
Abstract
Nephropathies arise from conditions that alter nephron development or trigger nephron damage during neonatal, juvenile, and adult stages of life. Much evidence suggests that a key role in maintaining kidney integrity, homeostasis, and regenerative capacity is played by a population of progenitor cells resident in the organ. Although the primary goals in the field of renal progenitor cells are understanding their ability to regenerate nephrons and to restore damaged kidney function, the discovery of these cells could also be used to elucidate the molecular and pathophysiological basis of kidney diseases. As a result, once the identification of a subset of progenitor cells capable of kidney regeneration has been obtained, the increasing knowledge about their characteristics and about the mechanisms of renal development had pointed out the possibility of understanding the molecular basis of kidney diseases, so that, nowadays, some renal disorders could also be related to renal progenitor dysfunction. In this review, we summarize the evidence on the existence of renal progenitors in fetal and adult kidneys and discuss their role in physiology as well as in the pathogenesis of renal disorders with a particular focus on childhood age.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kidney health depends on the complete integrity and functionality of the nephrons and their cellular components. There is a lot of evidence suggesting that a key role in maintaining tissues integrity, homeostasis, and regenerative capacity is played by stem or progenitors cells resident in the organs [1, 2]. A stem cell (SC) is defined as a cell that exhibits two properties: self-renewal, which is the ability to go through numerous cycles of cell division while maintaining indefinitely an undifferentiated state, and multi-potency, which is the ability to generate a progeny of differentiated cells or their precursors. Progenitor cells, instead, are characterized by the potential to differentiate along one or more particular cell lineages, but they display a limited or null self-renewal potential [3]. In this review, we summarize the evidence on the existence of renal progenitors in fetal and adult kidneys and discuss their role in physiology as well as in the pathogenesis of renal disorders with a particular focus on childhood age.
Renal progenitors from development to adult life
In mammals, the metanephros arises from the reciprocal interaction of two structures, the ureteric bud (UB) and the metanephric mesenchyme (MM). While growing, the UB progressively divides into several ramifications, generating the portion of the nephron from the renal papilla to the collecting ducts system of the mature kidneys. On the other hand, MM originates renal corpuscles (or glomeruli) and proximal tubules, the loop of Henle, distal tubule, and the connecting segment [4–6]. During MM and UB interactions, MM differentiates into two portions representing two distinct cell lineages: the cap mesenchyme (CM) and the stromal mesenchyme (SM). CM surrounds the tips of UB’s branches and undergoes epithelial transformation in its more peripheral portion, giving rise to renal vesicle, comma- and S-shaped bodies and, finally, mature nephrons. This cell population is characterized by the expression of the homeobox gene Six2 (sine oculis-related homeobox 2 homologue), Hox11 (homeobox 11) paralogues, Osr1 (odd-skipped related 1) and Pax2 (paired box gene 2), as well as Cited1 (CBP/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain 1), Eya1 (eyes absent homologue), Sall1 (salt-like 1), and WT1 (Wilms tumor). Six2 is essential to maintain SC characteristics such as self-renewal and multi-potency [4], so that the lack of its expression determines the interruption of nephron formation early in embryonic kidney organogenesis because the CM compartment is not maintained [7]. Moreover, during embryonic nephrogenesis, the self-renewing ability is lost by CM cells, but it is still not completely clear if this is determined by disappearance of self-renewal ability itself or by the progressive commitment of Six2+ cells toward a more differentiated phenotype. Six2 marks a nephron progenitor population that gives rise to the epithelial cells of cortical nephrons, including podocytes, proximal, distal and connecting tubular cells [7]. Recently, Lgr5 (leucine-rich repeat-containing G-protein-coupled receptor 5) has been identified in mice as a marker of a nephron-specific progenitor population in the early-patterned S-shaped bodies that originates the thick ascending limb, the distal convoluted tubule, and the connecting segment, suggesting that the commitment toward specific cell lineages occurs very early during kidney development [8]. On the other hand, SM is characterized by the expression of FoxD1 (forkhead box D1) and localizes between UB branches and growing nephrons, contributing to the interstitial population of mature kidneys [4].
In humans, all of the branches of the UB and the nephrons have been formed by the 32nd to 36th week of gestation. However, these structures are not yet mature and will continue to grow and differentiate even after birth, during the perinatal period, as the generation of Henle’s loop occurs [3]. The capacity of generating new nephrons is lost at the time of birth so that, once matured, human kidneys have an estimated number of nephrons of one million per kidney or more [9, 10]. This nephron endowment is roughly proportional to body mass [3].
