
Abstract
Treatment options for type 2 diabetes based on the action of the incretin hormone glucagon-like peptide-1 (GLP-1) were first introduced in 2005. These comprise the injectable GLP-1 receptor agonists solely acting on the GLP-1 receptor on the one hand and orally active dipeptidyl-peptidase inhibitors (DPP-4 inhibitors) raising endogenous GLP-1 and other hormone levels by inhibiting the degrading enzyme DPP-4. In adult medicine, both treatment options are attractive and more commonly used because of their action and safety profile. The incretin-based therapies stimulate insulin secretion and inhibit glucagon secretion in a glucose-dependent manner and carry no intrinsic risk of hypoglycaemia. GLP-1 receptor agonists allow weight loss, whereas DPP-4 inhibitors are weight neutral. This review gives an overview of the mechanism of action and the substances and clinical data available.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes is a disease that is increasing tremendously in prevalence worldwide, and is expected to more than double within the next 20 years and to affect 440 million people by 2030 [1]. Formerly mostly found in adults and the elderly, the prevalence of type 2 diabetes is also increasing in children and adolescents [2]. Effective and patient-orientated treatment is still a major task, since a large percentage of patients do not reach the therapeutic goal of a near normal HbA1c value as one criterion for an acceptable glycaemic control. Along with this dissatisfactory therapeutic effect, other important treatment goals, such as body weight reduction or the prevention of hypoglycaemic episodes, are not accomplished. Furthermore, type 2 diabetic patients have a significantly elevated cardiovascular mortality risk, which can be lowered by improved metabolic control [3, 4]. The established therapies are not able to halt the disease progression, which is caused by the continuous loss of function of the insulin-secreting beta cells in the islets of Langerhans. This loss of function is characterised by an increasing defect in the insulin response to glucose as well as a loss of beta cell mass over time. Furthermore, many established therapies are associated with an elevated incidence of hypoglycaemic events (sulphonylureas, glinides and insulin) or with an unwanted increase in body weight caused by the antidiabetic medications (sulphonylureas, glinides, glitazones and insulin) [5].
In the past decade, the pharmacological actions of the incretin hormone glucagon-like peptide-1 (GLP-1) were utilised to develop two novel substance classes for type 2 diabetes therapy: the GLP-1 receptor agonists (or “GLP-1 mimetics”) and the dipeptidyl-peptidase-IV inhibitors (DPP-4 inhibitors or “GLP-1 enhancers”) [6]. The incretin hormones GLP-1 and GIP (gastric inhibitory polypeptide, later often also referred to as glucose insulinotropic polypeptide) are secreted postprandially from the endocrine L- and K-cells in the intestinal mucosa respectively. Activation of promiscuous seven transmembrane receptors located in the intestinal wall and activated by nutrient components (“taste receptors”) may additionally contribute to incretin secretion after a meal [7]. These hormones are responsible for approximately 60% of the insulin secretion following a meal and for the so-called incretin effect. The incretin effect describes the phenomenon that oral glucose leads to a greater insulin response than an isoglycaemic intravenous glucose load [8, 9]. In patients with type 2 diabetes, the incretin effect is diminished. One important reason for this loss is that GIP does not act as an insulinotropic hormone under chronic hyperglycaemia in type 2 diabetes. GLP-1 on the other hand is still able to stimulate insulin secretion under hyperglycaemia in type 2 diabetes [10]. Conversely, hyperglycaemia acutely reduces the postprandial levels of GIP and GLP-1, possibly through a deceleration of gastric emptying. Therefore, the reduced incretin levels in some patients with type 2 diabetes could be a consequence rather than a cause of type 2 diabetes [11].
Increasing GLP-1 plasma concentrations to pharmacological levels by exogenous GLP-1 application leads to a normalisation of the incretin effect with an adequate insulin response under hyperglycaemic conditions [12].
Physiological actions of GLP-1
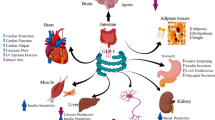
GLP-1 is a product of the glucagon gene and is post-translationally cleaved from preproglucagon in the neuroendocrine L-cells of the intestinal mucosa and in the central nervous system. It binds to highly specific GLP-1 receptors that belong to the G-protein coupled receptors [6]. GLP-1 shows numerous actions in different tissues and a broad therapeutic potential (see Fig. 1 for details).

GLP-1 stimulates insulin secretion of the beta cells and additionally inhibits glucagon secretion from the alpha cells. Both actions occur in a glucose-dependent manner and lead to a normalisation of postprandial and fasting hyperglycaemia. GLP-1 furthermore slows gastric emptying. This effect also contributes to a normalisation of postprandial hyperglycaemia (see below). Under hypoglycaemic conditions the counter-regulation by glucagon is not affected and insulin secretion is not stimulated. GLP-1 is therefore not able to elicit hypoglycaemia by itself [6].
Animal studies in rodents and studies in isolated human islets showed beneficial long-term actions of GLP-1: insulin synthesis is stimulated by GLP-1 and beta cell mass is increased [6, 13, 14]. Whether these findings are relevant for long-term type 2 diabetes therapy with a positive effect on halting disease progression is not known, since long-term study data are still not available and beta cell mass cannot yet be quantified in humans in a clinical setting.
GLP-1 binds to its receptor on hypothalamic neurons and stimulates satiety. In the gastrointestinal tract, GLP-1 has a direct effect on motility and slows gastric emptying. These two effects explain that long-term treatment with GLP-1 receptor agonists lead to weight loss in the long run [6].
