly in cohorts assembled by neurologists, i.e. patients presenting with these problems [18]. In renal cohorts, these complications have been reported only rarely, which may, of course reflect failure to ascertain this phenotype by nephrologists [60]. However, in a cohort of 39 children with HNF1B-related renal disease (26 with the 17.12 deletion and 13 with a HNF1B point mutation), no severe developmental delay was found, but one 17q12 deletion carrier had autistic spectrum disorder. Whilst overall IQ was lower in the deletion group, the difference was not statistically significant [18].
Oesophageal malformation
Whilst most reports found no renal manifestations in patients with a 17q12 duplication [17, 58], there is a case report of a man affected by renal hypoplasia with ESKD by the age of 27 years. His son was affected by a left cystic and hypodysplastic kidney with megaureter, as well as right low-grade vesicoureteric reflux and bladder diverticuli [61]. In both, a 17q12 duplication was identified; the son also suffered from oesophageal atresia. Oesophageal atresia had been previously reported in a patient with the 17q12 duplication (but without associated renal involvement) [58]. In addition, a missense mutation in HNF1B had been reported in a patient with VACTERL association, including a tracheoesophageal fistula [62]. However, in nine additional patients with the combination of oesophageal atresia and renal malformation, no abnormality in HNF1B could be identified [61].
Clinical consequences
Selecting patients for HNF1B mutation screening
The increasing knowledge of the spectrum of HNF1B-associated phenotypes has recently led to the development of algorithms to predict the likelihood of an HNF1B mutation in a patient with renal malformation. The highest likelihood (41 %) of HNF1B mutation identification is in patients with both MODY and renal malformations, but due to the onset of MODY typically occurring in adulthood, this form of diagnosis is not very practical in paediatric patients [63]. Recently, a more complicated score was developed based on 17 parameters, with renal hyperechogenicity and/or cysts, genital and pancreatic abnormalities scoring highest, followed by other features, such as abnormal fetal renal ultrasound, positive family history, renal hypo- or dysplasia and hypomagnesaemia [64]. Using this score, the authors achieved a sensitivity of 98.2 % and a specificity of 41.1 %. Perhaps not all clinicians will use this scoring system prior to requesting HNF1B mutation testing, but it provides a useful overview of the most common associated features.
Screening patients for potential extrarenal manifestations
No published guidelines exist on screening for potential associated extrarenal abnormalities in HNF1B mutation carriers. Thus, the following statements are based on the authors’ personal opinions:
-
Systematic screening for all potential abnormalities should likely be restricted to research cohorts to better determine the frequency and characteristics of these complications. In the routine clinic, clinician awareness of these complications is important so that patients and families can be counselled appropriately and those with suspicious symptoms can specifically be assessed.
-
Given the frequency of genital (especially in girls) and pancreatic abnormalities, an abdominal and pelvic ultrasound should be considered.
-
Gout can be prevented by appropriate treatment; thus monitoring of uric acid levels should be considered. So far, gout has only been reported in adult patients; therefore, monitoring may be indicated only in adolescent and older patients.
-
Not enough is known about the potential for delaying or even preventing the onset of diabetes using life style modifications, but discussion of this possibility with the patient/family should be considered.
-
There are only single case reports of NODAT in HNF1B mutation carriers, making a meaningful risk assessment impossible. Nevertheless, this should be discussed with patients/families, and avoidance/minimisation of prodiabetic drugs, such as glucocorticoids and tacrolimus, should be considered.
Conclusion
Since the discovery in 1997 of HNF1B as a disease gene, well over a 100 patients with HNF1B-related disorders have been reported in the literature, and the associated phenotypic spectrum is still expanding. Renal manifestations clearly appear to be most common, followed by pancreatic and genital abnormalities. Neurological complications appear to be restricted to patients with the 17q12 deletion. Some of the reported phenotypes have only been observed in single patients, making ascertainment of a true association difficult. Moreover, there is little, if any, data regarding complication prevention or treatment. To better understand this rare disease and develop potential interventions, good clinical registries are needed, such as the Rare Renal Disease Registry (RaDaR) initiative in the UK (http://rarerenal.org/rare-disease-groups/hnf1b-rdg/).