Human renal progenitors
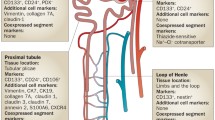
While developmental biology was elucidating the steps that characterize kidney organogenesis, allowing to expand our knowledge about stem and progenitor cells’ origin, determination, and commitment during embryogenesis, the search for renal stem/progenitor cells in mammalian kidney has been performed also using different research strategies. In rodents, lineage-tracing studies definitively proved the existence of a common renal embryonic progenitor for glomerular and tubular epithelial cells. These strategies could not be applied in humans, so that the identification of a similar population has been based on different criteria, such as the expression of specific cell-surface markers and the ability to self-renew and to give rise to one or more differentiated mature cell types. In human embryonic kidneys, renal multipotent progenitors are characterized by the expression of the surface molecules CD133, a marker of several different types of adult resident stem cells, and CD24, a marker of human MM, and by the expression of the transcription factors Bmi-1 (B lymphoma Mo-MLV insertion region 1 homolog) and Oct-4 (octamer-binding transcription factor 4), both typical of SC phenotype, in the absence of the expression of lineage-specific markers [11]. These cells share the expression of CD133 and CD24 surface markers with adult renal progenitors (Fig. 1) [11, 12]. The isolation of CD133+CD24+ cells allowed their phenotypical and functional characterization: in fact, they show self-renewal potential, high cloning efficiency, and the ability to generate either podocytes and cells with phenotypic features of proximal and distal tubular mature cells, when cultured in specific inductive medium (Fig. 1) [12, 13]. More importantly, when injected in mouse models of tubular or glomerular damage, these cells showed the potential to improve or restore the anatomic integrity of different structures of the nephron, as well as their function [12–14]. During renal organogenesis, these cells are first localized in condensed MM-derived primordial structures, then in renal vesicles, comma-shaped bodies, and in the proximal as well as in the distal loops of the S-shaped bodies, representing an elevated percentage of cells of these structures in the early phases of metanephros formation. Their number significantly reduces during development, when the mature structures of the kidney appear, representing only about 2 % of renal cells in the adult kidney. In this setting, they become selectively localized as a major cluster within the Bowman’s capsule, or as scattered cells in the elongating tubules [11, 15], suggesting that CD133+CD24+ adult renal progenitors represent a subset of multipotent embryonic progenitors that persist in human kidneys from early stages of nephrogenesis [11, 16]. During development, renal progenitors act as precursors to all renal epithelial cells of the cortical nephron, differentiating toward either glomerular either tubular epithelial cell lineages, through a graded series of committed progenitors, as demonstrated by clonal in vitro studies as well as in vivo transplantation experiments (Fig. 1) [11–13, 15, 17]. In adult human kidney, bipotent renal progenitors localize at the urinary pole of the Bowman’s capsule and are characterized by the expression of progenitor markers in absence of lineage markers (Fig. 1), while podocyte-committed progenitors localize along the Bowman’s capsule close to the vascular pole and are characterized by co-expression of progenitors and podocyte markers (Fig. 1). Finally, tubular-committed progenitors that are scattered within the proximal tubule, the thick ascending limb, the distal convoluted tubule, and the connecting segment and can generate cells of all these portions of the tubular compartment, are characterized by expression of renal progenitor markers in presence of low levels of tubular markers [11–13, 15, 17].

Schematic representation of the human “nephropoietic system”. Diagram showing the development of different types of nephron epithelial cells from multipotent progenitors to mature cells. From a single cell type, the multipotent progenitor, all mature nephron epithelial cells emerge through a hierarchical series of lineage decisions via different committed-progenitor cells. Thus, nephropoiesis can be depicted as a hierarchical differentiation tree, with a multipotent epithelial progenitor at the root and the mature epithelial cells as the leaves. The multipotent progenitor is characterized by expression of renal progenitor markers CD133 and CD24, as well of the adhesion molecule CD106, in absence of expression of lineage markers. The intermediate cellular states are the common tubular progenitor and the podocyte progenitor. The common tubular progenitor is characterized by co-expression of progenitor markers CD133 and CD24 and absence of CD106, in presence of low expression of tubular progenitor markers and can proliferate and differentiate into proximal tubular cells, cells of the loop of Henle, distal tubular cells, and cells of the connecting segment. The podocyte progenitor is characterized by co-expression of CD133, CD24, and CD106, as well as low levels of podocyte markers and can proliferate and differentiate into functional podocytes
Moreover, very recent findings suggest that tubular-committed progenitors scattered along the proximal and distal tubule are distinguishable from the subset of renal progenitors localized within the Bowman’s capsule by the expression of the surface marker vascular adhesion molecule 1 (VCAM1), or CD106. CD133+CD24+CD106− (Fig. 1) cells represent tubular-committed progenitors that display resistance to apoptotic stimuli and exert regenerative potential for injured tubular tissue [15]; this population has the same embryological origin of the more immature CD133+CD24+CD106+ cells localized to the urinary pole, remaining separated from that when the Henle’s loop forms after birth [3]. Taken together, these results strongly suggest that the adult kidney contains a “nephropoietic system”, a term that defines a hierarchical system of progenitor showing different commitment, and the capacity to generate all the epithelial cell subtypes of the cortical nephron (Fig. 1). Bowman’s capsule may therefore represent an SC niche, which is a specific site in adult tissues where SCs reside [16], and cellular regeneration probably represents the main regenerative strategy in humans and mammals [3, 12–15, 17]. Understanding the mechanisms that lead to determination, growth, proliferation, differentiation, activation, and recruitment after an injury of renal progenitors is essential to a deep comprehension of renal diseases and to their modulation for clinical and therapeutic purposes.
Renal progenitors and childhood nephropathies
Nephropathies arise from conditions that alter nephron development or trigger nephron damage during neonatal, juvenile, and adult stages of life [4]. The identification of a subset of progenitor cells capable of kidney regeneration and the increasing knowledge about their characteristics and about the molecular keys of kidney development had pointed out the possibility of understanding their role in kidney diseases, especially those related to childhood and adolescence. So nowadays, some renal disorders of infancy could be interpreted as renal progenitors’ dysfunction.