Furthermore, pharmacological application of GLP-1 receptor agonists lead to an improvement in cardiovascular parameters (reduction of systolic blood pressure, beneficial effects on myocardial ischaemia in animal models, positive effects on left ventricular function in heart failure) [15–17].
GLP-1 is degraded rapidly by the ubiquitous enzyme dipeptidyl-peptidase IV (DPP-4). An animal study in rodents even showed that DPP-4 expression in the intestine and kidneys increased with high-fat feeding and type 2 diabetes [18]. Intravenously administered GLP-1 has a biological half-time of approximately only 1–2 min [6]. Also, subcutaneous injections of GLP-1 do not lead to a sufficiently high and long-lasting elevation of GLP-1 concentrations to use native GLP-1 as a practical therapeutic agent. An adenoviral vectored gene therapeutic option transfecting the GLP-1 gene into hepatozytes of a diabetic rat strain showed positive effects in one study, but because of the present difficulties and safety concerns using these methods in humans, this theoretical option has not been followed up so far [19]. In order to utilise GLP-1 action for type 2 diabetes therapy, two options are presently available [6]:
-
1.
GLP-1-receptor agonists (or GLP-1 mimetics) as injectable compounds
-
2.
Dipeptidyl-peptidase-IV (DPP-4) inhibitors (or incretin enhancers) as orally active substances
The following paragraphs deal with the approved compounds.
GLP-1 receptor-agonists, exenatide and liraglutide
Exenatide (Byetta®, Eli Lilly Pharmaceuticals) was the first GLP-1 receptor agonist to be approved for the treatment of type 2 diabetes in combination with metformin and/or a sulphonylurea in patients failing to reach the therapeutic goals with this oral medication [20]. Exenatide is the synthetic form of exendin-4, a peptide first discovered in the saliva of the gila monster (heloderma suspectum) in 1992. It has a 53% amino acid sequence homology to human GLP-1 and is a strong GLP-1 receptor agonist [21]. It is administered subcutaneously twice daily. A slow release formulation for once-weekly administration (Exenatide LAR [long-acting release]) is presently in clinical phase III studies [22–24]. The long-acting human GLP-1 analogue liraglutide (Victoza®, Novo Nordisk Pharmaceuticals) for once-daily dosing has just been approved by the European Medicines Agency (EMEA) [25–27]. Further compounds for once-daily or once-weekly application are in development (taspoglutide, Roche Pharmaceuticals [28]; AVE0010, Sanofi-Aventis Pharmaceuticals [29]; albiglutide, GlaxoSmithKline Pharmaceuticals [30]).
Exenatide has a prolonged half-life in comparison to native GLP-1 of approximately 3.5 h. After subcutaneous injection sufficient plasma concentrations are reached for 4–6 hours [20, 31, 32]. Exenatide lowered the HbA1c by 0.8–1.1% in clinical studies [33–35]. The reduction of HbA1c was constant over a time period of 3 years in one study [16]. Comparative studies with insulin show that effects of exenatide on glycaemic parameters are comparable to the improvement seen with a newly started insulin therapy [16, 20, 36–39].
Patients receiving exenatide showed a significant drop in body weight by 1.5–3.0 kg after 30 weeks. The patients who continued to use exenatide lost further weight (5.3 kg after 3 years) [16, 40]. The comparative studies with insulin showed a difference in weight development of 4–5 kg in 30 weeks between the insulin- and exenatide-treated groups [36, 37, 39].
An improvement of beta cell function (measured with the clinical surrogate parameters insulin secretion rate HOMA B (homeostatic modelling assessment of beta cell function) and the proinsulin:insulin ratio) was also observed in the clinical studies. Furthermore, the first phase of insulin secretion is restored after an intravenous glucose bolus under treatment with exenatide [20, 40]. However, in a 1-year study with exenatide and a consecutive wash-out phase of 12 weeks, the improvements in the above-mentioned beta cell function parameters were no longer present after the end of the wash-out period [41].
Severe hypoglycaemic events were only observed in exenatide-treated patients who had received combination therapy with a sulphonylurea. In the comparative studies comparing exenatide with insulin treatment, the rates of hypoglycaemic episodes was similar in the patient group that had sulphonylurea–exenatide combination therapy. However, the incidence of nocturnal hypoglycaemic events was lower in the exenatide-treated patients [20, 40].
The most frequent adverse events were fullness and nausea. These were less pronounced when the exenatide dose was titrated from a small dose to the full dose at the beginning of treatment. Generally, nausea was mild to moderate occurring in the first weeks of treatment and ceasing with time. Nausea was the most common reason to stop therapy with 2–6.4% drop-outs in the clinical studies with exenatide [16, 20, 40].
In approximately 40% of exenatide-treated patients, anti-exenatide antibodies can be detected. Over a time period of at least 3 years, these antibody titres did not have any obvious effect on glycaemic control. Furthermore, the exenatide antibodies do not cross-react with human GLP-1 [24].
Since exenatide has been approved, cases of acute pancreatitis have been reported [42, 43]. The Food and Drugs Administration of the United States (FDA) in reaction then published a warning. In total, the incidence of pancreatitis is very low and rather corresponds to the elevated risk of pancreatitis in obese type 2 diabetic patients. Type 2 diabetic patients have an elevated pancreatitis risk due to a higher prevalence of gall stones, hypertriglyceridaemia and other factors, and a recent meta-analysis confirmed this [44].