References
Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, Reutter HM, Soliman NA, Bogdanovic R, Kehinde EO, Tasic V, Hildebrandt F (2014) Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int 85:1429–1433
Thomas R, Sanna-Cherchi S, Warady BA, Furth SL, Kaskel FJ, Gharavi AG (2011) HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 26:897–903
Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, Ghiggeri GM, Jankauskiene A, Mir S, Montini G, Peco-Antic A, Wuhl E, Zurowska AM, Mehls O, Antignac C, Schaefer F, Salomon R (2006) Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 17:2864–2870
Johnson PF (1990) Transcriptional activators in hepatocytes. Cell Growth Differ 1:47–52
Mendel DB, Hansen LP, Graves MK, Conley PB, Crabtree GR (1991) HNF-1 alpha and HNF-1 beta (vHNF-1) share dimerization and homeo domains, but not activation domains, and form heterodimers in vitro. Genes Dev 5:1042–1056
Lu P, Rha GB, Chi YI (2007) Structural basis of disease-causing mutations in hepatocyte nuclear factor 1beta. Biochemistry 46:12071–12080
Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV, Chen X, Cox NJ, Oda Y, Yano H, Le Beau MM, Yamada S, Nishigori H, Takeda J, Fajans SS, Hattersley AT, Iwasaki N, Hansen T, Pedersen O, Polonsky KS, Bell GI (1996) Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 384:455–458
Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O, Kuroki H, Kasahara T, Iwamoto Y, Bell GI (1997) Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet 17:384–385
Nishigori H, Yamada S, Kohama T, Tomura H, Sho K, Horikawa Y, Bell GI, Takeuchi T, Takeda J (1998) Frameshift mutation, A263fsinsGG, in the hepatocyte nuclear factor-1beta gene associated with diabetes and renal dysfunction. Diabetes 47:1354–1355
Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O (1999) A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet 8:2001–2008
Bingham C, Bulman MP, Ellard S, Allen LI, Lipkin GW, Hoff WG, Woolf AS, Rizzoni G, Novelli G, Nicholls AJ, Hattersley AT (2001) Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet 68:219–224
Kolatsi-Joannou M, Bingham C, Ellard S, Bulman MP, Allen LI, Hattersley AT, Woolf AS (2001) Hepatocyte nuclear factor-1beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. J Am Soc Nephrol 12:2175–2180
Edghill EL, Stals K, Oram RA, Shepherd MH, Hattersley AT, Ellard S (2013) HNF1B deletions in patients with young-onset diabetes but no known renal disease. Diabet Med 30:114–117
Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van’t Hoff W, Marks SD, Trompeter RS, Tullus K, Winyard PJ, Cansick J, Mushtaq I, Dhillon HK, Bingham C, Edghill EL, Shroff R, Stanescu H, Ryffel GU, Ellard S, Bockenhauer D (2009) HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 20:1123–1131
Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C (2015) HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol 11:102–112
Bellanne-Chantelot C, Clauin S, Chauveau D, Collin P, Daumont M, Douillard C, Dubois-Laforgue D, Dusselier L, Gautier JF, Jadoul M, Laloi-Michelin M, Jacquesson L, Larger E, Louis J, Nicolino M, Subra JF, Wilhem JM, Young J, Velho G, Timsit J (2005) Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54:3126–3132
Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, Pinkel D, Cooper GM, Ventura M, Ropers HH, Tommerup N, Eichler EE, Bellanne-Chantelot C (2007) Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet 81:1057–1069
