
Abstract
Background
The most common cause of end-stage renal disease in children can be attributed to congenital anomalies of the kidney and urinary tract (CAKUT). Despite this high incidence of disease, the genetic mutations responsible for the majority of CAKUT cases remain unknown.
Methods
To identify novel genomic regions associated with CAKUT, we screened 178 children presenting with the entire spectrum of structural anomalies associated with CAKUT for submicroscopic chromosomal imbalances (deletions or duplications) using single-nucleotide polymorphism (SNP) microarrays.
Results
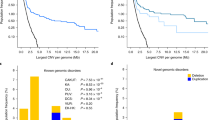
Copy-number variation (CNV) was detected in 10.1 % (18/178) of the patients; in 6.2 % of the total cohort, novel duplications or deletions of unknown significance were identified, and the remaining 3.9 % harboured CNV of known pathogenicity. CNVs were inherited in 90 % (9/10) of the families tested. In this cohort, patients diagnosed with multicystic dysplastic kidney (30 %) and posterior urethral valves (24 %) had a higher incidence of CNV.
Conclusions
The genes contained in the altered genomic regions represent novel candidates for CAKUT. This study has demonstrated that a significant proportion of patients with CAKUT harbour submicroscopic chromosomal imbalances, warranting screening in clinics for CNV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) account for one third of all congenital malformations detected by routine fetal ultrasonography [1]. Severity may vary from incidental clinical findings to chronic ill health and end-stage renal failure in childhood. In fact, 40–50 % of childhood renal failure worldwide can be attributed to CAKUT [2–4]. CAKUT comprises a broad spectrum of renal and lower urinary tract structural and functional abnormalities, including renal hypoplasia/dysplasia, renal agenesis, multicystic kidney, duplex kidney, hydroureter, hydronephrosis, vesicoureteric reflux (VUR) and obstruction at the vesicoureteric (VUJ) or ureteropelvic (UPJ) junction and posterior urethral valves (PUV) [5]. These anomalies can occur in isolation (nonsyndromic CAKUT) or as part of multiorgan malformation syndromes [6, 7].
Much of our understanding of CAKUT etiology comes through mouse studies in which single-gene mutations can result in kidney and urinary tract anomalies [8, 9]. The genes identified in these models have increased the range of genes available for genetic screening of families with CAKUT. Until recently, only a handful of CAKUT-causing genes had been screened in large cohorts, with mutations in HNF1B and PAX2 being the most prevalent [10, 11]. With the recent advancement in new and more affordable sequencing technologies, there has been a rapid increase in the number of CAKUT-causing genes identified [12–16]. Genes contained in altered genomic regions may also be responsible for CAKUT [17, 18]. Despite the fact that mutations in >20 genes have been shown to cause CAKUT, mutations responsible for the majority of CAKUT remain unknown [19]. In addition, variable CAKUT conditions have been reported in family members harbouring mutations in the same gene, demonstrating the complex genotype–phenotype relationship in CAKUT.
We used molecular karyotyping to detect submicroscopic chromosomal imbalances (microdeletions and microduplications) in a cohort of 178 patients presenting with a broad range of CAKUT as a first-tier approach prior to embarking on candidate-gene and exome sequencing approaches.
Methods
Participants
A consecutive series of patients with CAKUT presenting to tertiary referral paediatric nephrology and urology units at either The Royal Children’s Hospital or Monash Children’s Hospital, (Victoria, Australia) between September 2012 and August 2013 were invited to participate. A total of 201 patients from 195 unrelated families, aged newborn to 18 years old, were recruited. The patients presented with one or more of the following CAKUT conditions: renal hypoplasia/dysplasia, agenesis, multicystic or dysplastic disease, duplex kidney, VUR (grade 4 or 5), VUJ obstruction (>5 mm retrovesical ureter), ureteropelvic junction (UPJ) obstruction with hydronephrosis >10 mm, hydroureter and posterior urethral valve (PUV). Diagnoses were made following abnormal antenatal screening or early-onset urinary tract infection. Anomalies were further characterised by renal and/or bladder ultrasound (US), nuclear medicine studies [using mercaptoacetyltriglycine (MAG3), dimethylsulfoxide (DMSO) or diethylenetriamine pentaacetic acid (DTPA)], micturating cystourethrogram (MCUG), cystoscopy and retrograde pyelography, as clinically indicated. The diagnosis of PUV was made in all cases following cystoscopy and urethroscopy to visualise the valves. Patients with syndromic CAKUT as part of a diagnosed syndrome or who had more than three nonrenal congenital anomalies, suggesting a possible syndromic condition, were excluded. Extrarenal anomalies were detected in 20.5 % of patients: genital disorders (hypospadias, undescended testes, vaginal-cavity defect), macrocephaly, neurocognitive, neurobehavioural and learning problems, constipation, hypothyroidism and respiratory tract and cardiac problems. Patients were not screened for mutations in any known CAKUT-causing genes. Informed consent and/or assent were obtained from patients and/or parents, as appropriate. Parents of children determined to have CNVs were also invited to participate in the study, and consent was obtained. The study was approved by the Human Research Ethics Committee of the Royal Children’s Hospital, Monash Children’s Hospital and Monash University, and performed in accordance with the Declaration of Helsinki.
DNA extraction and molecular karyotyping by microarray
Molecular karyotyping was requested for all 201 patients; however, 23 tests were not performed, either because the patient did not present for blood collection or because the DNA quality or volume was insufficient. Genomic DNA was isolated from peripheral blood samples (171/178) (Janus/Chemagic MSI, Perkin Elmer, Waltham, MA, USA) or saliva (9/178) (Oragene OG-250), according to the manufacturer’s instructions. Molecular karyotyping was carried out on either the Illumina Human CytoSNP-12 v2.1 arrays (Illumina, San Diego, CA, USA), Illumina HumanCoreExome-12v1.0 (Illumina) or Affymetrix CytoScan 750 K (Affymetrix, Santa Clara, CA, USA). Microarray hybridisations for the Illumina platform were performed at the Australian Genome Research Facility (Melbourne, VIC, Australia) and for the Affymetrix platform at the Victorian Clinical Genetics Service, (Melbourne, VIC, Australia).
Data Analysis for CNV Detection
Raw data were processed using either Karyostudio (Illumina) or Chromosome Analysis Suite (Affymetrix) and probe-intensity measurements were normalised to a reference set of 100 and 150 clinical samples, respectively. CNVs that did not contain genes, were well-established polymorphisms or were <0.2 Mb (unless containing a gene of known pathogenic significance) were excluded. The significance of each CNV detected was determined by comparison with public CNV databases [i.e. Children’s Hospital of Philadelphia (CHOP), International Standards for Cytogenomic Arrays (ISCA), Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER), Database of Genomic Variants (DGV) and an in-house database of 30,000 Australian (multiethnic) samples analysed for CNVs locally]. Analysis was performed using UCSC Genome Browser March 2006 hg18 assembly for patients run on the Illumina Human CytoSNP-12 v2.1 platform, and the February 2009 hg19 assembly for patients run on the Affymetrix and Illumina HumanCoreExome-12v1.0 platforms.
CNVs were classified according to the following four criteria; Benign (well-established polymorphic variants); Pathogenic (well-established CNVs known to be associated with any genetic disease in humans, not just kidney disease); CNVs of uncertain significance (recurrent CNVs previously associated with phenotypic abnormality but with incomplete penetrance and/or variable expressivity); CNVs of unknown significance (novel CNVs that are not represented in the public databases, do not have a clear association with genetic disease or contain at least one gene of potential relevance but meet the laboratory reporting criteria). CNVs were reported according to these criteria to compare identical breakpoint boundaries. Long, continuous stretches of homozygosity (LCSH) >5 Mb long were also reported.
Results
A total of 201 patients were recruited for the study, 137 (68.2 %) male and 64 (31.8 %) female patients, representing the first report of an Australian CAKUT cohort to be analysed for genetic aberrations. No molecular karyotype result was available for 23 individuals, who either did not provide a blood/saliva sample (n = 21) or because DNA quality or volume was insufficient (n = 2). The most common primary anomalies displayed by the 178 patients who underwent molecular karyotyping were VUR (n = 29), PUV (n = 29), PUJ obstruction (n = 26), duplex kidney (n = 25) and renal hypodysplasia (n = 23) (Table 1). CNVs were detected in 18 of 178 patients (10.1 %) and LCSH in 4/178 (2.2 %). Within the anomaly groups, CNVs were identified at the highest frequency in patients with multicystic dysplastic kidney (MCDK) (3/10; 30 %), followed by patients with PUV (7/29; 24 %) (Table 1). Of the 18 patients with CNVs, patients diagnosed with PUV were the most frequent (n = 7; 39 %), followed by VUR (n = 4; 22 %), MCDK (n = 3; 17 %), hydronephroureterosis (n = 3) and dysplastic kidney (n = 1) (Table 1). Duplications were identified in 11 patients, deletions in five, both in one and one case was XYY (Table 2). The size of the rearrangements ranged from 0.2 Mb to 6.3 Mb (Table 2). CNVs >1,000 kb were the most common (44 %), followed by those between 500 and 999 kb (33 %), 250–499 kb (11 %) and 249–200 kb (11 %). Parents of 10/18 patients harbouring CNVs participated in the study; a de novo mutation was found in one case, and nine cases were inherited (Table 2). Interestingly, only 1/9 inherited CNVs was inherited maternally; the remainder were paternal. The cohort included six pairs of affected siblings in which both individuals underwent molecular karyotyping; a significant genomic alteration was identified in one of these families. The index patient (Case1 (CP1)) in this family has a brother who also has bilateral VUR and a deletion in 6q15q16.1 (Table 2). Thirty two families within the total cohort (32/195; 16.4 %) indicated that they had family members with CAKUT. Of the patients detected to have CNVs, only two (CP156 and CP88) reported family members with renal anomalies (Supplementary Table 1).
In the total cohort, seven patients (CP85, CP138, CP75, CP27, CP123, CP169, CP177) had pathogenic genomic disorders that have been described before (3.9 %) (Table 2). Of these seven, only two genomic regions involved genes previously associated with CAKUT; hepatocyte nuclear factor 1 homeobox B (HNF1B) (CP85) within the recurrent 17q12 deletion has been associated with renal cyst and diabetes (RCAD) syndrome (OMIM 137920) [20]; myosin, heavy chain 11, smooth muscle (MYH11) (CP75) within the recurrent 16p13.11 deletion has been described as a candidate gene in a patient with renal hypodysplasia [18] (Table 2 and Supplementary Table 1). The remaining 11 patients (6.2 %) had genomic disorders of unknown significance when compared with public databases and an in-house database of 30,000 Australian samples analysed for CNVs locally. Of these 11, four had duplications of 1q23.1, 4p16.1, 7q33 and 8q13.2q13.3, containing genes NEPH1, SLC2A9, AKR1B1 and EYA1, respectively, which have all been associated with renal anomalies or disease (Table 2 and Supplementary Table 1). Deletions or mutations in EYA1 have been associated with branchio-oto-renal syndrome (OMIM 113650) [7] and NEPH1 with nephrotic syndrome in mice [21]. Mutations in SLC2A9 have been associated with renal hypouricemia [22] and AKR1B1 with diabetic nephropathy [23]. To the best of our knowledge, there are no reported duplications of these genes being associated with renal anomalies; therefore, their clinical significance is unknown. Case 93 had a deletion (8q24.13) containing gene ZHX1, which has been associated with nephrotic syndrome in rats [24].
We identified only one genomic disorder in which the breakpoint occurred within a gene. CP138 carries a 0.3-Mb deletion of 7q31.1, resulting in loss of the first two exons of FOXP2 (NM_014491.3), which includes the ATG start site. There are no known reports of the role of FOXP2 in the urinary tract. In addition, 7/18 patients contained noncoding miRNA (Supplementary Table 1) within the altered regions. The most common anomaly within patients identified as having CNV was PUV (7/18; 39 %). The chromosomal regions affected were 3p25.1p25.2, 8q13.2q13.3, 9p24.2p24.1, 9q32, 11p15.2, 17p12 and Xq28 (see Supplementary Table 1 for list of genes).
Discussion
In this study using single-nucleotide polymorphism (SNP) microarrays, we identified CNVs in 10.1 % of patients with CAKUT. Using a similar approach, Sanna-Cherchi et al. reported that CNVs were identified in 16.6 % of individuals with CAKUT [18]. This difference in CNV frequencies between the two studies may be due to sample size (522 vs 178), differences in laboratory-reporting policies, demographic differences, proportion of individuals with multiple malformations versus isolated CAKUT, and the renal anomaly inclusion criteria of both studies. In particular, there are two notable differences: In our study, CNVs ≥200 kb were considered significant (commonly used in clinical practise), whereas Sanna-Cherchi et al. used a threshold of >100 kb, in which 47.9 % of their cohort had CNVs of 100–250 kb [18]. Secondly, our cohort represents an unselected, consecutive series of all cases of CAKUT presenting to tertiary referral units, and as such, includes a full range of CAKUT that is seen in clinical practise, whereas the cohort described by Sanna-Cherchi et al. is selected for renal agenesis, congenital solitary kidney and renal hypodysplasia only [18]. Using a different approach to detect genomic imbalances (array-based comparative genomic hybridization) in a relatively small cohort (n = 30), Weber et al., reported that 10 % of patients with syndromic CAKUT carried DNA microimbalances [17]. Although the frequency and range of CAKUT diagnoses are comparable with our study, Weber et al. focussed on syndromic CAKUT cases, whereas the majority of our cohort (80 %) consists of nonsyndromic CAKUT.
Approximately 6.2 % of the altered genomic regions in our CAKUT cohort have not been described before based on literature and database searches: i.e. contain identical breakpoint boundaries and represent the same CNV state (gain or loss). Some of these regions, however, contained genes that have been previously associated with CAKUT or kidney disease but in the context of being deleted or mutated rather than being duplicated, as was the case in patients in our study. For example, duplications were identified in our study that contained EYA1, NEPH1, SLC2A9 and AKR1B1 genes. These findings suggest that if these candidates are the causal genes, then overexpression (triplosensitivity) of these genes can also perturb normal kidney and lower urinary tract development, leading to CAKUT. Comparison of CNVs identified in our cohort with those described by Sanna-Cherchi et al. identified the region associated with RCAD (involving HNF1B) and velocardiofacial syndrome (VCFS) in both cohorts [18]. Two additional overlapping genomic regions were identified between the two studies involving 4p16.1 and 16p13.11. However, in our patients, CNV status of these regions was opposite to those described by Sanna-Cherchi et al. [18]. This further supports the idea that either a loss or gain of function of the most likely pathogenic genes within these regions may play a role in contributing to CAKUT.
To identify CAKUT-causing gene candidates within the altered chromosomal regions, literature and expression (GUDMAP www.gudmap.org) databases were searched to identify genes known to play a role in kidney and ureter development and/or disease and/or are spatially expressed in cell types that have been implicated in causing CAKUT if disrupted. For example, previous studies in mice and humans have demonstrated that disruption in ureter smooth muscle cell development and function can lead to VUR [25, 26]. Two of the four CNV patients (CP138 and CP75) who presented with VUR had very good candidate genes within the deleted regions that are expressed in smooth muscle cells in the ureter and bladder. CP138, a male patient who presented with bilateral grade V VUR, hydronephroureterosis and megacystis harboured a deletion of 7q31.1, resulting in deletion of the first two FOXP2 exons, which included one of the ATG start sites. Microarray expression and whole-mount RNA in situ hybridisation data demonstrate that Foxp2 is expressed specifically in smooth muscle cells of the ureter and bladder in the mouse (www.gudmap.org). Heterozygous mutations and deletions in FOXP2 have been associated with speech and language disorders (OMIM 602081) in humans and mice [27–30]. CP138 also presented with speech delay, as did his father who carries the same deletion. Loss of Foxp2 in mice also causes lung defects, leading to postnatal lethality. In addition, oesophageal smooth and skeletal muscle defects are seen in Foxp2 −/− ;Foxp1 +/− embryos [31]. There were no signs of these anomalies in our patient. There are no published reports of kidney defects in these mice or in patients with speech and language disorders associated with mutations in FOXP2. However, patients with congenital anomalies and an abnormal karyotype were excluded from these studies, and it is therefore possible that patients with renal anomalies and a possible FOXP2 mutation were excluded [28]. Although deletion within FOXP2 was also identified in the patient’s father, he has not been diagnosed as having a renal condition. However, we cannot rule out whether he or other family members have asymptomatic renal conditions. Given that the patient and his father have experienced speech delay suggests that FOXP2 function has been altered; however, it is equally possible that a separate genetic mechanism undetected by an SNP microarray is actually responsible for the urinary tract anomaly.
CP75, a male patient who also presented with bilateral high-grade VUR and a right scarred kidney had a 1.3-Mb deletion of region 16p13.11 containing MYH11, which is expressed in smooth muscle cells of the bladder and ureter (www.gudmap.org). A duplication of 16p13.11 spanning the 15.03–15.80 Mb region, which is within the de novo deleted region in CP75 (14.96–16.21 Mb), has recently been reported in a patient with renal hypodysplasia. Those authors also identified MYH11 as a likely candidate ([18] (Table 2). This region is a susceptibility locus for neurodevelopmental disorders, congenital anomalies and seizures [32] and shows incomplete penetrance and variable expressivity. There are also several reports of patients with renal anomalies with CNVs that span MYH11 (see Supplementary Table 1). It should be noted that this region has also been found at low frequency in healthy control individuals [18].
The aetiology of PUV is poorly understood. The condition is caused by remnant flaps of tissue (valves), located at the posterior urethra proximal to the verumontanum, that block bladder voiding. This results in dilation of the posterior urethra and a thick-walled trabeculated bladder [33]. Secondary to these conditions are VUR, hydronephrosis and chronic renal disease. Among the seven PUV patients with CNVs in our study, ~60 genes were contained in the altered genomic regions (Supplementary Table 1). Identifying causal genes within this subset of genes will aid in our understanding of how PUV develops.
Of the patients with PUV, patient 184 had only two genes in the affected genomic region (9p24.2p24.1 (4,390,991–4,606,385)). SLC1A1 is the major epithelial transporter of glutamate and aspartate in the kidney, and mutations in SLC1A1 result in human aminoacidurias [34]. In contrast, little is known about the role or expression of SPATA6L (spermatogenesis-associated 6-like), but either of these two genes could be considered a candidate for PUV. CP18 had a duplication in 8q13.2q13.3, which contains EYA1. As described above, mutations and deletions of EYA1 have been associated with branchio-oto-renal syndrome. However, there have been no reports of patients with a duplication spanning EYA1 resulting in renal anomalies. In addition, the codeletion of SULF1 and SLCO5A1, which are present in the 8q13.2q13.3 region duplicated in CP18, has been associated with mesomelia-syntostoses syndrome (MSS, OMIM 600383). MSS patients can also present with congenital hydronephrosis [35]. The genomic region deleted in these MSS patients does not include EYA1 (Supplementary Table 1). Mice deficient for Sulf1 and Sulf2 present with smaller kidneys [36]. In light of these findings SULF1, SLCO5A1 and EYA1 should be considered as possible CAKUT-causing candidates in CP18.
Three patients with CNVs presented with MCDK, and the altered genomic regions all contained genes known to be involved in either human or rodent models of kidney disease. CP85 contained a deletion within 17q12 containing HNF1B, which has been associated with RCAD (OMIM 137920). This patient also had a duplication of 22q11.21. Rearrangements in 22q11.21 have been associated with DiGeorge, VCFS [37] and vertebral defects, anal atresia, cardiac defects, tracheo-oesophageal fistula, renal anomalies and limb abnormalities (VACTERL) [38] syndrome, and patients with these syndromes can present with renal anomalies. Either one of these regions may have contributed to the renal phenotype in our patient CP85; however, HNF1B is one of the most prevalent CAKUT-causing genes [10, 11]. This patient was screened at 3 months of age and at that time did not present with any extrarenal anomalies suggesting syndromic CAKUT.
CP15 had a novel duplication within 1q23.1 containing NEPH1. Loss of function of Neph1 in mice leads to proteinuria and renal lesions resembling congenital nephrotic syndrome in humans [21]. Neph1 binds to nephrin, and mutations in NEPHRIN (NPHS1) are associated with nephrotic syndrome of the Finnish type [39]. However, to the best of our knowledge, no mutations have yet been identified in NEPH1 in patients with congenital nephrotic syndrome. Neph1 is expressed in podocytes and plays a critical role in maintaining filtration function of the glomerulus [40]. Interestingly, CP93, presenting with a dysplastic kidney, had a deletion in 8q24.13 containing Zhx1, which is also expressed in podocytes and is involved in regulating podocyte gene expression during nephrotic syndrome in the rat [24]. To the best of our knowledge, there are no reports of patients with a mutation in ZHX1 or loss-of-function mouse models. Although the roles of Neph1 and Zhx1 have been studied in podocytes, both genes are expressed in other cell types within the kidney and lower urinary tract (www.gudmap.org). Therefore, disruption of NEPH1 and ZHX1 in these cell types may play a role in the development of MCDK/DK.
CP88 with MCDK had a novel duplication in 7q33. This genomic region contains six genes, of which three belong to the aldo-keto reductase superfamily: AKR1B1, AKR1B10 and AKR1B15. An increase in AKR1B1 activity contributes to the development and progression of diabetic nephropathy [23]; and recently AKR1B10 has been implicated in diabetic nephropathy [41]. Given that this patient has an increased dose of both these enzymes may indicate diabetes.
CP72 presented with hydronephroureterosis and a 1-Mb duplication of 4p16.1 (9.35–9.68 Mb) containing three RefSeq genes: DRD5, SLC2A9 and WDR1. The most likely candidate gene in this region is SLC2A9. Loss-of-function mutations in SLC2A9 have been implicated in renal hypouricemia [42]. Sanna-Cherchi et al. identified two patients with renal hypodysplasia with a 17.23-Mb deletion (0.06–17.29 Mb) in 4p16, which spans the region duplicated in our patient [18]. The 17.23-Mb deletion is associated with Wolf-Hirschhorn syndrome [43]; the authors nominated FGFRL1, FGFR3 and SLC2A9 as likely candidates. These patients had extrarenal phenotypes. The genomic region affected in our patient excludes FGFRL1 and FGFR3. The difference in phenotype in the two patients in the Sanna-Cherchi et al. study compared with our patient, who has no extrarenal phenotypes, may be due to a SLC2A9 gain of function rather than loss of function or that FGFRL1 and FGFR3 were not disrupted in our patient [18].
Seven patients harbouring CNVs displayed miRNAs within the altered region. A literature search did not implicate any of these as playing a role in the kidney or ureter. In addition, a search of the target genes possibly regulated by these miRNAs generated an extensive list of possible targets that could in turn be involved in CAKUT. However, further analysis is required to validate these possibilities.
In our cohort 32/195 (16.4 %) families indicated that renal anomalies existed in their families. This self-reported information is likely to represent an underestimate, as other family members may have asymptomatic renal anomalies that would only be detected by renal ultrasound. In fact, it has recently been shown that there is a high frequency of CAKUT in asymptomatic first-degree relatives of patients with CAKUT [44]. For patients who presented with CNVs in our study, for which the mode of inheritance was determined, only one case (1/10) was sporadic and the remainder (9/10) inherited, demonstrating that pathogenesis of CAKUT is influenced by genetic factors.
Conclusion
In this study, we demonstrate that a significant proportion of patients with a broad spectrum of CAKUT harbour a CNV (10.1 %). This high frequency supports screening of future patients in the clinic for CNVs as a first-tier screen prior to sequencing approaches. Novel genomic regions were identified, providing a list of CAKUT-causing candidate genes. Future studies involving mice harbouring loss- or gain-of-function mutations in candidate CAKUT-causing genes and screening patient cohorts and families for mutations will determine genes that are pathogenic. Identifying these genes will aid in understanding the aetiology behind the spectrum of structural anomalies associated with CAKUT and aid in preventing the progression of end-stage renal disease in such patients.
References
Woolf AS, Price KL, Scambler PJ, Winyard PJ (2004) Evolving concepts in human renal dysplasia. J Am Soc Nephrol 15:998–1007
Neild GH (2010) Primary renal disease in young adults with renal failure. Nephrol Dial Transplant 25:1025–32
Australian and New Zealand Dialysis and Transplant Registry (ANZDATA). (2012) Annual Paediatric Report. http://www.anzdata.org.au/v1/report_2012.html.
North American Paediatric Renal Transplant Cooperative Study (NAPRTCS) (2008) Annual report. The EMMES Corporation, Rockville
Schedl A (2007) Renal abnormalities and their developmental origin. Nat Rev Genet 8:791–802
Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Ella M, Pierpont M, Sullivan MJ, Dobyns WB, Eccles MR (1995) Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 9:358–364
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C (1997) A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet 15:157–64
Airik R, Kispert A (2007) Down the tube of obstructive nephropathies: the importance of tissue interactions during ureter development. Kidney Int 72:1459–67
Yosypiv IV (2012) Congenital anomalies of the kidney and urinary tract: a genetic disorder? Int J Nephrol 2012, article ID 909083 http://dx.doi.org/10.1155/2012/909083
Renkema KY, Winyard PJ, Skovorodkin IN, Levtchenko E, Hindryckx A, Jeanpierre C, Weber S, Salomon R, Antignac C, Vainio S, Schedl A, Schaefer F, Knoers NV, Bongers EM; EUCAKUT consortium (2011) Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT). Nephrol Dial Transplant 26:3843–51
Thomas R, Sanna-Cherchi S, Warady BA, Furth SL, Kaskel FJ, Gharavi AG (2011) HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 26:897–903
Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, Innis JW, Hoskins BE, Hoefele J, Otto EA, Hildebrandt F (2012) Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int 81:196–200
Saisawat P, Kohl S, Hilger AC, Hwang D-Y, Gee HY, Dworschak GC, Tasic V, Pennimpede T, Natarajan S, Sperry E, Matassa DS, Stajić N, Bogdanovic R, de Blaauw MCL, Wijers CH, Bartels E, Schmiedeke E, Schmidt D, Märzheuser S, Grasshoff-Derr S, Holland-Cunz S, Ludwig M, Nöthen MM, Draaken M, Brosens E, Heij H, Tibboel D, Herrmann B, Solomon BD, de Klein A, van Rooij IA, Esposito F, Reutter HM, Hildebrandt F (2013) Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int 85(6):1310–1317
Sanna-Cherchi S, Sampogna RV, Papeta N, Burgess KE, Nees SN, Perry BJ, Choi M, Bodria M, Liu Y, Weng PL, Lozanovski VJ, Verbitsky M, Lugani F, Sterken R, Paragas N, Caridi G, Carrea A, Dagnino M, Materna-Kiryluk A, Santamaria G, Murtas C, Ristoska-Bojkovska N, Izzi C, Kacak N, Bianco B, Giberti S, Gigante M, Piaggio G, Gesualdo L, Kosuljandic Vukic D, Vukojevic K, Saraga-Babic M, Saraga M, Gucev Z, Allegri L, Latos-Bielenska A, Casu D, State M, Scolari F, Ravazzolo R, Kiryluk K, Al-Awqati Q, D’Agati VD, Drummond IA, Tasic V, Lifton RP, Ghiggeri GM, Gharavi AG (2013) Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med 369:621–629
Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, Reutter HM, Soliman NA, Bogdanovic R, Kehinde EO, Tasic V, Hildebrandt F (2014) Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. doi:10.1038/ki.2013.508
Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F (2014) Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol. doi:10.1007/s00467-013-2684-4
Weber S, Landwehr C, Renkert M, Hoischen A, Wühl E, Denecke J, Radlwimmer B, Haffner D, Schaefer F, Weber RG (2011) Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrol Dial Transplant 26:136–43
Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, Nees SN, Verbitsky M, Perry BJ, Sterken R, Lozanovski VJ, Materna-Kiryluk A, Barlassina C, Kini A, Corbani V, Carrea A, Somenzi D, Murtas C, Ristoska-Bojkovska N, Izzi C, Bianco B, Zaniew M, Flogelova H, Weng PL, Kacak N, Giberti S, Gigante M, Arapovic A, Drnasin K, Caridi G, Curioni S, Allegri F, Ammenti A, Ferretti S, Goj V, Bernardo L, Jobanputra V, Chung WK, Lifton RP, Sanders S, State M, Clark LN, Saraga M, Padmanabhan S, Dominiczak AF, Foroud T, Gesualdo L, Gucev Z, Allegri L, Latos-Bielenska A, Cusi D, Scolari F, Tasic V, Hakonarson H, Ghiggeri GM, Gharavi AG (2012) Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 91:987–97
Kohl S, Hwang DY, Dworschak GC, Hilger AC, Saisawat P, Vivante A, Stajic N, Bogdanovic R, Reutter HM, Kehinde EO, Tasic V, Hildebrandt F (2014) Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 25(9):1917–1922
Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O (1999) A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet 8:2001–8
Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, Mathur BN, Turner CA, Geske R, Montgomery CA, Starbuck M, Brandt M, Gupta A, Ramirez-Solis R, Zambrowicz BP, Powell DR (2001) Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol 21:4829–4836
Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, Wiriyasermkul P, Kikuchi Y, Oda T, Nishiyama J, Nakamura T, Morimoto Y, Kamakura K, Sakurai Y, Nonoyama S, Kanai Y, Shinomiya N (2008) Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet 83:744–51
Hodgkinson AD, Søndergaard KL, Yang B, Cross DF, Millward BA, Demaine AG (2001) Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int 60:211–218
Liu G, Clement LC, Kanwar YS, Avila-Casado C, Chugh SS (2006) ZHX proteins regulate podocyte gene expression during the development of nephrotic syndrome. J Biol Chem 281:39681–92
Radmayr C, Schwentner C, Lunacek A, Karatzas A, Oswald J (2010) Embryology and anatomy of the vesicoureteric junction with special reference to the etiology of vesicoureteral reflux. Ther Adv Urol 1:243–250
Chen F (2009) Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 24:1621–1632
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP (2001) A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 413:519–23
MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CS, Vernes SC, Vargha-Khadem F, McKenzie F, Smith RL, Monaco AP, Fisher SE (2005) Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet 76:1074–80
Turner SJ, Hildebrand MS, Block S, Damiano J, Fahey M, Reilly S, Bahlo M, Scheffer IE, Morgan AT (2013) Small intragenic deletion in FOXP2 associated with childhood apraxia of speech and dysarthria. Am J Med Genet A 161:2321–6
Groszer M, Keays DA, Deacon RM, de Bono JP, Prasad-Mulcare S, Gaub S, Baum MG, French CA, Nicod J, Coventry JA, Enard W, Fray M, Brown SD, Nolan PM, Pääbo S, Channon KM, Costa RM, Eilers J, Ehret G, Rawlins JN, Fisher SE (2008) Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol 18:354–62
Shu W, Lu MM, Zhang Y, Tucker PW, Zhou D, Morrisey EE (2007) Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development 134:1991–2000
Nagamani SC, Erez A, Bader P, Lalani SR, Scott DA, Scaglia F, Plon SE, Tsai C-H, Reimschisel T, Roeder E, Malphrus AD, Eng PA, Hixson PM, Kang SH, Stankiewicz P, Patel A, Cheung SW (2011) Phenotypic manifestations of copy number variation in chromosome 16p13.11. Eur J Hum Genet 19:280–6
Hodges S, Patel B, McLorie AA (2009) Posterior urethral valves. Sci World J 9:1119–1126
Bailey CG, Ryan RM, Thoeng AD, Ng C, King K, Vanslambrouck JM, Auray-Blais C, Vandenberg RJ, Bröer S, Rasko JE (2011) Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J Clin Invest 121:446–53
Isidor B, Pichon O, Redon R, Day-Salvatore D, Hamel A, Siwicka KA, Bitner-Glindzicz M, Heymann D, Kjelle’n L, Kraus C, Leroy JG, Mortier GR, Rauch A, Verloes A, David A, Le Caignec C (2010) Mesomelia-Synostoses Syndrome Results from Deletion of SULF1 and SLCO5A1 Genes at 8q13. Am J Hum Genet 87:95–100
Holst CR, Bou-Reslan H, Gore BB, Wong K, Grant D, Chalasani S, Carano RA, Frantz GD, Tessier-Lavigne M, Bolon B, French DM, Ashkenazi A (2007) Secreted sulfatases Sulf1 and Sulf2 have overlapping yet essential roles in mouse neonatal survival. PLoS One 2:e575
Shprintzen RJ (1994) Velocardiofacial syndrome and DiGeorge sequence. J Med Genet 31:423–424
Schramm C, Draaken M, Bartelsa E, Boemersc TM, Aretza S, Brockschmidt FF, Nöthena MM, Ludwig M, Reutter H (2011) De novo microduplication at 22q11.21 in a patient with VACTERL association. Eur J Med Gen 54:9–13
Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K (1998) Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1:575–582
Liu G, Kaw B, Kurfis J, Rahmanuddin S, Kanwar YS, Chugh SS (2003) Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J Clin Invest 112:209–221
Shaw N, Bingmei Y, Millward A, Demaine A, Hodgkinson A (2014) AKR1B10 is induced by hyperglycaemia and lipopolysaccharide in patients with diabetic nephropathy. Cell Stress Chaperones 19:281–287
Li J, Parker B, Martyn C, Natarajan C, Guo J (2013) The PMP22 gene and Its related diseases. Mol Neurobiol 47:673–698
Bergemann AD, Cole F, Hirschhorn K (2005) The etiology of Wolf-Hirschhorn syndrome. Trends Genet 21:188–95
Bulum B, Ozçakar ZB, Ustüner E, Düşünceli E, Kavaz A, Duman D, Walz K, Fitoz S, Tekin M, Yalçınkaya F (2013) High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr Nephrol 28:2143–7
Luo C, Yang YF, Yin BL, Chen JL, Huang C, Zhang WZ, Wang J, Zhang H, Yang JF, Tan ZP (2012) Microduplication of 3p25.2 encompassing RAF1 associated with congenital heart disease suggestive of Noonan syndrome. Am J Med Genet 158A:1918–23
Faivre L, Gosset P, Cormier-Daire V, Odent S, Amiel J, Giurgea I, Nassogne M-C, Pasquier L, Munnich A, Romana S, Prieur M, Vekemans M, De Blois MC, Turleau C (2002) Overgrowth and trisomy 15q26.1-qter including the IGF1 receptor gene: report of two families and review of the literature. Eur J Hum Genet 10:699–706
Rudnik-Schöneborn S, Schüler HM, Schwanitz G, Hansmann M, Zerres K (1996) Further arguments for non-fortuitous association of Potter sequence with XYY males. Ann Genet 39:43–6
Weiss AC, Airik R, Bohnenpoll T, Greulich F, Foik A, Trowe MO, Rudat C, Costantini F, Adams RH, Kispert A (2014) Nephric duct insertion requires EphA4/EphA7 signaling from the pericloacal mesenchyme. Development 141:3420–30
Acknowledgments
The authors thank the families for their participation in this study, which was supported by a National Health and Medical Research Council Project Grant (APP1021532).
URLs for data presented herein are as follows:
DECIPHER,
https://decipher.sanger.ac.uk/
GUDMAP Genitourinary Database Molecular Anatomy Project,
National Center for Biotechnology Information,
Online Mendelian Inheritance in Man (OMIM),
UCSC Genome Browser Home,
CHOPS, http://cnv.chop.edu/
International Standards for Cytogenomic Arrays (ISCA),
http://www.iscaconsortium.org/
Database of Genomic Variants (DGV)
http://dgv.tcag.ca/dgv/app/home
Funding
National Health and Medical Research Council Project Grant (APP1021532)
Note added in proof
Recently, Epha4 –/–; Epha7 –/–mice have been reported to display hydroureter, megaureter and hydronephrosis, therefore EPHA7 can be considered as a candidate for Case 1 (Table 2) [48].
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 26 kb)
Rights and permissions
About this article

Cite this article
Caruana, G., Wong, M.N., Walker, A. et al. Copy-number variation associated with congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 30, 487–495 (2015). https://doi.org/10.1007/s00467-014-2962-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-014-2962-9