Exenatide is predominantly eliminated by glomerular filtration followed by proteolytic degradation [45]. Its use is not recommended in patients with severe renal insufficiency (creatinine clearance <30 ml/min). In patients with end-stage renal disease on dialysis, exenatide (5 µg) has been poorly tolerated because of gastrointestinal side effects [20, 40]. Just recently, the FDA published a warning after having observed 78 cases of altered renal function under exenatide therapy in the time period from 2005 to 2008. These cases comprised 62 patients with acute renal failure and 16 cases of renal insufficiency. Some cases occurred in patients with pre-existing kidney disease or in patients with one or more risk factors for developing kidney problems [46]. These complications may have been associated with nausea, vomiting and consecutive dehydration and worsening of kidney function. According to the warning by the FDA, exenatide should not be used in patients with severe renal impairment (creatinine clearance <30 ml/min) or end-stage renal disease. Additionally, caution should be applied when initiating or increasing doses of exenatide in patients with moderate renal impairment (creatinine clearance 30–50 ml/min). Patients should be carefully observed for the development of kidney dysfunction under therapy.
There is one published paediatric study that investigated the pharmacology and tolerability of a single dose of exenatide in 13 adolescents (aged 10–16 years) with type 2 diabetes and a baseline HbA1c of 8.2% on an ongoing stable therapy with metformin, a sulphonylurea or a combination of both. Pharmacokinetics and the safety profile of exenatide were the primary endpoints. The secondary endpoints comprised postprandial plasma glucose, serum insulin as well as plasma glucagon concentrations. The exenatide AUC (area under the curve) expectedly appeared to be dose-dependent. However, exenatide was not quantifiable in all patients at the lower 2.5- µg dose. Single doses of 2.5 µg and 5.0 µg of exenatide were well tolerated and normalised postprandial glucose and glucagon concentrations compared with placebo (P < 0.01). Insulin plasma concentration did not differ significantly after exenatide and placebo. No hypoglycaemic events were recorded during the study [47].
Liraglutide
Liraglutide is the first human GLP-1 analogue. It has two modifications in the amino acid sequence of native GLP-1 and an attachment of a fatty acid side chain to the peptide. It is injected subcutaneously once daily [26].
In animal studies with diabetic rodents, liraglutide has been shown to increase beta cell mass. Liraglutide lowers blood glucose, body weight and food intake in a broad selection of animal models [48]. In clinical studies in type 2 diabetic patients involving approximately 4,200 patients receiving the drug, it is efficacious and safe in the treatment of type 2 diabetes across all stages of the natural course of the disease [25, 49–54].
In a clinical study using liraglutide in monotherapy in newly diagnosed type 2 diabetic patients it led to a sustained and stable HbA1c reduction of 0.9–1.1% in a dose of 1.2 or 1.8 mg once daily respectively, over a period of up to 2 years [55, 56].
In other studies, the same doses of liraglutide effectively lowered glycaemic parameters in various combinations with oral antidiabetic drugs by approximately 1.0–1.5%. Liraglutide treatment additionally led to a significant weight loss comparable to that previously observed in studies with exenatide [57, 58]. The weight loss was accompanied by a more pronounced loss in visceral fat than subcutaneous fat [57–59].
Furthermore, systolic blood pressure was lowered by 2–6 mmHg in the liraglutide-treated patients. This effect was independent of the weight loss, as the reduction of blood pressure was already observed early on in therapy, when weight loss had not yet occurred [54–56, 59].
The incidence of hypoglycaemic episodes was comparable to placebo in all studies and study arms, where no sulphonylurea was used in the combination with liraglutide [57, 58]. Gastrointestinal symptoms were also common in clinical studies with liraglutide, but nausea and vomiting were less frequent and only reported for a shorter period at the beginning of therapy compared with exenatide [60]. Antibody formation against liraglutide was only 8.6% in the clinical studies [55–58]. In a study directly comparing the clinical efficacy and safety of exenatide and liraglutide, liraglutide had an advantage with regard to lowering the glycaemic parameters HbA1c and fasting glucose and to improving HOMA-B [60]. Liraglutide improves the first phase of insulin secretion after intravenous glucose as well as the insulin response to a maximal stimulation with arginine [61].
Data on the pharmacokinetic profile of liraglutide in mild to moderate renal impairment show no alteration of the profile [57, 58]. So far, no studies of paediatric patients with type 2 diabetes and liraglutide therapy have been published.
DPP-4 inhibitors: sitagliptin, vildagliptin and saxagliptin
DPP-4 inhibitors are orally active and tolerated well. After once- or twice-daily dosing they effectively inhibit DPP-4 and lead to a postprandial elevation of endogenous GLP-1 concentrations 2–3 times normal physiological levels [62, 63]. The presently available compounds are sitagliptin (Januvia®, Merck Pharmaceuticals), vildagliptin (Galvus®, Novartis Pharmaceuticals) and saxagliptin (Onglyza®, AstraZeneca and Bristol-Myers Squibb Pharmaceuticals) [64]. They are approved in combination with metformin, a sulphonylurea or a glitazone or a combination of metformin and a sulphonylurea. Sitagliptin is also approved for monotherapy in the USA and in monotherapy for patients with metformin contraindications or intolerance in Europe [62]. There are fixed-dose combinations for both sitagliptin and vildagliptin with metformin (sitagliptin plus metformin: Janumet®, Merck Pharmaceuticals, vildaglitpin plus metformin: Eucreas®, Novartis Pharmaceuticals). Further DPP-4 inhibitors are in clinical studies (alogliptin, Takeda Pharmaceuticals [65]; linagliptin, Boehringer Ingelheim Pharmaceuticals [66] and others) [64].