Laffargue F, Bourthoumieu S, Llanas B, Baudouin V, Lahoche A, Morin D, Bessenay L, De Parscau L, Cloarec S, Delrue MA, Taupiac E, Dizier E, Laroche C, Bahans C, Yardin C, Lacombe D, Guigonis V (2015) Towards a new point of view on the phenotype of patients with a 17q12 microdeletion syndrome. Arch Dis Child 100:259–264
Barbacci E, Reber M, Ott MO, Breillat C, Huetz F, Cereghini S (1999) Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. Development 126:4795–4805
Coffinier C, Thepot D, Babinet C, Yaniv M, Barra J (1999) Essential role for the homeoprotein vHNF1/HNF1beta in visceral endoderm differentiation. Development 126:4785–4794
Naylor RW, Davidson AJ (2014) Hnf1beta and nephron segmentation. Pediatr Nephrol 29:659–664
De Vas MG, Kopp JL, Heliot C, Sander M, Cereghini S, Haumaitre C (2015) Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development 142:871–882
Lokmane L, Heliot C, Garcia-Villalba P, Fabre M, Cereghini S (2010) vHNF1 functions in distinct regulatory circuits to control ureteric bud branching and early nephrogenesis. Development 137:347–357
Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, Fiette L, Igarashi P, Yaniv M, Pontoglio M (2004) A transcriptional network in polycystic kidney disease. EMBO J 23:1657–1668
Massa F, Garbay S, Bouvier R, Sugitani Y, Noda T, Gubler MC, Heidet L, Pontoglio M, Fischer E (2013) Hepatocyte nuclear factor 1beta controls nephron tubular development. Development 140:886–896
Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, Clauin S, Deschenes G, Bouissou F, Bensman A, Bellanne-Chantelot C (2006) Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol 17:497–503
Edghill EL, Bingham C, Ellard S, Hattersley AT (2006) Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet 43:84–90
Adalat S, Bockenhauer D, Ledermann SE, Hennekam RC, Woolf AS (2010) Renal malformations associated with mutations of developmental genes: messages from the clinic. Pediatr Nephrol 25:2247–2255
Heidet L, Decramer S, Pawtowski A, Moriniere V, Bandin F, Knebelmann B, Lebre AS, Faguer S, Guigonis V, Antignac C, Salomon R (2010) Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol 5:1079–1090
Rasmussen M, Ramsing M, Petersen OB, Vogel I, Sunde L (2013) A description of a fetal syndrome associated with HNF1B mutation and a wide intrafamilial disease variability. Am J Med Genet A 161A:3191–3195
Decramer S, Parant O, Beaufils S, Clauin S, Guillou C, Kessler S, Aziza J, Bandin F, Schanstra JP, Bellanne-Chantelot C (2007) Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol 18:923–933
Madariaga L, Moriniere V, Jeanpierre C, Bouvier R, Loget P, Martinovic J, Dechelotte P, Leporrier N, Thauvin-Robinet C, Jensen UB, Gaillard D, Mathieu M, Turlin B, Attie-Bitach T, Salomon R, Gubler MC, Antignac C, Heidet L (2013) Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol 8:1179–1187
Tsatsaris V, Gagnadoux MF, Aubry MC, Gubler MC, Dumez Y, Dommergues M (2002) Prenatal diagnosis of bilateral isolated fetal hyperechogenic kidneys. Is it possible to predict long term outcome? BJOG 109:1388–1393
Bingham C, Ellard S, Cole TR, Jones KE, Allen LI, Goodship JA, Goodship TH, Bakalinova-Pugh D, Russell GI, Woolf AS, Nicholls AJ, Hattersley AT (2002) Solitary functioning kidney and diverse genital tract malformations associated with hepatocyte nuclear factor-1beta mutations. Kidney Int 61:1243–1251
Bingham C, Ellard S, van’t Hoff WG, Simmonds HA, Marinaki AM, Badman MK, Winocour PH, Stride A, Lockwood CR, Nicholls AJ, Owen KR, Spyer G, Pearson ER, Hattersley AT (2003) Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int 63:1645–1651
Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV (2000) Dominant isolated renal magnesium loss is caused by misrouting of the Na(+), K(+)-ATPase gamma-subunit. Nat Genet 26:265–266
Ferre S, de Baaij JH, Ferreira P, Germann R, de Klerk JB, Lavrijsen M, van Zeeland F, Venselaar H, Kluijtmans LA, Hoenderop JG, Bindels RJ (2014) Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 25:574–586
Owen K, Hattersley AT (2001) Maturity-onset diabetes of the young: from clinical description to molecular genetic characterization. Best Pract Res Clin Endocrinol Metab 15:309–323
Ryffel GU (2001) Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol 27:11–29
Haldorsen IS, Vesterhus M, Raeder H, Jensen DK, Sovik O, Molven A, Njolstad PR (2008) Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet Med 25:782–787
Body-Bechou D, Loget P, D’Herve D, Le Fiblec B, Grebille AG, Le Guern H, Labarthe C, Redpath M, Cabaret-Dufour AS, Sylvie O, Fievet A, Antignac C, Heidet L, Taque S, Patrice P (2014) TCF2/HNF-1beta mutations: 3 cases of fetal severe pancreatic agenesis or hypoplasia and multicystic renal dysplasia. Prenat Diagn 34:90–93
Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S (2005) Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci U S A 102:1490–1495
Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, Shishido S, Hasegawa Y, Nakahata T (2004) Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J Clin Endocrinol Metab 89:2905–2908
Zuber J, Bellanne-Chantelot C, Carette C, Canaud G, Gobrecht S, Gaha K, Mallet V, Martinez F, Thervet E, Timsit J, Legendre C, Dubois-Laforgue D (2009) HNF1B-related diabetes triggered by renal transplantation. Nat Rev Nephrol 5:480–484
Tudorache E, Sellier-Leclerc AL, Lenoir M, Tubiana-Rufi N, Bensman A, Bellanne-Chantelot C, Ulinski T (2012) Childhood onset diabetes posttransplant in a girl with TCF2 mutation. Pediatr Diabetes 13:e35–e39
Tjora E, Wathle G, Erchinger F, Engjom T, Molven A, Aksnes L, Haldorsen IS, Dimcevski G, Raeder H, Njolstad PR (2013) Exocrine pancreatic function in hepatocyte nuclear factor 1beta-maturity-onset diabetes of the young (HNF1B-MODY) is only moderately reduced: compensatory hypersecretion from a hypoplastic pancreas. Diabet Med 30:946–955
Oram RA, Edghill EL, Blackman J, Taylor MJ, Kay T, Flanagan SE, Ismail-Pratt I, Creighton SM, Ellard S, Hattersley AT, Bingham C (2010) Mutations in the hepatocyte nuclear factor-1beta (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am J Obstet Gynecol 203(364):e361–e365
Faguer S, Decramer S, Chassaing N, Bellanne-Chantelot C, Calvas P, Beaufils S, Bessenay L, Lengele JP, Dahan K, Ronco P, Devuyst O, Chauveau D (2011) Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 80:768–776
Bellanne-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Beaufils S, Wilhelm JM, Boitard C, Noel LH, Velho G, Timsit J (2004) Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med 140:510–517
Beckers D, Bellanne-Chantelot C, Maes M (2007) Neonatal cholestatic jaundice as the first symptom of a mutation in the hepatocyte nuclear factor-1beta gene (HNF-1beta). J Pediatr 150:313–314
Kitanaka S, Miki Y, Hayashi Y, Igarashi T (2004) Promoter-specific repression of hepatocyte nuclear factor (HNF)-1 beta and HNF-1 alpha transcriptional activity by an HNF-1 beta missense mutant associated with Type 5 maturity-onset diabetes of the young with hepatic and biliary manifestations. J Clin Endocrinol Metab 89:1369–1378