Renal hypoplasia
Renal hypoplasia is defined as the presence of congenitally small kidneys with a reduced number of nephrons but normal architecture. Pure renal hypoplasia is a rare condition, so it is frequently found associated to renal dysplasia, defined as the presence of malformed renal tissue elements, including primitive tubules, interstitial fibrosis, and/or the presence of cartilage in the renal parenchyma [18]. Renal hypoplasia, particularly the oligomeganephronic form, results from developmental arrest of the MM between the 14th and the 20th week of fetal life: this causes a decrease in the number of developing nephrons, leading to hypertrophy of the remaining ones [18]. As previously described, during intrauterine life, signals originating from the branching UB induce surrounding progenitor cells of the MM to differentiate and form individual nephrons. In mammals, nephron number varies widely due to genetic or environmental abnormalities altering production and/or function of factors that regulate not only the process of UB branching and subsequent mesenchyme-to-epithelium transition (MET) but also the progenitor’s pool size, growth, and/or differentiation. In mice, the complete loss of GDNF (glial cell-derived neurotrophic factor) and RET (REarranged during Transfection) or GFRA1 (GDNF family receptor alpha 1), which are critical components of the GDNF/Ret pathway, results in failure to form an UB, with agenesis of the kidneys and ureters and prenatal death [19]. Similarly, PAX2 mice mutants, which are unable to respond to inductive signals, lack GDNF production from MM and therefore the generation and the arborization of the UB [4]. Interestingly, in humans, allelic variants of RET and PAX2 genes are associated with a significant decrease in newborn kidney size, probably influencing nephron number through an effect on UB arborization [20]. Consistently, heterozygous mutations in PAX2 lead to the renal-coloboma syndrome, also known as papillo-renal syndrome, an autosomal dominant disease characterized by ocular abnormalities and renal hypoplasia [21]. Moreover, mutations in EYA1 and SIX1 or SIX5 genes are responsible for brachio-oto-renal syndrome, a complex phenotype comprising bilateral renal hypo-dysplasia [22]: Eya1 interacts with members of the SIX family genes, which encode for transcription factors that control the expression of Pax2 and GDNF in the renal progenitors of MM. Other pathways and molecules, including sonic hedgehog (Shh), bone morphogenic protein 7 (BMP-7), and fibroblast growth factor 2 (FGF-2), which control UB branching, as well as p53, which has a demonstrated role in the regulation of metanephric development, have been involved [23]. A recent study has reported a cooperative role for Dlg1 (discs-large homolog 1) and Cask (calcium/calmodulin-dependent serine protein kinase) scaffolding proteins in maintaining the nephron progenitor population, potentially via a mechanism involving effects on FGF signaling, as suggested by DLG1. CASK double-knockout mice, whose kidneys were severely hypoplastic and dysplastic, demonstrated rapid, premature depletion of nephron progenitors/stem cells, reduced proliferation, and increased apoptosis of cells in the nephrogenic zone and a progressive decrease in the number of cells expressing Six2, in association with reduced levels of components of the Ras pathway, which is activated by FGF [24].
Recent evidence that Wnt (wingless-type MMTV integration site family) signaling alterations in renal progenitors can alter nephron growth and induce renal hypoplasia comes from the observation that WNT9B and WNT4 knockout mice lack UB branching and MET, resulting in rudimentary, non-functioning kidneys [25]. A similar phenotype has been described in β-catenin loss-of-function mutant mice and β-catenin is thought to have an important function in the early phases of MET, acting as an intracellular mediator of Wnt9b and Wnt4 [25]. However, in a mouse model of conditional β-catenin knockout, with specific deficiency of expression in renal epithelial cells at the S-shaped body stage, hypoplastic kidneys with a thin cortex and reduced renal function were described. At a more detailed level, kidneys presented a high proportion of glomeruli with underdeveloped capillary tuft and cystic dilatation of the Bowman’s capsule; to be noted, these alterations did not affect juxtamedullary glomeruli that represent the earlier developing nephrons. Moreover, the authors described the substitution of progenitors of the Bowman’s capsule with mature, fully differentiated podocytes, demonstrating a switch in differentiation of progenitors toward the podocyte lineage when they become deficient for β-catenin expression, thus conclusively proving that β-catenin/Wnt signaling is crucial during the late stages of nephrogenesis and for renal progenitor lineage specification [26].
The identification of OSR1 as an essential gene for the maintenance of the renal progenitor pool during primary nephrogenesis allowed to clearly demonstrate that in humans the final nephron number might also be set by genes that establish or sustain the pool of CD133+CD24+ renal progenitor cells during development [20]. In fact, kidney SCs of embryonic intermediate mesoderm are critically dependent on the expression of OSR1, which is one of their earliest genetic markers, becoming expressed long before that morphological signs of MM specification occur [4]; in mice, homozygous inactivation of OSR1 induce failure to form MM, leading to renal agenesis [27]. Finally, the identification of an allelic variant of OSR1 in children with reduced newborn kidney size and function strongly supports the fundamental role of this gene in establishing renal progenitor pool in embryonic kidney and in opposing to their differentiation, thus contributing to the pool maintenance in adult life [20]. In addition, OSR1 mutants completely lack the expression of Eya1 and Pax2, as well as that of Six2 and Cited1 in the metanephric region. Indeed, OSR1 lies genetically upstream of the Pax2/Eya1/Hox11 and the Six2/Cited1 complexes: the first one drives GDNF expression and thus regulates the growth of the UB, while the latter is thought to be critical in the maintenance of self-renewal of the nephron progenitor/stem cell population and in delaying the differentiation cascade following an inductive signal [20], as loss of Six2 results in premature loss of this cellular compartment due to differentiation [28]. Osr1 is also required for the expression of other transcriptional factors, including Lim1, Six1 and Six4, Sall1, WT1, and GDNF, which are essential to UB outgrowth and are required for nephron development, as the loss of their functions in the developing MM results in either renal agenesis or hypoplasia [28]. All these genes are nowadays well-recognized markers of early renal progenitors cells specification: it is easy to comprehend how mutations in genes critical for the conversion of MM to epithelia are responsible for a reduction in the endowment of renal progenitors cells, finally leading to childhood-onset renal hypoplasia.