Sitagliptin is the first DPP-4 inhibitor to be approved. In mono- as well as in combination therapy it lowers HbA1c by 0.6–1.1% compared with placebo in a standard dose of 100 mg once daily [62, 67–69]. Likewise, it reduces fasting plasma glucose and postprandial glucose significantly. Sitagliptin was weight-neutral in all studies [62, 67–69]. As an add-on to an existing metformin therapy, it was able to lower the HbA1c by 0.7%. In a primary combination therapy with metformin, a constant and sustained reduction of HbA1c and fasting plasma glucose was observed over a time period of 2 years [70]. The incidence of hypoglycaemic episodes observed under sitagliptin was comparable to that under placebo [69]. An improvement of the proinsulin:insulin ratio as a surrogate parameter of beta cell function was observed in clinical studies in sitagliptin-treated patients [62, 67, 68, 70]. The most common side effects of sitagliptin were unspecific, like headache, arthritis, nasopharyngitis, respiratory or urinary tract infections and rarely skin reactions [69]. Adverse events concerning infections showed the largest difference compared with placebo and were more frequent in the sitagliptin-treated patients (sitagliptin 34.5%, placebo 32.9%; 95% confidence interval −0.8 to 4.0) [71].
The elimination and excretion of sitagliptin is mainly renal (75% of an oral dose is found in the urine as unchanged drug); the elimination half-time is 12–14 h [72–74]. Sitagliptin was also generally well tolerated and effective in patients with impaired renal function. In this study, a dose of 25 mg/day was chosen for patients with a creatinine clearance of <30 ml/min or end-stage renal disease, a dose of 50 mg/dl was given to patients with a creatinine clearance between 30 and 50 ml/min [75, 76].
Vildaglitpin is the second available compound of the DPP-4 inhibitors with approval in Europe and many other countries. Its dosage is 50 mg twice daily. In clinical studies testing vildaglitpin in monotherapy or combination therapy with metformin, glimepiride, pioglitazone or insulin, vildagliptin was able to decrease the HbA1c by approximately 0.5–1.0% [62, 67, 68]. As an add-on therapy to metformin, it decreased the HbA1c by 0.65–1.1% [77]. Vildagliptin has a good safety and tolerability profile and the most common adverse events are unspecific (flu-like symptoms, headache, dizziness, rarely liver enzyme elevations during the initiation of therapy). The incidence of hypoglycaemic episodes is also comparable to that of placebo. Vildagliptin, like the other DPP-4 inhibitors, is also weight-neutral. Study data with vildagliptin demonstrated a positive influence on acute and medium-term parameters for insulin secretion under vildagliptin treatment [62, 68, 77]. In this respect, HOMA B improved as well as the proinsulin:insulin ratio and the first phase of insulin secretion after intravenous glucose [78]. Vildagliptin has been tested in an elderly population, where it was shown to be efficacious and safe [79].
Long-term studies investigating cardiovascular outcomes and a possible positive influence on disease progression of type 2 diabetes are being carried on with DPP-4 inhibitors.
Saxagliptin was subjected to a large phase III study program. A dose-range study showed a dose-dependent reduction in HbA1c by 0.7–0.9% (baseline HbA1c 7.9%). Fasting plasma glucose was also lowered dose-dependently [80]. In a study with drug-naïve patients, saxagliptin lowered glycaemic parameters (HBA1c, fasting plasma glucose and postprandial glucose) significantly [81]. As an add-on medication to a therapy with either metformin or a glitazone, saxagliptin also led to significant metabolic improvements comparable to other DPP-4 inhibitors [64, 82–85]. Saxagliptin did not cause hypoglycaemia, was well-tolerated and was weight-neutral. A meta-analysis of the clinical phase III studies showed favourable data on the development of cardiovascular events [86].
Common characteristics of and differences between incretin-based therapies
The incretin-based therapies offer a good alternative to the established antidiabetic compounds due to their satisfying antihyperglycaemic efficacy, their lack of risk of causing hypoglycaemia and their positive effects on body weight development. A further advantage is their positive effect on the insulin response of the beta cells. At this time, however, it is not yet clear, whether incretin-based therapies will lead to a sustained and durable positive effect on beta cell function and mass under clinical conditions in patients with type 2 diabetes. Animal data suggest that the novel compounds may lead to a retardation or halting of the progression of type 2 diabetes.
The most patient-relevant and striking difference between the incretin-based therapies is that GLP-1 receptor agonists are injectable agents, while DPP-4 inhibitors are effective orally (Table 1). Glycaemic control seems to be improved more effectively by GLP-1 receptor agonists in comparison to DPP-4 inhibitors, but the data of a study directly comparing the efficacy and safety of liraglutide with sitagliptin are not yet available. Also, only GLP-1 receptor agonists lead to a reduction in body weight, whereas DPP-4 inhibitors are weight-neutral. Nausea, the most common adverse event observed with GLP-1 receptor agonist therapy is not observed in treatment with DPP-4 inhibitors. So far, no characteristic pattern of adverse events has been observed with the DPP-4 inhibitors. DPP-4 is also expressed on the plasma membrane of T-lymphocytes, where it was first described as a CD-26 receptor. However, no immunological alterations have been observed with DPP-4 inhibitor therapy. Furthermore, DPP-4 has multiple substrates (all peptides with a penultimate alanine or proline in the N-terminal position); the physiological effect of DPP-4 inhibition on all substrates has not been characterised in full detail as yet. Further long-term studies should clarify the long-term effects and safety of DPP-4 inhibitors.