Ferre S, Bongers EM, Sonneveld R, Cornelissen EA, van der Vlag J, van Boekel GA, Wetzels JF, Hoenderop JG, Bindels RJ, Nijenhuis T (2013) Early development of hyperparathyroidism due to loss of PTH transcriptional repression in patients with HNF1beta mutations? J Clin Endocrinol Metab 98:4089–4096
Hoorn EJ, Zietse R (2013) Disorders of calcium and magnesium balance: a physiology-based approach. Pediatr Nephrol 28:1195–1206
Lebrun G, Vasiliu V, Bellanne-Chantelot C, Bensman A, Ulinski T, Chretien Y, Grunfeld JP (2005) Cystic kidney disease, chromophobe renal cell carcinoma and TCF2 (HNF1 beta) mutations. Nat Clin Pract Nephrol 1:115–119
Edghill EL, Oram RA, Owens M, Stals KL, Harries LW, Hattersley AT, Ellard S, Bingham C (2008) Hepatocyte nuclear factor-1beta gene deletions–a common cause of renal disease. Nephrol Dial Transplant 23:627–635
Nik-Zainal S, Strick R, Storer M, Huang N, Rad R, Willatt L, Fitzgerald T, Martin V, Sandford R, Carter NP, Janecke AR, Renner SP, Oppelt PG, Oppelt P, Schulze C, Brucker S, Hurles M, Beckmann MW, Strissel PL, Shaw-Smith C (2011) High incidence of recurrent copy number variants in patients with isolated and syndromic Mullerian aplasia. J Med Genet 48:197–204
Bernardini L, Gimelli S, Gervasini C, Carella M, Baban A, Frontino G, Barbano G, Divizia MT, Fedele L, Novelli A, Bena F, Lalatta F, Miozzo M, Dallapiccola B (2009) Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports. Orphanet J Rare Dis 4:25
Nagamani SC, Erez A, Shen J, Li C, Roeder E, Cox S, Karaviti L, Pearson M, Kang SH, Sahoo T, Lalani SR, Stankiewicz P, Sutton VR, Cheung SW (2010) Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur J Hum Genet 18:278–284
Kasperaviciute D, Catarino CB, Chinthapalli K, Clayton LM, Thom M, Martinian L, Cohen H, Adalat S, Bockenhauer D, Pope SA, Lench N, Koltzenburg M, Duncan JS, Hammond P, Hennekam RC, Land JM, Sisodiya SM (2011) Uncovering genomic causes of co-morbidity in epilepsy: gene-driven phenotypic characterization of rare microdeletions. PLoS One 6, e23182
Loirat C, Bellanne-Chantelot C, Husson I, Deschenes G, Guigonis V, Chabane N (2010) Autism in three patients with cystic or hyperechogenic kidneys and chromosome 17q12 deletion. Nephrol Dial Transplant 25:3430–3433
Faguer S, Chassaing N, Bandin F, Prouheze C, Arveiler B, Rooryck C, Nogier MB, Chauveau D, Calvas P, Decramer S (2011) A 17q12 chromosomal duplication associated with renal disease and esophageal atresia. Eur J Med Genet 54:e437–e440
Hoskins BE, Cramer CH 2nd, Tasic V, Kehinde EO, Ashraf S, Bogdanovic R, Hoefele J, Pohl M, Hildebrandt F (2008) Missense mutations in EYA1 and TCF2 are a rare cause of urinary tract malformations. Nephrol Dial Transplant 23:777–779
Chen YZ, Gao Q, Zhao XZ, Chen YZ, Bennett CL, Xiong XS, Mei CL, Shi YQ, Chen XM (2010) Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J (Engl) 123:3326–3333
Faguer S, Chassaing N, Bandin F, Prouheze C, Garnier A, Casemayou A, Huart A, Schanstra JP, Calvas P, Decramer S, Chauveau D (2014) The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int 86:1007–1015
Acknowledgments
DB is a HEFCE Clinical Reader and supported by European Union, FP7 [grant agreement 2012–305608, “European Consortium for High-Throughput Research in Rare Kidney Diseases (EURenOmics)”]
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bockenhauer, D., Jaureguiberry, G. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol 31, 707–714 (2016). https://doi.org/10.1007/s00467-015-3142-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-015-3142-2