Environmental factors have also been linked to the development of renal hypoplasia: maternal malnutrition, stress, diseases such as diabetes, utero-placental insufficiency, and maternal and neonatal drugs have been associated with a reduced nephron endowment in the newborn. In addition, premature birth has been recognized as a risk factor independent from low birth weight for reduced nephron number in neonates and it has been associated, both in animals and in humans, with abnormal glomerulogenesis, probably related to accelerated nephrogenesis after birth, podocytopenia, and increased risk of focal segmental glomerulosclerosis (FSGS) and chronic kidney disease (CKD) later in life [29, 30]. As nephrogenesis takes place entirely before birth, intrauterine growth restriction or retardation (IUGR) is the most widely studied environmental phenomenon influencing renal development [31]. In rodents and in other animal models, maternal protein restriction during pregnancy and lactation lead to IUGR and to a significant reduction in nephron number in the offspring, resulting in renal hypoplasia. This effect could be antagonized by the administration, during embryonic life, of retinoic acid (RA), the active metabolite of vitamin A (retinol), which has been demonstrated to have a stimulatory effect on nephrogenesis in both in vivo and in vitro studies [31, 32]. It is in fact well recognized that vitamin A mild-to-severe deficiency during intrauterine life directly affects nephron number, leading to renal hypoplasia, which can be prevented by the administration of vitamin A or RA [9]. A direct relationship between circulating vitamin A levels during pregnancy and nephron number has been demonstrated either in animals or in humans, supporting the hypothesis that retinoids modulate nephron number in a dose-dependent manner and that an optimal level is required in fetal kidneys for normal nephrogenesis [9]. Moreover, in rodents, a severe deficit of vitamin A during pregnancy determines renal congenital abnormalities similar to those described in retinoic acid nuclear receptors (RARs) and retinoid X receptors (RXRs) knock-out mice [9, 32]. Furthermore, renal agenesis was described in mice deficient in the RA-generating enzyme RALDH2, while variants in human ALDH1A2 gene, encoding for an enzyme that catalyzes the synthesis of RA from retinaldehyde, is associated with an increased umbilical cord level of RA and increased newborn kidney size [20], thus demonstrating the role of RA for proper kidney development. RA influences nephrogenesis through an effect on UB branching, probably stimulating mesenchymal cells expressing RARs to release branching factors. Ret plays a key role: genetic deletion of RARs leads to impaired UB growth and branching and to downregulation of Ret in the UB, while forced expression of Ret in mice deficient for RARs genetically rescues renal development, restoring UB growth [33]. Finally, RA regulates several genes expressed during renal organogenesis, such as transcriptional factors of the Hox family, hepatic nuclear factor 1, Lim1, epidermal growth factor (EGF), transferrin receptor, and Ret, strongly suggesting the need for RA for proper kidney organogenesis and branching nephrogenesis. Consistently, in zebrafish RA has been reported to be required to modulate renal progenitor differentiation: RA signaling is necessary for podocyte formation and/or survival, as well as for establishing the normal pattern of nephron segmentation, inducing, in particular, proximal segment fates determination and preventing the expansion of distal segment fates [34].
These experiments show how important this molecule is for renal progenitors during kidney development and the dramatic effects of disturbing the RA signaling pathway, thus further suggesting that renal hypoplasia could also result from perturbations in renal progenitor cells physiology [35, 36].
Although the evidence for a role of number and properties of renal progenitors in the pathogenesis of renal hypoplasia are still limited, the above-reported examples and, in particular, the role of OSR1 and RA alterations in inducing renal hypoplasia through alteration of renal progenitors suggest that, as already described for other organs, alterations of nephron progenitors during kidney development may represent a critical determinant of disease.
Glomerular disorders
Several results suggest that an altered growth and/or differentiation ability of renal progenitors may also be responsible for the generation of kidney disorders, a concept that is widely accepted in several SC systems [37, 38]. Indeed, in crescentic glomerulonephritis, a glomerular disorder characterized by an unfavorable prognosis and rapid progression towards glomerulosclerosis and end-stage renal disease (ESRD), an abnormal response to podocyte injury causes aberrant renal progenitor proliferation, the formation of hypercellular lesions, and the obliteration of Bowman’s space, leading to the generation of pseudo-crescents and crescents [39–42]. Recent results suggest that these hyperplastic glomerular lesions may be initiated by glomerular vascular injury and glomerular basement-membrane breakage, which cause plasma leakage, and trigger the proliferation of renal progenitors and loss of their polarity [43]. Interestingly, an inadequate regenerative response of renal progenitors to podocyte injury associated with extracellular matrix production by renal progenitors was also proposed as the possible cause of FSGS lesions [43]. Indeed, podocyte damage is a key event initiating progression towards glomerulosclerosis [44]. Podocyte represents a terminally differentiated and post-mitotic cell, highly specialized in the maintenance of the integrity and selectivity of the glomerular filtration barrier (GFB), and therefore provided with only limited proliferation abilities. However, recent studies demonstrated that new podocytes could be recruited from renal progenitors localized on the surface of the Bowman’s capsule [11–14, 45]. Renal progenitors of the Bowman’s capsule can proliferate and amplify in response to injury, a capacity that no longer exists once they are committed to differentiate into podocytes. Indeed, in three distinct mouse models of FSGS, as well as in human biopsies, podocyte injury was associated with focal activation of renal progenitors, which invaded the affected segment of the glomerular tuft and deposited extracellular matrix [43]. These observations suggest that the response of renal progenitors to podocyte injury is a crucial determinant of progression towards glomerulosclerosis. The recruitment of renal progenitors to podocytes needs to be tightly regulated. Indeed, renal progenitors that differentiate into podocytes start the expression of the cell-cycle inhibitors p21, p27, and p53 and of other podocyte-specific markers, while decrease that of Notch that seems to be essential to permit the correct differentiation [46–48]. In addition, recent studies suggest that blockade of the chemokine stromal-derived factor-1 (SDF-1/CXCL12) with a specific antagonist also increases podocyte number, reduces proteinuria, and ameliorates renal function, in mouse models of diabetic nephropathy, by enhancing renal progenitor differentiation towards the podocyte phenotype [49].
It appears as a consequence that any condition that impairs renal progenitors proliferation or their ability to correctly activate and differentiate in response to injury to restore glomerular podocytes endowment could be responsible for the progressively irreversible podocytes depletion and, as previously described, glomerular scarring and glomerulosclerosis development. In fact, it was previously reported that renal progenitors could be found in sclerotic areas of glomeruli affected by FSGS [50]. Moreover, recent studies reported that activated renal progenitors expressing CD44 acquire the ability to produce extra-cellular matrix (ECM), both in mice and in humans [43, 51, 52]. These matrix molecules initially accumulate along Bowman’s basement membrane, leading to its thickening, and subsequently form “bridge” connections from the capsule and the glomerular capillary tuft, which are usually referred to as synechiae, a histological hallmark of FSGS. These structures, finally, are thought to facilitate the passage of ECM-producing renal progenitors from Bowman’s capsule inside the glomeruli, where they ultimately lead to advanced sclerotic lesion formation [46]. Moreover, in human biopsies from patients with minimal change disease that is characterized by the absence of glomerular scarring, CD44 was barely expressed [51], underlying a different pathogenic process for this condition. Interestingly, collapsing glomerulopathy, initially considered as a variant of FSGS and now usually treated as a separate entity, has recently been related to an abnormal and dysregulated renal progenitor proliferation in response to massive podocyte injury [46]. Segmental to global collapse of the capillary tuft and pronounced epithelial cell hyperplasia, often referred to as pseudo-crescents, are the histological characteristic of collapsing glomerulopathy. Recent studies demonstrated that these hyperplastic lesions also mainly consist of renal progenitors at different stages of differentiation toward the podocyte lineage [39].
Interestingly, very recent results suggest that strong proteinuria may lead to an impairment of the capacity of renal progenitors to differentiate into mature podocytes, thus generating FSGS and other types of glomerular cellular lesions when strong proteinuria occurs. Proteinuria is a common sign of glomerular disease and characterizes FSGS as well as nephrotic syndrome and other glomerulopathies. We recently demonstrated that proteinuria represents not only a consequence but also a key determinant of glomerular injury and damage progression by suppressing podocyte regeneration. Indeed, different components of proteinuria exert distinct effects on renal progenitor survival and differentiation towards a podocyte lineage. In particular, albumin, the main constituent of nephrotic proteinuria, prevents human renal progenitor differentiation into podocytes in vitro by sequestering RA, which is a critical driver of progenitors differentiation in multi-organ systems, thus impairing retinoic acid response element (RARE)-mediated transcription of podocyte-specific genes. In adriamycin nephropathy, a mouse model of human FSGS blocking endogenous RA synthesis increased proteinuria and worsened glomerulosclerosis by reducing podocyte number, while treatment with RA reverted RARE activity and induced expression of podocyte markers in renal progenitors, resulting in a decrease of proteinuria and an increase in podocytes number [53]. Our results also suggest that, when in addition to albuminuria, larger proteins, like IgG and transferrin, are lost, a combined toxic effect not only blocks renal progenitors’ differentiation into podocytes but also promotes their death. This provides a possible explanation for the observation that when non-selective proteinuria occurs, renal function declines faster. These findings may explain why reducing proteinuria delays CKD progression, and provide a biological rationale for the clinical use of pharmacological agents to induce regression of glomerular diseases.
Wilms tumor
Wilms tumor (WT), also known as nephroblastoma, is the most common pediatric renal malignancy, accounting for 6 % of all childhood cancers [54]. It represents an useful setting for studying the relation between development and tumorigenesis: indeed, it is viewed as a prototype of differentiation failure in human neoplasia, as it recapitulates the histology of the nephrogenic zone of the growing fetal kidney [54, 55]. In fact, recent studies point to an early renal stem/progenitor multipotent population that undergoes malignant transformation as the source for WT. Indeed, WT cells show similarities in terms of genetic, epigenetic, and phenotypic properties to human fetal mesenchymal progenitors and are therefore likely to be derivatives of the same lineage [54, 55]. These cells probably represent an upstream progenitor for CD133+CD24+ cells that are already committed to an epithelial phenotype. In fact, recent results suggest that CD133+CD24+ population may instead represent the cell source of origin of papillary human renal cell carcinoma that nevertheless represents a malignancy of adult life [56].
Acute kidney injury
Acute kidney injury (AKI) represents a complication of many clinical settings in children. Following AKI, kidney undergoes a regenerative response leading in most cases to complete recovery of renal function. However, a glowing debate has been raised about the cell source for tubule regenerating cells. A great amount of evidence, either in humans or in animal models of AKI, supports a role for surviving, less injured intrinsic cells localized within the tubular epithelium, which proliferate and migrate to replace the neighboring dead cells and reline denuded tubules, restoring the structural and functional integrity of the kidney [57, 58]. It has recently been demonstrated that in adult human kidney, CD133+CD24+CD106− cells represent tubular-committed progenitors scattered within the proximal and distal convoluted tubule, but mostly localize in the S3 segment of the proximal tubule. This tubular portion is highly susceptible to ischemia and toxic insults, typical of AKI from different causes, but also has a remarkable capacity to repair its structure and function [57]. Interestingly, CD133+CD24+CD106− tubular progenitors share with bipotent CD133+CD24+CD106+ progenitors of the Bowman’s capsule resistance to apoptotic stimuli and regenerative potential for injured tubular tissue [15, 58]. As a consequence, although tubular progenitors represent only a small amount of all tubular cells in healthy kidneys, following injury they are the cells that preferentially survive, being enriched proportionally to the severity of damage [15]. In addition, this population proliferates and represents the predominant regenerating population in the kidney of patients affected by acute and chronic tubular disorders [59]. Similar regenerative properties have been demonstrated in evolutionary distant animal classes such as insects or fish [3, 58]. Moreover, only tubular progenitors, but not differentiated tubular cells, engrafted, generated novel tubular cells and improved renal function when injected into mice models of AKI, further confirming that the phenotype of these cells is stable and distinct from that of differentiated tubular cells, at least in humans [15]. What the real mechanisms of tubular regeneration are, and what the contribution of progenitor cells versus differentiated tubular cells is, is still a matter of debate. In fact, the existence of a compartment of tubular progenitors does not rule out the contribution of differentiated tubular cells to repair tubular injury in adult mammalian kidneys, as suggested by evolutionary studies that demonstrated that usually more than one mechanism is involved in regeneration of a tissue because this confers an advantage during evolution. Further studies are encouraged in order to understand whether different mechanisms of regeneration contribute to tubular repair and regeneration during AKI [57, 58].
Conclusions
Kidney health depends on the complete integrity and functionality of the nephrons and their components parts developing in the early phases of life. As already described, a large body of evidence has recently suggested that renal progenitors of adult human kidney can regenerate injured podocytes as well as damaged tubular epithelial cells, suggesting that they represent a source for nephron regeneration. However, there is a long way to go in our understanding of molecular mechanisms that drive pathologic behaviors and their differences with what happens in physiological conditions. We founded in the hope that studies on renal progenitor cells will eventually lead to the creation of innovative treatments for kidney diseases. For this, it is of striking importance to acquire detailed information about how self-renewal and fate decision of renal progenitor cells in physiological conditions as well as in response to injury occurs and may be perturbed or modulated, to the aim of obtaining novel tools to prevent and treat kidney diseases.
References
Romagnani P (2010) From Proteus to Prometeus: learning from fish to modulate regeneration. J Am Soc Nephrol 21:726–728
Shahragim T (2009) Stem cell: what’s in the name? Nat Rep Stem Cells. doi:10.1038/stemcells.2009.90
Romagnani P, Lasagni L, Remuzzi G (2013) Renal progenitors: an evolutionary conserved strategy for kidney regeneration. Nat Rev Nephrol 9(3):137–146
Mc Campbell KK, Wingert RA (2012) Renal stem cells: fact or science fiction? Biochem J 444:153–168
Dressler GR (2006) The cellular basis of kidney development. Annu Rev Cell Dev Biol 22:509–529
Schedl A (2007) Renal abnormalities and their developmental origin. Nat Rev Genet 8:791–802
Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP (2008) Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3(2):169–181
Barker N, Rookmaaker MB, Kujala P, Ng A, Leushacke M, Snippert H, van de Wetering M, Tan S, Van Es JH, Huch M, Poulsom R, Verhaar MC, Peters PJ, Clevers H (2012) Lgr5(+ve) stem/progenitor cells contribute to nephron formation during kidney development. Cell Rep 2(3):540–552
Bhat PV, Manolescu DC (2008) Role of vitamin A in determining nephron mass and possible relationship to hypertension. J Nutr 138(8):1407–1410
Bertram JF, Douglas-Denton RN, Diouf B, Hughson MD, Hoy WE (2011) Human nephron number: implications for health and disease. Pediatr Nephrol 26:1529–1533
Lazzeri E, Crescioli C, Ronconi E, Mazzinghi B, Sagrinati C, Netti GS, Angelotti ML, Parente E, Ballerini L, Cosmi L, Maggi L, Gesualdo L, Rotondi M, Annunziato F, Maggi E, Lasagni L, Serio M, Romagnani S, Vannelli GB, Romagnani P (2007) Regenerative potential of embryonic renal multipotent progenitors in acute renal failure. J Am Soc Nephrol 18(12):3128–3138
Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, Maggi E, Annunziato F, Lasagni L, Serio M, Romagnani S, Romagnani P (2006) Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol 17(9):2443–2456
Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P (2009) Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20(2):322–332
Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ (2009) Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20(2):333–343
Angelotti ML, Ronconi E, Ballerini L, Peired A, Mazzinghi B, Sagrinati C, Parente E, Gacci M, Carini M, Rotondi M, Fogo AB, Lazzeri E, Lasagni L, Romagnani P (2012) Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 30(8):1714–1725
Romagnani P (2009) Toward the identification of a “renopoietic system”? Stem Cells 27(9):2247–2253
Mazzinghi B, Ronconi E, Lazzeri E, Sagrinati C, Ballerini L, Angelotti ML, Parente E, Mancina R, Netti GS, Becherucci F, Gacci M, Carini M, Gesualdo L, Rotondi M, Maggi E, Lasagni L, Serio M, Romagnani S, Romagnani P (2008) Essential but differential role for CXCR4 and CXCR7 in the therapeutic homing of human renal progenitor cells. J Exp Med 205(2):479–490
Sanna-Cherchi S, Caridi G, Weng PL, Scolari F, Perfumo F, Gharavi AG, Ghiggeri GM (2007) Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr Nephrol 22(10):1675–1684
Davies JA, Fisher CE (2002) Genes and proteins in renal development. Exp Nephrol 10:102–113
Zhang Z, Iglesias D, Eliopoulos N, El Kares R, Chu L, Romagnani P, Goodyer P (2011) A variant OSR1 allele which disturbs OSR1 mRNA expression in renal progenitor cells is associated with reduction of newborn kidney size and function. Hum Mol Genet 20(21):4167–4174
McCarroll MN, Lewis ZR, Culbertson MD, Martin BL, Kimelman D, Nechiporuk AV (2012) Graded levels of Pax2a and Pax8 regulate cell differentiation during sensory placode formation. Development 139(15):2740–2750
Kochhar A, Fischer SM, Kimberling WJ, Smith RJ (2007) Branchio-oto-renal syndrome. Am J Med Genet A 143A(14):1671–1678
Aboudehen K, Hilliard S, Saifudeen Z, El-Dahr SS (2012) Mechanisms of p53 activation and physiological relevance in the developing kidney. Am J Physiol Renal Physiol 302(8):F928–F940
Ahn SY, Kim Y, Kim ST, Swat W, Miner JH (2013) Scaffolding proteins DLG1 and CASK cooperate to maintain the nephron progenitor population during kidney development. J Am Soc Nephrol 24:1127–1138
Park JS, Valerius MT, McMahon AP (2007) Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134:2533–2539
Grouls S, Iglesias DM, Wentzensen N, Moeller MJ, Bouchard M, Kemler R, Goodyer P, Niggli F, Gröne HJ, Kriz W, Koesters R (2012) Lineage specification of parietal epithelial cells requires β-catenin/Wnt signaling. J Am Soc Nephrol 23(1):63–72
Wang Q, Lan Y, Cho ES, Maltby KM, Jiang R (2005) Odd-skipped related 1 (Odd 1) is an essential regulator of heart and urogenital. Dev Biol 288:582–594
Chai OH, Song CH, Park SK, Kim W, Cho ES (2013) Molecular regulation of kidney development. Anat Cell Biol 46(1):19–31
Black MJ, Sutherland MR, Gubhaju L, Kent AL, Dahlstrom JE, Moore L (2013) When birth comes early: effects on nephrogenesis. Nephrology 18(3):180–182
Schreuder MF (2012) Safety in glomerular numbers. Pediatr Nephrol 27(10):1881–1887
Makrakis J, Zimanyi MA, Black MJ (2007) Retinoic acid enhances nephron endowment in rats exposed to maternal protein restriction. Pediatr Nephrol 22:1861–1867
Lelièvre-Pégorier M, Vilar J, Ferrier ML, Moreau E, Freund N, Gilbert T, Merlet-Bénichou C (1998) Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int 54(5):1455–1462
Batourina E, Gim S, Bello N, Shy M, Clagett-Dame M, Srinivas S, Costantini F, Mendelsohn C (2001) Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat Genet 27(1):74–78
Wingert RA, Selleck R, Yu J, Song HD, Chen Z, Song A, Zhou Y, Thisse B, Thisse C, McMahon AP, Davidson AJ (2007) The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet 3(10):1922–1938
Vaughan MR, Pippin JW, Griffin SV, Krofft R, Fleet M, Haseley L, Shankland SJ (2005) ATRA induces podocyte differentiation and alters nephrin and podocin expression in vitro and in vivo. Kidney Int 68(1):133–144
Zhang J, Pippin JW, Vaughan MR, Krofft RD, Taniguchi Y, Romagnani P, Nelson PJ, Liu ZH, Shankland SJ (2012) Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron Exp Nephrol 121(1–2):e23–e37
Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 129(6):1097–1110
Young NS, Maciejewski J (1997) The pathophysiology of acquired aplastic anemia. N Engl J Med 336(19):1365–1372
Smeets B, Angelotti ML, Rizzo P, Dijkman H, Lazzeri E, Mooren F, Ballerini L, Parente E, Sagrinati C, Mazzinghi B, Ronconi E, Becherucci F, Benigni A, Steenbergen E, Lasagni L, Remuzzi G, Wetzels J, Romagnani P (2009) Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol 20(12):2593–2603
Smeets B, Uhlig S, Fuss A, Mooren F, Wetzels JF, Floege J, Moeller MJ (2009) Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol 20(12):2604–2615
Ryu M, Migliorini A, Miosge N, Gross O, Shankland S, Brinkkoetter PT, Hagmann H, Romagnani P, Liapis H, Anders HJ (2012) Plasma leakage through glomerular basement membrane ruptures triggers the proliferation of parietal epithelial cells and crescent formation in non-inflammatory glomerular injury. J Pathol. doi:10.1002/path.4046
Sicking EM, Fuss A, Uhlig S, Jirak P, Dijkman H, Wetzels J, Engel DR, Urzynicok T, Heidenreich S, Kriz W, Kurts C, Ostendorf T, Floege J, Smeets B, Moeller MJ (2012) Subtotal ablation of parietal epithelial cells induces crescent formation. J Am Soc Nephrol 23(4):629–640
Smeets B, Kuppe C, Sicking EM, Fuss A, Jirak P, van Kuppevelt TH, Endlich K, Wetzels JF, Gröne HJ, Floege J, Moeller MJ (2011) Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol 22(7):1262–1274
Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC (2005) Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16(10):2941–2952
Peti-Peterdi J, Sipos A (2010) A high-powered view of the filtration barrier. J Am Soc Nephrol 21:1835–1841
Shankland SJ, Anders HJ, Romagnani P (2013) Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr Opin Nephrol Hypertens. doi:10.1097/MNH.0b013e32835fefd4
Lasagni L, Ballerini L, Angelotti ML, Parente E, Sagrinati C, Mazzinghi B, Peired A, Ronconi E, Becherucci F, Bani D, Gacci M, Carini M, Lazzeri E, Romagnani P (2010) Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells 28(9):1674–1685
Lasagni L, Lazzeri E, Shankland SJ, Anders HJ, Romagnani P (2013) Podocyte mitosis—a catastrophe. Curr Mol Med 13(1):13–23
Darisipudi MN, Kulkarni OP, Sayyed SG, Ryu M, Migliorini A, Sagrinati C, Parente E, Vater A, Eulberg D, Klussmann S, Romagnani P, Anders HJ (2011) Dual blockade of the homeostatic Chemokine CXCL12 and the proinflammatory Chemokine CCL2 has additive protective effects on diabetic kidney disease. Am J Pathol 179(1):116–124
Ohtaka A, Ootaka T, Sato H, Soma J, Sato T, Saito T, Ito S (2002) Significance of early phenotypic change of glomerular podocytes detected by Pax2 in primary focal segmental glomerulosclerosis. Am J Kidney Dis 39(3):475–485
Fatima H, Moeller MJ, Smeets B, Yang HC, D’Agati VD, Alpers CE, Fogo AB (2012) Parietal epithelial cell activation marker in early recurrence of FSGS in the transplant. Clin J Am Soc Nephrol 7(11):1852–1858
Hodgin JB, Borczuk AC, Nasr SH, Markowitz GS, Nair V, Martini S, Eichinger F, Vining C, Berthier CC, Kretzler M, D’Agati VD (2010) A molecular profile of focal segmental glomerulosclerosis from formalin-fixed, paraffin-embedded tissue. Am J Pathol 177(4):1674–1686
Peired A, Angelotti ML, Ronconi E, la Marca G, Mazzinghi B, Sisti A, Lombardi D, Giocaliere E, Della Bona M, Villanelli F, Parente E, Ballerini L, Sagrinati C, Wanner N, Huber TB, Liapis H, Lazzeri E, Lasagni L, Romagnani P (2013) Proteinuria impairs podocyte regeneration by sequestering retinoic acid. J Am Soc Nephrol 24(11):1756–1768
Pode-Shakked N, Dekel B (2011) Wilms tumor–a renal stem cell malignancy? Pediatr Nephrol 26(9):1535–1543
Pode-Shakked N, Shukrun R, Mark-Danieli M, Tsvetkov P, Bahar S, Pri-Chen S, Goldstein RS, Rom-Gross E, Mor Y, Fridman E, Meir K, Simon A, Magister M, Kaminski N, Goldmacher VS, Harari-Steinberg O, Dekel B (2013) The isolation and characterization of renal cancer initiating cells from human Wilms tumour xenografts unveils new therapeutic targets. EMBO Mol Med 5(1):18–37
Lindgren D, Boström AK, Nilsson K, Hansson J, Sjölund J, Möller C, Jirström K, Nilsson E, Landberg G, Axelson H, Johansson ME (2011) Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am J Pathol 178(2):828–837
Romagnani P (2011) Family portrait: renal progenitor of Bowman’s capsule and its tubular brothers. Am J Pathol 178(2):490–493
Romagnani P (2013) Of mice and men: the riddle of tubular regeneration. J Pathol 229:641–644
Smeets B, Boor P, Dijkman H, Sharma SV, Jirak P, Mooren F, Berger K, Bornemann J, Gelman IH, Floege J, van der Vlag J, Wetzels JF, Moeller MJ (2013) Proximal tubular cells contain a phenotypically distinct, scattered cell population involved in tubular regeneration. J Pathol 229(5):645–659
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Becherucci, F., Lazzeri, E., Lasagni, L. et al. Renal progenitors and childhood: from development to disorders. Pediatr Nephrol 29, 711–719 (2014). https://doi.org/10.1007/s00467-013-2686-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-013-2686-2