Indications for incretin-based therapies and their placement in treatment guidelines for type 2 diabetes
The DPP-4 inhibitors sitagliptin, vildagliptin and saxagliptin are approved in many countries for an oral combination therapy, when therapeutic goals are not reached with life-style intervention and metformin monotherapy. The DPP-4 inhibitors play a role in this indication in the German guidelines and a recommendation by the British National Institute for Health and Clinical Excellence (NICE) for patients who should not be treated with sulphonylureas in order to prevent hypoglycaemia or further weight gain [87, 88]. A recent retrospective study has shown that a higher incidence of hypoglycaemia is associated with the development of symptoms of dementia [89]. In this respect, hypoglycaemia avoidance as stated by NICE, is an important therapeutic goal. It should be noted that DPP-4 inhibitors lower the HbA1c by approximately 1.0% and that other treatment options (namely insulin) should be considered, if the HbA1c is elevated by more than 1.0% or if metabolic control has decompensated. The combination of metformin with DPP-4 inhibitors combined two synergistic treatment principles, metformin acting on insulin resistance and the DPP-4 inhibitor acting on the glucose-dependent stimulation of insulin secretion and inhibition of glucagon secretion (the same synergistic principle of action applies to the combination of a glitazone and a DPP-4 inhibitor). DPP-4 inhibitors are not inferior to sulphonylureas in the combination with metformin regarding glycaemic parameters [90]. Theoretically, DPP-4 inhibitors may succeed sulphonylureas as insulinotropic agents, if the above-mentioned advantages are underlined by positive outcomes in long-term studies concerning glycaemic and other relevant endpoints as well as safety outcomes.
Therapy with a GLP-1 receptor agonist is a favourable treatment option when oral therapy with metformin or a combination therapy with metformin and a sulphonylurea are insufficient and a simultaneous loss of body weight is another therapeutic goal (e.g. obesity-associated complications and concomitant morbidity) or hypoglycaemia strictly has to be avoided (see above). Therapy with a GLP-1 receptor agonist at this stage may be a favourable alternative to initiating insulin treatment. If sulphonylureas are used before initiation of GLP-1 receptor agonist therapy, the sulphonylurea dose should be at least reduced when adding the GLP-1 receptor agonist. In a large proportion of patients, the sulphonylurea treatment can even be stopped.
Incretin-based therapies may help to bring a larger percentage of patients to their glycaemic goals. Fixed-dose combinations of a DPP-4 inhibitor with metformin may be a favourable alternative as the patient does not have to take more tablets when intensifying oral antidiabetic therapy with a DPP-4 inhibitor. Obese patients with weight loss as another important therapeutic goal may profit from therapy with a GLP-1 receptor agonist. The higher price of the novel incretin-based therapies is outweighed in some respects by the possibility of reducing the cost of blood glucose monitoring that is not necessary for safety reasons as long as the patient is not simultaneously treated with a sulphonylurea.
Prevention of hypoglycaemic events and prevention of further weight gain are important therapeutic goals considering the results of the ACCORD trial, which showed increased mortality in patients with type 2 diabetes who were allocated to the intensified treatment arm with an HbA1c goal <6.0% and who were treated with multiple combinations of the classical antidiabetic agents [91]. The increased mortality rate in this group may be explained by the higher gain in body weight and by the increased incidence of hypoglycaemic episodes. On the other hand, the 10-year follow up data of the UKPDS show that an early and effective diabetes treatment not only lowers microvascular complications, but also lowers macrovascular endpoints significantly [4]. With respect to these study results, patients with a newly diagnosed type 2 diabetes should have a treatment that enables them to reach normoglycaemia in a safe way without the risk of hypoglycaemia or weight gain.
A consensus statement published in 2008 by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) separates the existing antidiabetic compounds and treatment algorithms into well-validated therapies (“tier 1”, comprising metformin, sulphonylureas and insulin) and less well-validated therapies (“tier 2”, comprising pioglitazone and GLP-1 receptor agonists). In this statement, the established substances are preferred according to their published endpoint and safety data as well as pharmaco-economic data. In the less well-validated therapies, GLP-1 receptor agonists, however, have their place as second therapeutic escalation after metformin failure in the same line as the widely used therapy with pioglitazone [92]. In the German guidelines, DPP-4 inhibitors and GLP-1 receptor agonists are placed in second line after metformin failure, if the HbA1c does not exceed >7.5% [87].
Both incretin-based therapies may also play a role in the earlier or later stages of type 2 diabetes, when effectiveness is shown at these stages. Preliminary data show, that the addition of a DPP-4 inhibitor to existing insulin therapy further reduces HbA1c and may have a positive effect on hypoglycaemic events [93, 94]. Combination studies with insulin and GLP-1 receptor agonists are also being carried out and should bring results soon. Furthermore, long-term studies are underway, investigating the effect of incretin-based therapies on disease progression. Results of these studies are pending should be available by approximately the middle of the next decade. If these studies show an effect on disease progression, another argument in favour of using incretin-based therapies early in the disease will be supported by study data. Recently, animal and human studies showed a positive influence of GLP-1 or GLP-1 receptor agonists on the cardiovascular system and on the nervous system, describing neuroprotective effects [17, 95–99]. These fields may also open novel indications for incretin-based therapies [17]. However, long-term studies on hard cardiovascular endpoints and safety finally have to reveal important data in clarifying the efficacy, safety and placement of incretin-based therapies in type 2 diabetes therapy.
Incretin hormones, incretin-based therapy and renal failure
GLP-1 has natriuretic actions that have been observed in several studies. These actions are in parallel to a diminished proton secretion. GLP-1 receptor activation most likely influences the activity of the sodium-proton exchanger 3 (NHE3 = natrium-proton [H+] exchanger 3) located in the brush border membrane of the proximal tubular cells by activation pathways involving cAMP (cyclic adenosine mono-phosphate) and protein kinase A [100]. The GLP-1 receptor is expressed at the mRNA level and protein level in proximal tubular kidney cells [101].
In an animal model of salt-sensitive obese db/db mice GLP-1 receptor activation by exendin-4 administration inhibited the development of hypertension. After a high salt load, urinary sodium excretion was delayed and hypertension was present in these animals. Exendin-4 was able to attenuate the latter effect. Exendin-4 also prevented angiotensin II-induced hypertension through the attenuation of angiotensin II-induced high-salt sensitivity [102].
Changes in the secretion, in vivo degradation and elimination of the incretin hormones GLP-1 and GIP in patients with chronic renal insufficiency were characterised in a small study involving 10 patients with moderate renal failure (serum creatinine 2.18 ± 0.86 mg/dl) receiving an oral glucose tolerance test and GIP- and GLP-1 infusions on separate occasions. After an oral glucose load, plasma concentrations of intact GLP-1 and intact GIP are comparable in patients with renal insufficiency and healthy controls. The concentrations of the degradation products GIP [3–42] and GLP-1 (9–36 amide) are significantly higher in the patients than in the control subjects after oral glucose or after an exogenous infusion of either GIP or GLP-1. The insulin plasma concentrations were slightly lower in the patients during all experiments, whereas C-peptide levels tended to be increased. These data underline the importance of the kidneys for the final elimination of GIP and GLP-1. The initial DPP-4-mediated degradation of both hormones is almost unaffected by impairment of renal function. Delayed elimination of GLP-1 and GIP in renal insufficiency may influence the pharmacokinetics and pharmacodynamics of DPP-4-resistant incretin derivatives to be used for the treatment of patients with type 2 diabetes [103].
Incretin-based therapies and type 1 diabetes
Since GLP-1 has a positive effect on beta cell mass in rodents as well as a beneficial effect on the survival of isolated human islets in cell culture [6, 104, 105], it is imaginable that incretin-based therapies may also be advantageous in type 1 diabetes, possibly in combination with immune therapy. First preclinical studies suggest that theoretically such a combination therapy may be feasible in the future [106]. Besides the positive effect on the beta cells, GLP-1 may also influence glycaemic parameters in a favourable way by slowing gastric emptying and affecting glucagon secretion in type 1 diabetes [107]. A small mechanistic study in type 1 diabetic individuals showed that an intravenous GLP-1 infusion reduced fasting hyperglycemia in the morning, which was provoked by omitting the basal insulin injection at night [108]. Preclinical and animal studies should be undertaken to clarify the potential influence of GLP-1 on beta cell mass in type 1 diabetes and on possible alterations of the autoimmune process. Clinical studies could then be implemented to investigate the metabolic effects of incretin-based therapies in type 1 diabetes, provided there are no unwanted side effects on immune function in the preclinical studies.
References
International Diabetes Federation (IDF) (2009) Diabetes Atlas. Available at http://www.diabetesatlas.org
Zeitler P (2009) Update on nonautoimmune diabetes in children. J Clin Endocrinol Metab 94:2215–2220
Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O (2003) Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 348:383–393
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359:1577–1589
Nathan DM, Buse JB, Davidson MB, Heine RJ, Holman RR, Sherwin R, Zinman B (2006) Management of hyperglycaemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. A consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 49:1711–1721
Drucker DJ, Nauck MA (2006) The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368:1696–1705
Wellendorph P, Johansen LD, Brauner-Osborne H (2009) Molecular pharmacology of promiscuous seven transmembrane receptors sensing organic nutrients. Mol Pharmacol 76:453–465
Creutzfeldt W (1979) The incretin concept today. Diabetologia 16:75–85
Nauck M, Stockmann F, Ebert R, Creutzfeldt W (1986) Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29:46–52
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W (1993) Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91:301–307
Vollmer K, Gardiwal H, Menge BA, Goetze O, Deacon CF, Schmidt WE, Holst JJ, Meier JJ (2009) Hyperglycemia acutely lowers the postprandial excursions of glucagon-like Peptide-1 and gastric inhibitory polypeptide in humans. J Clin Endocrinol Metab 94:1379–1385
Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W (1993) Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 36:741–744
Brubaker PL, Drucker DJ (2004) Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 145:2653–2659
Fehmann HC, Habener JF (1992) Insulinotropic hormone glucagon-like peptide-I(7–37) stimulation of proinsulin gene expression and proinsulin biosynthesis in insulinoma beta TC-1 cells. Endocrinology 130:159–166
Courreges JP, Vilsboll T, Zdravkovic M, Le-Thi T, Krarup T, Schmitz O, Verhoeven R, Buganova I, Madsbad S (2008) Beneficial effects of once-daily liraglutide, a human glucagon-like peptide-1 analogue, on cardiovascular risk biomarkers in patients with Type 2 diabetes. Diabet Med 25:1129–1131
Klonoff DC, Buse JB, Nielsen LL, Guan X, Bowlus CL, Holcombe JH, Wintle ME, Maggs DG (2008) Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr Med Res Opin 24:275–286
Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP (2006) Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail 12:694–699
Yang J, Campitelli J, Hu G, Lin Y, Luo J, Xue C (2007) Increase in DPP-IV in the intestine, liver and kidney of the rat treated with high fat diet and streptozotocin. Life Sci 81:272–279
Lee Y, Kwon MK, Kang ES, Park YM, Choi SH, Ahn CW, Kim KS, Park CW, Cha BS, Kim SW, Sung JK, Lee EJ, Lee HC (2008) Adenoviral vector-mediated glucagon-like peptide 1 gene therapy improves glucose homeostasis in Zucker diabetic fatty rats. J Gene Med 10:260–268
Gallwitz B (2006) Exenatide in type 2 diabetes: treatment effects in clinical studies and animal study data. Int J Clin Pract 60:1654–1661
Eng J, Kleinman WA, Singh L, Singh G, Raufman JP (1992) Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem 267:7402–7405
Gedulin BR, Smith P, Prickett KS, Tryon M, Barnhill S, Reynolds J, Nielsen LL, Parkes DG, Young AA (2005) Dose-response for glycaemic and metabolic changes 28 days after single injection of long-acting release exenatide in diabetic fatty Zucker rats. Diabetologia 48:1380–1385
Kim D, MacConell L, Zhuang D, Kothare PA, Trautmann M, Fineman M, Taylor K (2007) Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care 30:1487–1493
Drucker DJ, Buse JB, Taylor K, Kendall DM, Trautmann M, Zhuang D, Porter L (2008) Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet 372:1240–1250
McGill JB (2009) Insights from the liraglutide clinical development program—the Liraglutide Effect and Action in Diabetes (LEAD) studies. Postgrad Med 121:16–25
Agerso H, Jensen LB, Elbrond B, Rolan P, Zdravkovic M (2002) The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia 45:195–202
Chang AM, Jakobsen G, Sturis J, Smith MJ, Bloem CJ, An B, Galecki A, Halter JB (2003) The GLP-1 derivative NN2211 restores beta-cell sensitivity to glucose in type 2 diabetic patients after a single dose. Diabetes 52:1786–1791
Retterstol K (2009) Taspoglutide: a long acting human glucagon-like polypeptide-1 analogue. Expert Opin Investig Drugs 18:1405–1411
Werner U (2008) Preclinical pharmacology of the new GLP-1 receptor agonist AVE0010. Ann Endocrinol (Paris) 69:164–165
Rosenstock J, Reusch J, Bush M, Yang F, Stewart M (2009) The potential of albiglutide, a long-acting GLP-1 receptor agonist, in type 2 diabetes: a randomized controlled trial exploring weekly, biweekly, and monthly dosing. Diabetes Care 32:1880–1886
Barnett AH (2005) Exenatide. Drugs Today (Barc) 41:563–578
Kolterman OG, Kim DD, Shen L, Ruggles JA, Nielsen LL, Fineman MS, Baron AD (2005) Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm 62:173–181
DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD (2005) Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 28:1092–1100
Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD (2004) Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 27:2628–2635
Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, Baron AD (2005) Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 28:1083–1091
Heine RJ, van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG (2005) Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 143:559–569
Nauck MA, Duran S, Kim D, Johns D, Northrup J, Festa A, Brodows R, Trautmann M (2007) A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia 50:259–267
Zinman B, Hoogwerf BJ, Duran Garcia S, Milton DR, Giaconia JM, Kim DD, Trautmann ME, Brodows RG (2007) The effect of adding exenatide to a thiazolidinedione in suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 146:477–485
Barnett AH, Burger J, Johns D, Brodows R, Kendall DM, Roberts A, Trautmann ME (2007) Tolerability and efficacy of exenatide and titrated insulin glargine in adult patients with type 2 diabetes previously uncontrolled with metformin or a sulfonylurea: a multinational, randomized, open-label, two-period, crossover noninferiority trial. Clin Ther 29:2333–2348
Barnett A (2007) Exenatide. Expert Opin Pharmacother 8:2593–2608
Bunck MC, Diamant M, Corner A, Eliasson B, Malloy JL, Shaginian RM, Deng W, Kendall DM, Taskinen MR, Smith U, Yki-Jarvinen H, Heine RJ (2009) One-year treatment with exenatide improves beta-cell function, compared with insulin glargine, in metformin-treated type 2 diabetic patients: a randomized, controlled trial. Diabetes Care 32:762–768
Ahmad SR, Swann J (2008) Exenatide and rare adverse events. N Engl J Med 358:1970–1971
Cure P, Pileggi A, Alejandro R (2008) Exenatide and rare adverse events. N Engl J Med 358:1969–1970
Dore DD, Seeger JD, Arnold Chan K (2009) Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 25:1019–1027
Yoo BK, Triller DM, Yoo DJ (2006) Exenatide: a new option for the treatment of type 2 diabetes. Ann Pharmacother 40:1777–1784
Food and Drug Administration (FDA) (2009) Byetta (exenatide)—renal failure. Available at http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm188703.htm. Accessed on 2 November 2009
Malloy J, Capparelli E, Gottschalk M, Guan X, Kothare P, Fineman M (2009) Pharmacology and tolerability of a single dose of exenatide in adolescent patients with type 2 diabetes mellitus being treated with metformin: a randomized, placebo-controlled, single-blind, dose-escalation, crossover study. Clin Ther 31:806–815
Sturis J, Gotfredsen CF, Romer J, Rolin B, Ribel U, Brand CL, Wilken M, Wassermann K, Deacon CF, Carr RD, Knudsen LB (2003) GLP-1 derivative liraglutide in rats with beta-cell deficiencies: influence of metabolic state on beta-cell mass dynamics. Br J Pharmacol 140:123–132
Garber A, Klein E, Bruce S, Sankoh S, Mohideen P (2006) Metformin-glibenclamide versus metformin plus rosiglitazone in patients with type 2 diabetes inadequately controlled on metformin monotherapy. Diabetes Obes Metab 8:156–163
Garber AJ, Spann SJ (2008) An overview of incretin clinical trials. J Fam Pract 57(Suppl):S10–S18
Nauck MA, Ratner RE, Kapitza C, Berria R, Boldrin M, Balena R (2009) Treatment with the human once-weekly glucagon-like peptide-1 analog taspoglutide in combination with metformin improves glycemic control and lowers body weight in patients with type 2 diabetes inadequately controlled with metformin alone: a double-blind placebo-controlled study. Diabetes Care 32:1237–1243
Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, Zdravkovic M, During M, Matthews DR (2009) Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 32:84–90
Marre M, Shaw J, Brandle M, Bebakar WM, Kamaruddin NA, Strand J, Zdravkovic M, Le Thi TD, Colagiuri S (2009) Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with Type 2 diabetes (LEAD-1 SU). Diabet Med 26:268–278
Zinman B, Gerich J, Buse JB, Lewin A, Schwartz S, Raskin P, Hale PM, Zdravkovic M, Blonde L (2009) Efficacy and safety of the human glucagon-like peptide-1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD-4 Met+TZD). Diabetes Care 32:1224–1230
Garber A, Henry R, Ratner R, Garcia-Hernandez PA, Rodriguez-Pattzi H, Olvera-Alvarez I, Hale PM, Zdravkovic M, Bode B (2008) Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet 373:473–481
Garber A, Henry RR, Ratner RE, Hale P, Chang CT, Bode B (2009) Monotherapy with liraglutide, a once-daily human GLP-1 analog, provides sustained reductions in A1C, FPG, and weight compared with glimepiride in type 2 diabetes: LEAD-3 mono 2-year results. Diabetes 58(Suppl 1):162, OR
Deacon CF (2009) Potential of liraglutide in the treatment of patients with type 2 diabetes. Vasc Health Risk Manag 5:199–211
Vilsboll T (2009) Liraglutide: a new treatment for type 2 diabetes. Drugs Today (Barc) 45:101–113
Nauck M, Marre M (2009) Adding liraglutide to oral antidiabetic drug monotherapy: efficacy and weight benefits. Postgrad Med 121:5–15
Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH, Zychma M, Blonde L (2009) Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 374:39–47
Vilsboll T, Brock B, Perrild H, Levin K, Lervang HH, Kolendorf K, Krarup T, Schmitz O, Zdravkovic M, Le-Thi T, Madsbad S (2008) Liraglutide, a once-daily human GLP-1 analogue, improves pancreatic B-cell function and arginine-stimulated insulin secretion during hyperglycaemia in patients with Type 2 diabetes mellitus. Diabet Med 25:152–156
Ahren B (2008) Emerging dipeptidyl peptidase-4 inhibitors for the treatment of diabetes. Expert Opin Emerg Drugs 13:593–607
Mest HJ (2006) Dipeptidyl peptidase-IV inhibitors can restore glucose homeostasis in type 2 diabetics via incretin enhancement. Curr Opin Investig Drugs 7:338–343
Gallwitz B (2008) Saxagliptin, a dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. IDrugs 12:906–917
Pratley RE, Reusch JE, Fleck PR, Wilson CA, Mekki Q (2009) Efficacy and safety of the dipeptidyl peptidase-4 inhibitor alogliptin added to pioglitazone in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled study. Curr Med Res Opin 25:2361–2371
Heise T, Graefe-Mody EU, Huttner S, Ring A, Trommeshauser D, Dugi KA (2009) Pharmacokinetics, pharmacodynamics and tolerability of multiple oral doses of linagliptin, a dipeptidyl peptidase-4 inhibitor in male type 2 diabetes patients. Diabetes Obes Metab 11:786–794
Pratley RE (2008) Overview of glucagon-like peptide-1 analogs and dipeptidyl peptidase-4 inhibitors for type 2 diabetes. Medscape J Med 10:171
Barnett AH (2009) New treatments in type 2 diabetes: a focus on the incretin-based therapies. Clin Endocrinol (Oxf) 70:343–353
Karasik A, Aschner P, Katzeff H, Davies MJ, Stein PP (2008) Sitagliptin, a DPP-4 inhibitor for the treatment of patients with type 2 diabetes: a review of recent clinical trials. Curr Med Res Opin 24:489–496
Green J, Feinglos M (2008) New combination treatments in the management of diabetes: focus on sitagliptin-metformin. Vasc Health Risk Manag 4:743–751
Williams-Herman D, Round E, Swern AS, Musser B, Davies MJ, Stein PP, Kaufman KD, Amatruda JM (2008) Safety and tolerability of sitagliptin in patients with type 2 diabetes: a pooled analysis. BMC Endocr Disord 8:14
Herman GA, Bergman A, Yi B, Kipnes M (2006) Tolerability and pharmacokinetics of metformin and the dipeptidyl peptidase-4 inhibitor sitagliptin when co-administered in patients with type 2 diabetes. Curr Med Res Opin 22:1939–1947
Herman GA, Bergman A, Liu F, Stevens C, Wang AQ, Zeng W, Chen L, Snyder K, Hilliard D, Tanen M, Tanaka W, Meehan AG, Lasseter K, Dilzer S, Blum R, Wagner JA (2006) Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects. J Clin Pharmacol 45:876–886
Herman GA, Bergman A, Stevens C, Kotey P, Yi B, Zhao P, Dietrich B, Golor G, Schrodter A, Keymeulen B, Lasseter KC, Kipnes MS, Snyder K, Hilliard D, Tanen M, Cilissen C, De Smet M, de Lepeleire I, Van Dyck K, Wang AQ, Zeng W, Davies MJ, