
Abstract
Nephronophthisis (NPHP) is an autosomal recessive cystic kidney disease and the most frequent genetic cause of end-stage renal disease up to the third decade of life. It is caused by mutations in 11 different genes, denoted nephrocystins (NPHP1–11, NPHP1L). As an increasing number of these genes are identified, our knowledge of nephronophthisis is changing, thereby improving our understanding of the pathomechanisms in NPHP. Recent publications have described ciliary expression of nephrocystins together with other cystoproteins, such as polycystins 1 and 2 and fibrocystin. These findings have shifted our focus to a pathomechanism involving defects in ciliary function (ciliopathy) and planar cell polarity (PCP). In addition, discoveries of new nephrocystin genes have shown that the disease spectrum of NPHP is much broader than previously anticipated. Different forms of mutations within the same NPHP gene can cause different disease severity. In this review, we highlight the different hypotheses on the pathomechanisms for NPHP and underline the clinical variability of this disease. The clinical spectrum has become even more complex with the possibility of oligogenicity in NPHP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
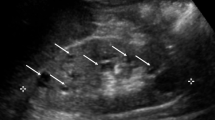
Nephronophthisis (NPHP) was first described in 1945 by Smith and Graham and 6 years later by Fanconi et al. [1, 2]. Whereas Smith and Graham called this disease “medullary cystic kidney disease”, Fanconi et al. introduced later the term “familial juvenile nephronophthisis” [1, 2]. The term “nephronophthisis” derives from the Greek and means “disintegration of nephrons”, which is one aspect of the histopathology. NPHP is an autosomal recessive tubulointerstitial nephropathy and one of the most frequent genetic disorders causing end-stage renal disease (ESRD) in children and adolescents [3]. The most frequent form of NPHP, called NPHP type 1, is characterized by ESRD at a mean age of 13 years [4]. The symptoms, primarily polyuria, polydipsia, secondary enuresis, growth retardation, and anemia, are very subtle and may start appearing in patients as young as 6 years of age [5]. In addition, NPHP has a rare infantile form, with onset of ESRD in patients younger than 4 years of age, and an adolescent form, in which the median age at onset of ESRD is 19 years [6]. Renal ultrasound scans at the initial stage of the disease show normally sized kidneys with increased echogenicity, poor corticomedullary differentiation, and corticomedullary cysts (Fig. 1), while imaging at a later stage reveals smaller, atrophic kidneys with increased echogenicity and more prominent cyst development [7]. Histological findings in NPHP are tubular atrophy with thickened or thinned tubular basement membrane, cysts at the corticomedullary border, and diffuse interstitial fibrosis (Fig. 2) [8, 9]. The histological characteristics of the infantile form of NPHP differ from those seen in juvenile NPHP, with the former combining features of NPHP (e.g. tubular cell atrophy, tubular cysts, and interstitial fibrosis) with features of polycystic kidney disease (PKD; e.g. enlarged kidneys, widespread cyst development) [10, 11]. Renal biopsy or mutation analysis is required for a definitive diagnosis of NPHP. Over 300 cases of NPHP have been published [8]. Between 10 and 15% of NPHP patients show extrarenal symptoms, including retinal degeneration (Senior–Loken syndrome), cerebellar vermis aplasia [Joubert syndrome (JS)], liver fibrosis, oculomotor apraxia (Cogan syndrome), and cone-shaped epiphysis [12]. A large variety of different syndromes have been published in association with NPHP (Table 1). One of the more prominent of these is the severe perinatal lethal Meckel–Gruber syndrome (MKS), which includes occipital encephalocele, polydactyly, microphthalmia, and liver fibrosis among other developmental abnormalities [13]. The incidence of NPHP varies largely, from 1:50,000 in Canada to approximately 1 in 1 million in the USA. In Finland, the incidence of NPHP is reported as 1 in 61,800 [5, 7, 14]. Finally, NPHP has also been diagnosed in adults, with renal failure occurring later in life [15].

Renal ultrasound in nephronophthisis (NPHP). The renal ultrasound shows smaller bilateral kidneys, increased echogenicity (compare to abnormally lower echogenicity of liver), decreased cortico-medullary differentiation, and cortico-medullary cyst formation

Renal histopathology in NPHP. Renal histopathology in NPHP is characterized by the triad of tubular cysts, tubular basement membrane disruption, and interstitial fibrosis with interstitial cell infiltration. Periodic acid-Schiff (PAS) staining, magnification 20×
The first patients with NPHP were published under the term “medullary cystic kidney disease” (MCKD). Today MCKD refers to an autosomal dominant cystic kidney disease with hypertension and hyperuricemia that shares the histology of NPHP [3]. Therefore, NPHP and MCKD have been combined to form the “nephronophthisis–MCKD disease complex” [8]. MCKD type 2 is caused by mutations in the Uromodulin (UMOD) gene [16]. For NPHP eleven different genes have been identified by positional cloning these genes are denoted nephrocystins (NPHP1–11, NPHP1L) (Table 2). The most frequent mutation in NPHP is a homozygous deletion of NPHP1, which causes approximately 20% of cases of the isolated renal form of NPHP, whereas the mutations in the other genes contribute to less than 3% each [12]. However, the causative gene is still unknown in approximately 70% of all individuals with NPHP [12]. Two mutations in a single recessive gene of any of these 11 NPHP genes are sufficient to cause NPHP. Both the type of gene mutated and the nature of the mutation(s) determine the severity of the phenotype in terms of age of onset and extent of organ involvement. Modifier effects have also been suggested [17].
The aim of this review is to emphasize the changes that have taken place in our understanding of the pathophysiology of NPHP. We will also outline the widening spectrum of phenotypes.
Additional phenotypes associated with nephronophthisis
Oculomotor apraxia type Cogan
Oculomotor apraxia (OMA) type Cogan [OMIM%257550] is characterized by an impaired horizontal gaze and nystagmus, resulting in the affected individual having to move the head by jerky head movements in order to follow objects. OMA is a rare ocular sign found in NPHP patients with NPHP1 and NPHP4 mutations [12, 18]. It is also encountered in JS. A study linking cerebellar vermis aplasia and OMA has also been published [19].
NPHP with retinitis pigmentosis (Senior-Loken syndrome)
About 10–15% of patients with NPHP have retinal degeneration, also called retinitis pigmentosa (RP) [20, 21], which can result in early and severe visual impairment. Early onset of RP resembles Leber’s congenital amaurosis (LCA), whereas late onset is characterized by night blindness and progressive visual loss. RP is diagnosed on the basis of fundoscopy and electroretinography findings. The association of retinitis pigmentosa and NPHP is called Senior–Loken syndrome (SLSN) [OMIM #266900, %606995, #606996, #609254, #610189]. The pathomechanism of the retinopathy is currently unknown but may be related to the function of the connecting cilium and centrosomes of photoreceptors where nephrocystin proteins are expressed [12, 17, 22, 23]. The frequency of RP with NPHP can range from 6 to 100%, depending on the NPHP mutation (e.g. 6% for NPHP1, 10% for NPHP2/INV, 100% for NPHP5 and NPHP6) [12]. Retinal symptoms due to NPHP1 deletions usually present with a milder phenotype. SLSN has also been found in a few patients with NPHP1 to NPHP4 mutations. Several genes causing NPHP (NPHP1, NPHP8/RPGRIP1L) and NPHP-related phenotypes (AHI1 in JS) are involved in photoreceptor development and act as modifiers of retinal degeneration [24–26].
Cerebellar vermis aplasia with NPHP (Joubert syndrome)
Joubert syndrome [OMIM%213300] is an autosomal recessive developmental disorder consisting of cerebellar vermis aplasia [revealed in magnetic resonance brain imaging (MRI) as “molar tooth sign”] (Fig. 3), cerebellar ataxia, hypotonia, oculomotor apraxia, neonatal tachypnea, mental retardation, and retinal degeneration [27]. NPHP is found in 17–27% of JS patients [28]. Additional associated symptoms include liver fibrosis, ocular coloboma, and polydactyly [27]. JS is also called CORS (cerebello-oculo-renal syndrome). This syndrome is caused by mutations in NPHP6/CEP290, which encodes nephrocystin-6 and NPHP8/RPGRIP1L, which encodes nephrocystin-8 [29–31]. Additional mutations in JS have been found in other genes, including AHI1, MKS3, ARL13B, CC2DA2, INPP5E, and TMEM216 [32–38]. Rare mutations in NPHP1 and NPHP4 have been reported in patients with JS [39, 40]. Cerebral symptoms due to NPHP1 deletions usually present with a milder phenotype.

Brain magnetic resonance imaging (MRI) axial image at the level of the superior cerebellar peduncles of a Joubert syndrome (JS) patient. The “molar tooth sign” (boxed) is a classical neuroradiological characteristic of JS and is characterized by cerebellar vermis aplasia, thickened and elongated superior cerebellar peduncles, and a deepened interpeduncular fossa
Meckel–Gruber syndrome
Meckel–Gruber syndrome is characterized by renal cystic dysplasia, occipital encephalocele, microphthalmia and other central nervous system malformations, polydactyly, situs inversus, bile duct proliferation, and pulmonary hypoplasia. Like all other forms of NPHP, MKS is inherited in an autosomal recessive mode. Newborns with MKS rarely survive longer than 2 weeks. A strong allelism has recently been described in MKS where two truncating mutations (nonsense, frame-shift or splice site mutations) in the genes MKS1, MKS3, NPHP3, NPHP6/CEP290, and NPHP8/RPGRIP1L cause MKS, whereas the presence of at least one missense mutation causes the milder phenotype of JS or SLSN [13, 17, 30, 41, 42]. The defects in MKS represent developmental defects, whereas in NPHP and SLSN, defects of the retina and kidney are degenerative in nature.
Liver fibrosis
A few cases of NPHP-like ciliopathies together with periportal liver fibrosis have been described. Hepatomegaly, portal fibrosis, and bile duct proliferation were described in a patient with the NPHP3 mutation [43]. Liver fibrosis is also found in Arima syndrome (cerebro-oculo-hepato-renal syndrome) and Meckel syndrome. Mutations in MKS3/TMEM67 have very recently been found to represent a major gene mutated in NPHP-like ciliopathies that exhibit a liver fibrosis phenotype [44]. In this context, two truncating mutations cause MKS with biliary duct dysplasia, whereas the presence of at least one missense mutation among the two alleles cause only NPHP-like degenerative liver fibrosis. A similar genotype–phenotype correlation has been described for NPHP6/CEP290 and MKS1 [12].
Skeletal defects
Skeletal symptoms associated with NPHP are rare. The most frequent skeletal manifestation of NPHP is the appearance of cone-shaped epiphyses of the phalanges, also called Mainzer–Saldino syndrome [45]. There can also be an association with cerebellar ataxia, retinal degeneration, and polydactyly [46]. Jeune syndrome (asphyxiating thoracic dysplasia), with the patients having short limbs and a small thorax, and Ellis van Creveld syndrome, with the patients having short stature, short extremities, and polydactyly, can also occur in association with NPHP [47, 48].
Cardiac defects
Rare cases of cardiac defects (e.g. ventricular septal defect) have been published in association with infantile NPHP and mutations in NPHP2/inversin and NPHP3 [42, 49]. Animal models (in zebrafish and mice) for NPHP2/inversin confirmed the association of cystic kidney disease with cardiac septal defects [49].
Diagnosing NPHP
Symptoms and signs of NPHP manifest slowly and are subtle. The medical history may reveal polyuria, polydipsia, or secondary enuresis usually starting around 6 years of age. General symptoms of renal failure, such as fatigue, pruritus, nausea, vomiting, uremic gastritis, anemia, and growth retardation, may be present. A family history of consanguity may hint at an autosomal recessive disease, and the physical exam may reveal any of the associated extrarenal phenotypes or may be unremarkable with the exception of pallor and short stature. Urinalysis may reveal a renal concentration defect (<400 mosm/kg in morning urine). Renal function should be evaluated and a complete blood count (CBC), liver function tests, and coagulation tests performed. Renal ultrasound may show small kidneys with poor corticomedullary differentiation and corticomedullary cysts and, if present, liver fibrosis. A renal biopsy can be performed if the kidneys are not too small and atrophic at the timepoint of diagnosis. However, molecular genetic analysis is currently the mainstay for making a definitive diagnosis of an NPHP-like ciliopathy. If there is any indication of cerebellar involvement, an MRI may be indicated to rule out the “molar tooth sign”, which indicates JS. If a diagnosis of NPHP is being considered, an ophthalmological exam should be performed to rule out retinal degeneration. There are only two ways to obtain a definitive diagnosis of NPHP: renal biopsy or mutation analysis (www.renalgenes.org). Mutation analysis should be initiated in the context of genetic counseling.
Genes mutated in NPHP
NPHP1 is located at cell contacts and ciliary transition zone
Homozygous deletions of NPHP1 on chromosome 2q13 cause NPHP type 1, the most frequent form of NPHP, accounting for about 20% of all cases [50, 51]. The homozygous NPHP1 deletion is also found in NPHP patients with OMA [18], Senior–Loken syndrome [52], and, very rarely, JS, which may be due to an epistatic effect by the AHI1 gene [39, 53]. A heterozygous deletion of NPHP1 has been associated with a NPHP1 point mutation in a few patients.
NPHP1 encodes nephrocystin-1, which is located at the adherens junctions and focal adhesions of renal epithelial cells. In the human kidney, nephrocystin-1 is expressed primarily in collecting duct cells [54]. Nephrocystin-1 has been reported to interact with p130cas, focal adhesion kinase 2, tensin, and filamin A and B [55–57]. In addition, it has also been shown to interact with nephrocystin-2/inversin, nephrocystin-3, nephrocystin-4, and Jouberin, indicating that there is a protein complex of nephrocystins [40, 43, 49, 58]. This complex of proteins may function in multiple intracellular compartments, including the cilium, cell–cell adherens junctions, and focal adhesions [49, 55, 56]. When the ciliary localization of nephrocystin-2/inversin was discovered, nephrocystin-1 was also identified in cilia [49]. The primary ciliary localization was later refined to the transition zone (e.g. at the base of the cilium) in respiratory and renal epithelium and to the connecting cilium of the photoreceptor [59]. PACS-1 and casein kinase 2 phosphorylation are required for the targeting of nephrocystin-1 to the transition zone [60]. Due to the expression pattern of nephrocystin-1 in the adherens junctions and focal adhesions and its interaction with integral components of these structures (e.g. p130CAS), nephrocystin-1 was initially thought to result in a defective cell–cell and cell-matrix signaling—which resulted in the “adherens junction/focal adhesion hypothesis” [3]. This hypothesis was later linked to the “ciliary hypothesis” by the finding that nephrocystin-4, an interaction partner of nephrocystin-1, co-localizes with β-catenin at cell–cell contact sites and to primary cilia in polarized renal epithelial cells and is found in centrosomes in dividing cells [61].
Mutations in nephrocystin-2 cause infantile NPHP, situs inversus, and cardiac defects
Recessive mutations of nephrocystin-2/inversin were identified as the cause of NPHP2 based on a candidate gene approach and positional cloning [10, 49]. Characteristics of NPHP2 are: (1) age of onset of ESRD in children younger than 5 years of age, (2) a renal ultrasound finding of normal or enlarged kidneys, (3) possible antenatal presentation with oligohydramnios, (4) renal histology showing an overlap of features characteristic of NPHP and autosomal dominant PKD (ADPKD), and (5) possible association with situs inversus and cardiac abnormalities (e.g. ventricular septal defects, VSDs) [11]. Retinitis pigmentosa is a rare finding in patients with NPHP2/inversin mutations [62]. Even though nephrocystin-2/inversin mutations are rare (1% of all NPHP patients), the identification of nephrocystin-2/inversin mutations as causing NPHP2 resulted in a major breakthrough in our understanding of NPHP: nephrocystin-2/inversin was found to be co-expressed in primary cilia of renal tubular cells with nephrocystin-1 and interacts with nephrocystin-1 and β-tubulin [49]. β-tubulin represents a major protein of the microtubule axoneme of primary cilia. This discovery was one of the first hints towards a unifying theory of renal cystogenesis, which implies that all genes causing cystic kidney disease are expressed in primary cilia, basal bodies, or centrosomes [3, 63]. Nephrocystin-2/inversin was recently shown to function as an anchor for NPHP3 and NPHP9/Nek8 in cilia [64], while other studies have revealed a cell cycle-dependent expression of nephrocystin-2/inversin in the mitotic spindle in mitosis, the mid-body in cytokinesis, and in cilia, the basal body, and centrosomes in the interphase [65]. Cell-cycle-specific expression of nephrocystin-2/inversin in these organelles supports the development of the “planar cell polarity” (PCP) hypothesis of the pathogenesis of NPHP (see section on Planar cell polarity). This hypothesis was supported by Simons et al., who demonstrated a role for nephrocystin-2/inversin in the Wnt signaling pathway, which is involved in planar cell polarity [66]. If nephrocystin-2/inversin is defective, the canonical pathway of the Wnt signaling will dominate over the non-canonical form, thereby disrupting apical–basolateral polarity of the renal epithelial cells. In addition to mutations in NPHP2/inversin, mutations in NPHP3 and NPHP9/NEK8 have also been identified in patients with infantile NPHP [67, 68].
NPHP3 mutations are a rare cause of NPHP but may cause a wide spectrum of disease
NPHP3 was mapped and identified in one large Venezuelan kindred with NPHP [43]. It encodes nephrocystin-3, which interacts with nephrocystin-1 and inversin [42, 43]. Nephrocystin-3, like inversin, may inhibit the canonical Wnt signaling pathway [42]. Moreover, mutations in the murine ortholog Nphp3 cause the renal cystic mouse mutant pcy, which generates a hypomorphic Nphp3 allele [43]. Interestingly, the pcy mouse model responds very well to treatment with a vasopressin-2 receptor antagonist [69]. The Nphp3 knockout mouse model shows situs inversus, congenital heart defects, and embryonic lethality, a phenotype very similar to MKS, thus confirming that complete loss-of-function mutations cause the developmental phenotype of MKS, whereas missense mutations cause primarily degenerative phenotypes [42]. In humans, mutations in NPHP3 result in a variety of phenotypes ranging from adolescent NPHP, NPHP with liver fibrosis, NPHP with RP, infantile NPHP to MKS, dependent on the nature of the mutated alleles [42, 43, 67]. Truncating mutations result in developmental, early-onset phenotypes resembling MKS, whereas non-truncating mutations result in milder degenerative phenotypes with a later age of onset.
Nephrocystin-4: combining the cilia and the cell-junction hypothesis
NPHP4 mutations were identified on chromosome 1p36 by positional cloning [40, 70]. NPHP4 encodes nephrocystin-4, which localizes to primary cilia, basal bodies, and centrosomes [61], interacts with nephrocystin-1 and nephrocystin-8/RPGRIP1L, and forms complexes with α-tubulin [30, 40]. Nephrocystin-4 and nephrocystin-1 have recently been shown to associate with PALS1/PATJ and Par6, which are required for epithelial morphogenesis [71]. Mutations in NPHP4 account for about 2% of NPHP cases and can result in isolated NPHP and in NPHP with OMA and SLSN.
NPHP5 mutations cause a retinal–renal phenotype
Homozygous truncating mutations of NPHP5/IQCB1 cause SLSN with early-onset RP in association with NPHP [72]. NPHP5/IQCB1 encodes nephrocystin-5, which contains two IQ calmodulin binding sites and a coiled-coil domain. Nephrocystin-5 interacts directly with calmodulin via the IQ domains and forms a complex with the retinitis pigmentosa GTPase regulator (RPGR) [72]. Mutations in RPGR result in X-linked retinitis pigmentosa. Prior to the “ciliary hypothesis”, the pathologic basis for retinal involvement in SLSN was not well understood. The strong association of NPHP5/IQCB1 mutations prompted further expression studies, and nephrocystin-5 was found to be expressed in the connecting cilia of photoreceptors [72]. This finding supported the ciliary hypothesis and provided a potential pathologic basis for the retinal–renal phenotype of SLSN. The primary cilium of renal epithelial cells corresponds to the connecting cilia of the photoreceptors of the retina [73]. In addition to nephrocystin-5, the expression of nephrocystin-6 has also been shown in the connecting cilium of the photoreceptors, and nephrocystin-5 and -6 have also been shown to interact with each other [29, 74].
NPHP6 mutations cause JS
NPHP6/CEP290 mutations were found to cause JS [29, 75]. The gene product, nephrocystin-6, activates and interacts with ATF4 (activating transcription factor 4), a transcription factor which may be involved in cAMP-dependent renal cyst formation [69]. Nephrocystin-6 constitutes a part of the centrosomal proteome [29, 76]. Similar to the NPHP2/INV and NPHP4 gene products, nephrocystin-6 is localized at centrosomes and at the mitotic spindle [29]. Knockdown of the nphp6 ortholog in zebrafish resulted in renal cysts, retinal degeneration, cerebellar malformation, and a defect of planar cell polarity, thereby recapitulating the human JS phenotype [29]. NPHP6/CEP290 mutations can also result in JS without renal involvement and in a broader variety of phenotypes ranging from isolated NPHP, SLSN, and JS to MKS and Bardet–Biedl syndrome (BBS) [17, 29, 77–79]. Interestingly, mutations of NPHP6/CEP290 also cause isolated LCA, accounting for 21% of the reported cases of this disease [80]. The mouse model rd16 has an inframe deletion of 300 amino acids in Nphp6/Cep290, which mimics the RP phenotype without showing brain or kidney abnormalities, resulting in a hypomorphic allele [23].
Increased apoptosis and fibrosis results in NPHP7
NPHP7/GLIS2 mutations were identified as causing isolated NPHP in a large Cree Indian kindred. Affected individuals developed renal failure prior to 8 years of age [81]. NPHP7/GLIS2 encodes the Kruppel-like zinc-finger transcription factor “Gli-similar protein 2”. NPHP7/GLIS2 localizes to the primary cilia and the nucleus. A mouse knockout model of Glis2 revealed severe renal atrophy and fibrosis [81]. The kidneys of the Glis2 mutant mice showed upregulation of genes that promote epithelial-to-mesenchymal transition and fibrosis [81]. NPHP7/GLIS2 is related to GLI transcription factors and thereby links the pathogenesis of NPHP to the sonic hedgehog pathway, which is involved in cell fate determination, tissue patterning and maintenance of stem cell pools in postembryonic tissues.
NPHP8/RPGRIP1L mutations cause JS and MKS
NPHP8/RPGRIP1L mutations were identified by positional cloning as causing JS-like phenotype [cerebro-oculo-renal syndrome (CORS)] [30]. NPHP8/RPGRIP1L encodes the protein RPGRIP1L (retinitis pigmentosa GTPase regulator interacting protein 1-like) which co-localizes with NPHP4 and NPHP6 at centrosomes and basal bodies [30]. Two missense mutations result in the CORS phenotype, whereas one or more truncating mutations cause the more severe phenotype of Meckel-Gruber syndrome [30, 31]. RPGRIP1L was shown to interact with nephrocystin-4 and missense mutations in NPHP8/RPGRIP1L of affected patients reduced the RPGRIP1L interaction with nephrocystin-4 [30, 82]. Additional characteristics of affected patients included polydactyly, scoliosis, pituitary agenesis, and partial growth deficiency. The corresponding Rpgrip1l (Ftm forfused-toesmouse) knockout mouse exhibits cerebral, renal and hepatic defects similar to CORS and Meckel-Gruber syndrome. Recently, a genotype-phenotype correlation became evident for NPHP3, NPHP6 and NPHP8, in which the presence of two truncating mutations causes the severe, early-onset developmental dysplastic phenotype of MKS with broad organ involvement, whereas at least one missense mutation (of the two recessive mutations) causes a milder, late-onset, degenerative phenotype with more restricted organ involvement.
NPHP8/RPGRIP1L mutations were recently shown to cause retinal degeneration [26]. Missense mutations and the sequence variant (A229T) were found in patients with LCA and retinal degeneration combined with other ciliopathies as BBS, SLSN, JS and MKS.
Linking cilia and cell-cycle defects in NPHP
NPHP9/NEK8 encodes the NEK8 protein (never in mitosis A-related kinase 8), which if mutated causes NPHP type 9. Three highly conserved missense mutations were found in three different individuals [68]. One patient with a homozygous NPHP9/NEK8 mutation developed infantile NPHP at age of 3 years [68]; in two other patients the second recessive mutation was not identified. One of these two latter patients had an additional homozygous NPHP5/IQCB1 mutation and RP in addition to NPHP [68], with one of the mutations found in the RCC1 domain of NEK8. The corresponding jck mouse model, which is characterized by cystic renal disease, is caused by a missense mutation (G448V) in the RCC1 domain [83]. Expression studies of all three mutated proteins in medullary collecting duct cells showed defects of centrosomal and ciliary localization of NEK8 [68]. NPHP9/NEK8 is important in the regulation of the cell cycle, providing a link between nephrocystins and the role of centrosomes for cell-cycle regulation. Interestingly, polycystin-1 and polycystin-2 (the two genes mutated in ADPKD, which are also expressed in primary renal cilia) signaling has also been linked to cell-growth regulation involving the JAK–STAT pathway [84–86]. The jck and cpk mice, which represent models for PKD, were successfully treated by the cyclin-dependent kinase inhibitor roscovitine, which underlines the involvement of cell-cycle regulation in renal cystic disease [87].
NPHP11/MKS3 may cause JS or MKS
Mutations in NPHP11/MKS3/TMEM67 result in a wide spectrum of NPHP-like ciliopathies ranging from NPHP with liver disease, to JS and Meckel syndrome. NPHP11/MKS3/TMEM67 encodes the protein Meckelin, which was found to be expressed in the primary cilia and the plasma membrane [88]. Missense mutations in NPHP11/MKS3/TMEM67 were discovered in a population characterized by NPHP and liver fibrosis [44, 89]. Four new missense mutations were found in five kindreds, resulting in a hypomorphic allele and leading to a milder phenotype than the truncating mutations [44]. Doherty et al. also identified some patients with COACH syndrome [cerebellar vermis hypoplasia, oligophrenia (developmental delay/mental retardation), ataxia, coloboma, and hepatic fibrosis], which is a JS-related disorder, and found MKS3/TMEM67 mutations in 19/23 families (83% of the cohort) [89]. Because of the strong association of MKS3/TMEM67 mutations and the NPHP plus liver fibrosis phenotype, MKS3/TMEM67 is now also called NPHP11 [44].
NPHP1L–a nephronophthisis-like phenotype
NPHP1L/XPNPEP3 was identified by homozygosity mapping in two consanguineous kindreds on chromosome 22. The renal histopathology was consistent with NPHP, and a splice site mutation and a 4-bp deletion, causing two loss-of-function mutations, were discovered [90]. The phenotype included hypertension, cardiomyopathy, renal failure, and seizures [90]. A complex-I-defect mitochondropathy with decreased NADH-CoQ-oxireductase activity was discovered. NPHP1L/XPNPEP3 isoform 1 has a N-terminal 79 amino acid sequence that is responsible for mitochondrial localization and suggests a mitochondrial function of this protein [90]. Because this is the first gene that is not consistent with the cilia hypothesis, it may only cause a phenocopy of NPHP but may not belong to the family of ciliopathies [90].
The “ciliary hypothesis” of NPHP
Ciliary expression of nephrocystins may explain organ involvement in NPHP
To date, all of the proteins of genes that cause cystic kidney disease are expressed in the primary renal cilium, basal bodies, centrosomes, or the mitotic spindle in a cell-cycle-dependent fashion [3, 63] (Fig. 4). Even Uromodulin, the gene altered in autosomal dominant medullary cystic kidney disease type 2 (MCKD2), which shares a similar histopathology with autosomal recessive nephronophthisis, was found to be expressed in cilia [91]. The primary cilium is an organelle of almost every cell and projects like an antenna from the cell surface. It contains an axoneme, which consists of 9 + 0 microtubular doublets (in contrast to motile cilia which contain 9 + 2 microtubular doublets) [3]. The axoneme is assembled by “intraflagellar transport” (IFT) because no protein biosynthesis occurs within the cilium [3]. Cilia are involved in photosensation, mechanosensation, osmotic, olfactory, and temperature sensation [3]. The basal body from which the cilium is assembled is located at the root of the cilium and derives from the mother centriole [3].

Subcellular localization of the nephrocystins. Nephrocystins are detected in the primary cilia, basal bodies, the mitotic spindle, focal adhesions, and adherens junctions. Most nephrocystins are expressed in the primary cilium (PC, enlarged box), the basal body (BB), and centrosomes (Cen) in a cell cycle-dependent manner. NPHP1 is expressed in the transition zone (TZ), focal adhesion plaques (FAP), adherens junctions (AJ), and tight junctions (TJ). Arrows in the cilium show the directions of the anterograde and retrograde transport along the microtubule transport. The intraflagellar transport is mediated by kinesin 2, a heterotrimeric protein that is composed of two motor units (Kif3a and Kif3b) and one nonmotor unit (KAP3). Sensory cilia transfer external stimuli. Wnt and hedgehog (Shh) signaling interfere with planar cell polarity by affecting the orientation of the centrosomes and mitotic spindles (Adapted by permission from Macmillan Publishers Ltd: Nature Genetics 34:355-356, 2003 [63])
Nephrocystin-1 and nephrocystin-4 are evolutionary conserved proteins in the nematode C. elegans, with expression of the nephrocystin-1 and nephrocystin-4 orthologs found in ciliated neurons of the head (amphids) and tail (phasmids) [92]. The expression pattern showed significant overlap with the localization of other cystoprotein orthologs in C. elegans, such as polycystin-1 (lov-1), polycystin-2 (pkd-2), or multiple orthologs of the BBS proteins [92, 93]. Knockdown of the nephrocystin-1 and nephrocystin-4 orthologs resulted in a phenotype that was very similar to that of the knockout nematodes of the polycystin-1 and polycystin-2 orthologs (lov-1 and pkd-2, respectively) [92]. Nephrocystin-1 and nephrocystin-4 orthologs were found to be required for morphologic integrity, and nephrocystin-4 contributes to the regulation of the life span of the nematode [94, 95]. For some nephrocystins (nephrocystin-2, -4, and -6), evolutionary conservation reaches back more than 1.5 billion years to a unicellular organism called Chlamydomonas reinhardtii. Nephrocystin-4 and a minimum of six other proteins of the BBS complex are part of the basal body proteome in Ch. reinhardtii, which if mutated causes impaired IFT and defective flagellar propulsion [86, 93].
The function of cilia in NPHP has still not been completely resolved. Renal cilia may sense the tubular flow of urine [96]. Polycystin-1 and polycystin-2 have been shown to be capable of sensing flow, resulting in intracellular calcium signaling [96]. Other phenotypes associated with NPHP can also be explained by the ciliary hypothesis. Nephrocystin-5 and nephrocystin-6 were found to be expressed in the connecting cilium of the photoreceptor [29, 72], which is responsible for the daily transport of rhodopsin [3]. Impaired rhodopsin transport results in retinitis pigmentosa. Ciliary expression of nephrocystins has also been reported to be present in the central nervous system and the cholangiocytes of the liver, which could explain the association with JS and liver fibrosis, respectively [43, 44]. Ciliary involvement has also been shown for Jeune syndrome by the identification of mutations in the component of intraflagellar transport IFT80 [97].
Planar cell polarity
The term planar cell polarity (PCP) refers to the orientation of cells in a plane perpendicular to apico-basal polarity which, in epithelial cells, would be the plane parallel to the basement membrane. PCP is achieved by the correct orientation of the mitotic spindle and centrosomes [12, 98]. The maintenance of normal tubular development and morphology is dependent on proper PCP [98]. The PCP hypothesis of renal cystic ciliopathies is based on the finding that the mitotic angle in cells with mutated cystoproteins is altered, resulting in abnormal cell divison [98] (Fig. 5). The result of abnormal PCP is that the tubules do not extend longitudinally but at a certain angle to the longitudinal axis, resulting in a dilatation of the tubule and, thereby, in a cystic structure [98] (Fig. 5).

Altered planar cell polarity causes cyst formation. Correct orientation of the mitotic spindle and centrosomes of renal tubular epithelial cells are especially important during development for the proper growth of the longitudinal axis of the tubule (a). If the apical–basolateral polarity is disrupted, a dilated tubule or cyst would develop (b). Non-canonical Wnt signaling is involved in proper cell orientation. Urinary flow in the renal tubules could provide signaling on cellular orientation via the cilia (Adapted by permission from Macmillan Publishers Ltd: Nature Genetics 37:455-457, 2005 [103])
Involvement of the non-canonical Wnt pathway is important for the maintenance of PCP [66]. If the elongation of tubulues is disrupted postnatally by PCP defects, aberrant morphogenesis leads to tubule cyst formation (Fig. 5). PCP defects due to malorientation of the mitotic spindle have been shown in the pck rat model of human ARPKD, the Hnf1β knockout mouse, and the Kif3a knockout mouse—three rodent models for cystic kidney disease [98, 99].
Modifier genes in NPHP
There is evidence for modifier genes of NPHP [13, 26, 54, 61]. Individuals with a homozygous NPHP1 deletion and an additional heterozygous NPHP6 mutation have been identified [53, 100], and modifier genes have also been reported for JS and MGS. Tory et al. reported a combination of mutations in either NPHP1 and AHI1, NPHP6 and AHI1, or NPHP1 and NPHP6 in 28 kindreds with JS [53]. In both publications, the authors point out that the additional heterozygous mutation in a second gene may modulate the phenotype of the two recessive mutations in a primary gene in an epistatic way.
Possible approaches to the treatment of NPHP
Currently, the treatment of NPHP has to focus on the conservative approach of treating ESRD and providing dialysis and renal transplantation. Even though there is no approved specific treatment available for NPHP at this point in time, there have been some promising developments. Possible future treatments might include a vasopressin V2 receptor antagonist because in the pcy mouse, a model of NPHP type 3, cystogenesis, and progression of disease were altered profoundly by treatment with OPC31260 via the reduction of cAMP [69]. In addition, there is growing evidence that rapamycin (an mTOR inhibitor) alleviates cystogenesis [101, 102]. Moreover, roscovitine has shown improvement of cyst growth in jck (the mouse model of NPHP type 9) and cpk mice, which are models for human cystic kidney disease [87].
Outlook
Our understanding of NPHP has improved significantly from a solely histopathological entity to the discovery of the NPHP-causing genes and molecular mechanisms. Only about 30% of patients with NPHP have an identifiable mutation. This means that many more NPHP genes are expected to be found. The identification of new genes will provide additional insight into the pathomechanism of NPHP and how cilia are linked to cyst development. New therapeutic approaches are promising and will hopefully succeed in starting alternative treatment options in addition to conservative treatment and renal replacement therapy.
References
Smith C, Graham J (1945) Congenital medullary cysts of the kidneys with severe refractory anemia. Am J Dis Child 69:369–377
Fanconi G, Hanhart E, von Albertini A, Uhlinger E, Dolivo G, Prader A (1951) Familial, juvenile nephronophthisis (idiopathic parenchymal contracted kidney). Helv Paediatr Acta 6:1–49
Hildebrandt F, Otto E (2005) Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet 6:928–940
Hildebrandt F, Strahm B, Nothwang H-G, Gretz N, Schnieders B, Singh-Sawhney I, Kutt R, Vollmer M, Brandis M, members of the APN study group (1997) Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. Kidney Int 51:261–269
Ala-Mello S, Sankila EM, Koskimies O, de la Chapelle A, Kääriäinen H (1998) Molecular studies in Finnish patients with familial juvenile nephronophthisis exclude a founder effect and support a common mutation causing mechanism. J Med Genet 35:279–283
Omran H, Fernandez C, Jung M, Häffner K, Fargier B, Villaquiran A, Waldherr R, Gretz N, Brandis M, Rüschendorf F, Reis A, Hildebrandt F (2000) Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am J Hum Genet 66:118–127
Blowey DL, Querfeld U, Geary D, Warady BA, Alon U (1996) Ultrasound findings in juvenile nephronophthisis. Pediatr Nephrol 10:22–24
Waldherr R, Lennert T, Weber HP, Födisch HJ, Schärer K (1982) The nephronophthisis complex a clinicopathologic study in children. Virchows Arch A Pathol Anat Histol 394:235–254
Zollinger HU, Mihatsch MJ, Edefonti A, Gaboardi F, Imbasciati E, Lennert T (1980) Nephronophthisis (medullary cystic disease of the kidney). A study using electron microscopy, immunofluorescence, and a review of the morphological findings. Helv Paediatr Acta 35:509–530
Haider NB, Carmi R, Shalev H, Sheffield VC, Landau D (1998) A Bedouin kindred with infantile nephronophthisis demonstrates linkage to chromosome 9 by homozygosity mapping. Am J Hum Genet 63:1404–1410
Gagnadoux MF, Bacri JL, Broyer M, Habib R (1989) Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol 3:50–55
Hildebrandt F, Attanasio M, Otto E (2009) Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol 20:23–35
Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, Algazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH 2nd, Harris PC, Johnson CA (2006) The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet 38:191–196
Ala-Mello S, Koskimies O, Rapola J, Kääriäinen H (1999) Nephronophthisis in Finland: epidemiology and comparison of genetically classified subgroups. Eur J Hum Genet 7:205–211
Bollée G, Fakhouri F, Karras A, Noël LH, Salomon R, Servais A, Lesavre P, Morinière V, Antignac C, Hummel A (2006) Nephronophthisis related to homozygous NPHP1 gene deletion as a cause of chronic renal failure in adults. Nephrol Dial Transplant 21:2660–2663
Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, Bleyer AJ (2002) Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 39:882–892
Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, Dauge MC, Roume J, Le Merrer M, Meiner V, Meir K, Menez F, Beaufrère AM, Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, Munnich A, Lyonnet S, Gubler MC, Génin E, Johnson CA, Vekemans M, Encha-Razavi F, Attié-Bitach T (2007) Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet 81:170–179
Betz R, Rensing C, Otto E, Mincheva A, Zehnder D, Lichter P, Hildebrandt F (2000) Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr 136:828–831
Harris CM, Hodgkins PR, Kriss A, Chong WK, Thompson DA, Mezey LE, Shawkat FS, Taylor DS, Wilson J (1998) Familial congenital saccade initiation failure and isolated cerebellar vermis hypoplasia. Dev Med Child Neurol 40:775–779
Senior B, Friedmann AI, Braudo JL (1961) Juvenile familial nephropathy with tapetoretinal degeneration: a new oculorenal dystrophy. Am J Ophthalmol 52:625–633
Loken AC, Hanssen O, Halvorsen S, Jolster NJ (1961) Hereditary renal dysplasia and blindness. Acta Paediatr 50:177–184
Adams NA, Awadein A, Toma HS (2007) The retinal ciliopathies. Ophthalmic Genet 28:113–125
Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, Sayer JA, Otto EA, Attanasio M, O'Toole JF, Jin G, Shou C, Hildebrandt F, Williams DS, Heckenlively JR, Swaroop A (2006) In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet 15:1847–1857
Jiang ST, Chiou YY, Wang E, Chien YL, Ho HH, Tsai FJ, Lin CY, Tsai SP, Li H (2009) Essential role of nephrocystin in photoreceptor intraflagellar transport in mouse. Hum Mol Genet 18:1566–1577
Louie CM, Caridi G, Lopes VS, Brancati F, Kispert A, Lancaster MA, Schlossman AM, Otto EA, Leitges M, Gröne HJ, Lopez I, Gudiseva HV, O'Toole JF, Vallespin E, Ayyagari R, Ayuso C, Cremers FP, den Hollander AI, Koenekoop RK, Dallapiccola B, Ghiggeri GM, Hildebrandt F, Valente EM, Williams DS, Gleeson JG (2010) AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat Genet 42:175–180
Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, Macdonald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, Inglehearn CF, Lewis RA, Jacobson SG, Bergmann C, Beales PL, Attié-Bitach T, Johnson CA, Otto EA, Bhattacharya SS, Hildebrandt F, Gibbs RA, Koenekoop RK, Swaroop A, Katsanis N (2009) A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet 41:739–745
Parisi MA, Doherty D, Chance PF, Glass IA (2007) Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet 15:511–521
Valente EM, Brancati F, Silhavy JL, Castori M, Marsh SE, Barrano G, Bertini E, Boltshauser E, Zaki MS, Abdel-Aleem A, Abdel-Salam GM, Bellacchio E, Battini R, Cruse RP, Dobyns WB, Krishnamoorthy KS, Lagier-Tourenne C, Magee A, Pascual-Castroviejo I, Salpietro CD, Sarco D, Dallapiccola B, Gleeson JG, International JSRD Study Group (2006) AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol 59:527–534
Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F (2006) The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 38:674–681
Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Berthélémé JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Rüther U, Schneider-Maunoury S, Attié-Bitach T, Saunier S (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 39:875–881
Wolf MT, Saunier S, O'Toole JF, Wanner N, Groshong T, Attanasio M, Salomon R, Stallmach T, Sayer JA, Waldherr R, Griebel M, Oh J, Neuhaus TJ, Josefiak U, Antignac C, Otto EA, Hildebrandt F (2007) Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int 72:1520–1526
Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA (2004) Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 36:1008–1013
Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason OS, Dobyns WB, Bennett CL, Chance PF, Glass IA (2006) AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet 43:334–339
Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, D'Arrigo S, Emma F, Fazzi E, Gallizzi R, Gentile M, Loncarevic D, Mejaski-Bosnjak V, Pantaleoni C, Rigoli L, Salpietro CD, Signorini S, Stringini GR, Verloes A, Zabloka D, Dallapiccola B, Gleeson JG, Valente EM, International JSRD Study Group (2009) MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat 30:E432–442
Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, Alswaid AF, Ozyurek H, Dibooglu S, Otto EA, Liu Y, Davis EE, Hutter CM, Bammler TK, Farin FM, Dorschner M, Topçu M, Zackai EH, Rosenthal P, Owens KN, Katsanis N, Vincent JB, Hildebrandt F, Rubel EW, Raible DW, Knoers NV, Chance PF, Roepman R, Moens CB, Glass IA, Doherty D (2008) CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet 83:559–571
Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attié-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, Valente EM, Woods CG, Gleeson JG, International Joubert Syndrome Related Disorders Study Group (2008) Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet 83:170–179
Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, Kayserili H, Swistun D, Scott LC, Bertini E, Boltshauser E, Fazzi E, Travaglini L, Field SJ, Gayral S, Jacoby M, Schurmans S, Dallapiccola B, Majerus PW, Valente EM, Gleeson JG (2009) Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41:1032–1036
Edvardson S, Shaag A, Zenvirt S, Erlich Y, Hannon GJ, Shanske AL, Gomori JM, Ekstein J, Elpeleg O (2010) Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet 86:93–97
Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, McDonald R, Eddy A, Chance PF, Glass IA (2004) The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet 75:82–91
Mollet G, Salomon R, Gribouval O, Silbermann F, Bacq D, Landthaler G, Milford D, Nayir A, Rizzoni G, Antignac C, Saunier S (2002) The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat Genet 32:300–305
Kyttälä M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestilä M (2006) MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet 38:155–157
Bergmann C, Fliegauf M, Brüchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kränzlin B, Nürnberg G, Becker C, Grimm T, Girschick G, Lynch SA, Kelehan P, Senderek J, Neuhaus TJ, Stallmach T, Zentgraf H, Nürnberg P, Gretz N, Lo C, Lienkamp S, Schäfer T, Walz G, Benzing T, Zerres K, Omran H (2008) Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet 82:959–970
Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, Gretz N, Walz G, Schermer B, Benzing T, Hildebrandt F, Omran H (2003) Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet 34:455–459
Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Utsch B, Wolf MT, Becker C, Nürnberg G, Nürnberg P, Nayir A, Saunier S, Antignac C, Hildebrandt F (2009) Hypomorphic mutations in Meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet 46:663–670
Ellis DS, Heckenlively JR, Martin CL, Lachman RS, Sakati NA, Rimoin DL (1984) Leber's congenital amaurosis associated with familial juvenile nephronophthisis and cone-shaped epiphyses of the hands (the Saldino-Mainzer syndrome). Am J Ophthalmol 97:233–239
Mainzer F, Saldino RM, Ozonoff MB, Minagi H (1970) Familial nephropathy associated with retinitis pigmentosa, cerebellar ataxia and skeletal abnormalities. Am J Med 49:556–562
Donaldson MD, Warner AA, Trompeter RS, Haycock GB, Chantler C (1985) Familial juvenile nephronophthisis, Jeune's syndrome, and associated disorders. Arch Dis Child 60:426–434
Moudgil A, Bagga A, Kamil ES, Rimoin DL, Lachman RS, Cohen AH, Jordan SC (1998) Nephronophthisis associated with Ellis-van Creveld syndrome. Pediatr Nephrol 12:20–22
Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA, Benzing T, Hildebrandt F (2003) Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet 34:413–420
Hildebrandt F, Otto E, Rensing C, Nothwang HG, Vollmer M, Adolphs J, Hanusch H, Brandis M (1997) A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet 17:149–153
Saunier S, Calado J, Heilig R, Silbermann F, Benessy F, Morin G, Konrad M, Broyer M, Gubler MC, Weissenbach J, Antignac C (1997) A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum Mol Genet 6:2317–2323
Otto EA, Helou J, Allen SJ, O'Toole JF, Wise EL, Ashraf S, Attanasio M, Zhou W, Wolf MT, Hildebrandt F (2008) Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat 29:418–426
Tory K, Lacoste T, Burglen L, Morinière V, Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, Antignac C, Salomon R, Saunier S (2007) High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol 18:1566–1575
Eley L, Moochhala SH, Simms R, Hildebrandt F, Sayer JA (2008) Nephrocystin-1 interacts directly with Ack1 and is expressed in human collecting duct. Biochem Biophys Res Commun 371:877–882
Benzing T, Gerke P, Höpker K, Hildebrandt F, Kim E, Walz G (2001) Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers phosphorylation of Pyk2. Proc Natl Acad Sci USA 98:9784–9789
Donaldson JC, Dempsey PJ, Reddy S, Bouton AH, Coffey RJ, Hanks SK (2000) Crk-associated substrate p130(Cas) interacts with nephrocystin and both proteins localize to cell-cell contacts of polarized epithelial cells. Exp Cell Res 256:168–178
Donaldson JC, Dise RS, Ritchie MD, Hanks SK (2002) Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J Biol Chem 277:29028–29035
Eley L, Gabrielides C, Adams M, Johnson CA, Hildebrandt F, Sayer JA (2008) Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int 74:1139–1149
Fliegauf M, Horvath J, von Schnakenburg C, Olbrich H, Müller D, Thumfart J, Schermer B, Pazour GJ, Neumann HP, Zentgraf H, Benzing T, Omran H (2006) Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol 17:2424–2433
Schermer B, Höpker K, Omran H, Ghenoiu C, Fliegauf M, Fekete A, Horvath J, Köttgen M, Hackl M, Zschiedrich S, Huber TB, Kramer-Zucker A, Zentgraf H, Blaukat A, Walz G, Benzing T (2005) Phosphorylation by casein kinase 2 induces PACS-1 binding of nephrocystin and targeting to cilia. EMBO J 24:4415–4424
Mollet G, Silbermann F, Delous M, Salomon R, Antignac C, Saunier S (2005) Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum Mol Genet 14:645–656
O'Toole JF, Otto EA, Frishberg Y, Hildebrandt F (2006) Retinitis pigmentosa and renal failure in a patient with mutations in INVS. Nephrol Dial Transplant 21:1989–1991
Watnick T, Germino G (2003) From cilia to cyst. Nat Genet 34:355–356
Shiba D, Manning DK, Koga H, Beier DR, Yokoyama T (2010) Inv acts as a molecular anchor for Nphp3 and Nek8 in the proximal segment of primary cilia. Cytoskeleton 67:112–119
Morgan D, Eley L, Sayer J, Strachan T, Yates LM, Craighead AS, Goodship JA (2002) Expression analyses and interaction with the anaphase promoting complex protein Apc2 suggest a role for inversin in primary cilia and involvement in the cell cycle. Hum Mol Genet 11:3345–3350
Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37:537–543
Tory K, Rousset-Rouvière C, Gubler MC, Morinière V, Pawtowski A, Becker C, Guyot C, Gié S, Frishberg Y, Nivet H, Deschênes G, Cochat P, Gagnadoux MF, Saunier S, Antignac C, Salomon R (2009) Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int 75:839–847
Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F (2008) NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol 19:587–592
Gattone VH 2nd, Wang X, Harris PC, Torres VE (2003) Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9:1323–1326
Otto E, Hoefele J, Ruf R, Mueller AM, Hiller KS, Wolf MT, Schuermann MJ, Becker A, Birkenhäger R, Sudbrak R, Hennies HC, Nürnberg P, Hildebrandt F (2002) A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet 71:1161–1167
Delous M, Hellman NE, Gaudé HM, Silbermann F, Le Bivic A, Salomon R, Antignac C, Saunier S (2009) Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum Mol Genet 18:4711–4723
Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O'Toole JF, Helou J, Attanasio M, Utsch B, Sayer JA, Lillo C, Jimeno D, Coucke P, De Paepe A, Reinhardt R, Klages S, Tsuda M, Kawakami I, Kusakabe T, Omran H, Imm A, Tippens M, Raymond PA, Hill J, Beales P, He S, Kispert A, Margolis B, Williams DS, Swaroop A, Hildebrandt F (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 37:282–288
Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol 157:103–113
Schäfer T, Pütz M, Lienkamp S, Ganner A, Bergbreiter A, Ramachandran H, Gieloff V, Gerner M, Mattonet C, Czarnecki PG, Sayer JA, Otto EA, Hildebrandt F, Kramer-Zucker A, Walz G (2008) Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum Mol Genet 17:3655–3662
Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E, Bertini E, Dallapiccola B, Gleeson JG, International Joubert Syndrome Related Disorders Study Group (2006) Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet 38:623–625
Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426:570–574
Frank V, den Hollander AI, Brüchle NO, Zonneveld MN, Nürnberg G, Becker C, Du Bois G, Kendziorra H, Roosing S, Senderek J, Nürnberg P, Cremers FP, Zerres K, Bergmann C (2008) Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum Mutat 29:45–52
Helou J, Otto EA, Attanasio M, Allen SJ, Parisi MA, Glass I, Utsch B, Hashmi S, Fazzi E, Omran H, O'Toole JF, Sayer JA, Hildebrandt F (2007) Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior-Løken syndrome. J Med Genet 44:657–663
Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N (2008) Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet 40:443–448
den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP (2006) Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 79:556–561
Attanasio M, Uhlenhaut NH, Sousa VH, O'Toole JF, Otto E, Anlag K, Klugmann C, Treier AC, Helou J, Sayer JA, Seelow D, Nürnberg G, Becker C, Chudley AE, Nürnberg P, Hildebrandt F, Treier M (2007) Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet 39:1018–1024
Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Märker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R (2007) Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet 39:882–888
Liu S, Lu W, Obara T, Kuida S, Lehoczky J, Dewar K, Drummond IA, Beier DR (2002) A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 129:5839–5846
Sohara E, Luo Y, Zhang J, Manning DK, Beier DR, Zhou J (2008) Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J Am Soc Nephrol 19:469–476
Natoli TA, Gareski TC, Dackowski WR, Smith L, Bukanov NO, Russo RJ, Husson H, Matthews D, Piepenhagen P, Ibraghimov-Beskrovnaya O (2008) Pkd1 and Nek8 mutations affect cell-cell adhesion and cilia in cysts formed in kidney organ cultures. Am J Physiol Ren Physiol 294:F73–83
Mykytyn K, Sheffield VC (2004) Establishing a connection between cilia and Bardet-Biedl Syndrome. Trends Mol Med 10:106–109
Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O (2006) Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 444:949–952
Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA (2007) The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet 16:173–186
Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA (2009) Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet 47:8–21
O'Toole JF, Liu Y, Davis EE, Westlake CJ, Attanasio M, Otto EA, Seelow D, Nurnberg G, Becker C, Nuutinen M, Kärppä M, Ignatius J, Uusimaa J, Pakanen S, Jaakkola E, van den Heuvel LP, Fehrenbach H, Wiggins R, Goyal M, Zhou W, Wolf MT, Wise E, Helou J, Allen SJ, Murga-Zamalloa CA, Ashraf S, Chaki M, Heeringa S, Chernin G, Hoskins BE, Chaib H, Gleeson J, Kusakabe T, Suzuki T, Isaac RE, Quarmby LM, Tennant B, Fujioka H, Tuominen H, Hassinen I, Lohi H, van Houten JL, Rotig A, Sayer JA, Rolinski B, Freisinger P, Madhavan SM, Herzer M, Madignier F, Prokisch H, Nurnberg P, Jackson P, Khanna H, Katsanis N, Hildebrandt F (2010) Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J Clin Invest 120:791–802
Zaucke F, Boehnlein JM, Steffens S, Polishchuk RS, Rampoldi L, Fischer A, Pasch A, Boehm CW, Baasner A, Attanasio M, Hoppe B, Hopfer H, Beck BB, Sayer JA, Hildebrandt F, Wolf MT (2010) Uromodulin is expressed in renal primary cilia and UMOD mutations result in decreased ciliary uromodulin expression. Hum Mol Genet 19:1985–1997
Wolf MT, Lee J, Panther F, Otto EA, Guan KL, Hildebrandt F (2005) Expression and phenotype analysis of the nephrocystin-1 and nephrocystin-4 homologs in Caenorhabditis elegans. J Am Soc Nephrol 16:676–687
Badano JL, Teslovich TM, Katsanis N (2005) The centrosome in human genetic disease. Nat Rev Genet 6:194–205
Jauregui AR, Nguyen KC, Hall DH, Barr MM (2008) The Caenorhabditis elegans nephrocystins act as global modifiers of cilium structure. J Cell Biol 180:973–988
Winkelbauer ME, Schafer JC, Haycraft CJ, Swoboda P, Yoder BK (2005) The C. elegans homologs of nephrocystin-1 and nephrocystin-4 are cilia transition zone proteins involved in chemosensory perception. J Cell Sci 118:5575–5587
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33:129–137
Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, Johnson C, Irving M, Elcioglu N, Winey M, Tada M, Scambler PJ (2007) IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet 39:727–729
Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M (2006) Defective planar cell polarity in polycystic kidney disease. Nat Genet 38:21–23
Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 100:5286–5291
Hoefele J, Wolf MT, O'Toole JF, Otto EA, Schultheiss U, Dêschenes G, Attanasio M, Utsch B, Antignac C, Hildebrandt F (2007) Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol 18:2789–2795
Tobin JL, Beales PL (2008) Restoration of renal function in zebrafish models of ciliopathies. Pediatr Nephrol 23:2095–2099
Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T (2006) The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 103:5466–5471
Germino GG (2005) Linking cilia to Wnts. Nat Genet 37:455–457
Acknowledgments
We thank Dr. Sandy Cope-Yokoyama (Department of Pathology, Children's Medical Center Dallas and UT Southwestern Medical Center, Dallas) for her contribution of the images of renal pathology in nephronophthisis and Dr. Michael Craig Morris (Department of Radiology, Children's Medical Center Dallas and UT Southwestern Medical Center, Dallas) for his contribution of the Joubert syndrome image.
F.H. is an investigator of the Howard Hughes Medical Institute, the Frederick G.L. Huetwell professor, and a Doris Duke Distinguished Clinical Scientist. He is supported by grants from the NIH (DK068306, DK064614 and DK069274). M.T.W. is a fellow of the Pediatric Scientist Development Program (PSDP) and was supported by grants from the Koeln Fortune Program Faculty of Medicine, University of Cologne (184/2004), the German Kidney Fund (Deutsche Nierenstiftung), the German Research Foundation (DFG WO 1229/2-1), and a T32 training grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wolf, M.T.F., Hildebrandt, F. Nephronophthisis. Pediatr Nephrol 26, 181–194 (2011). https://doi.org/10.1007/s00467-010-1585-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-010-1585-z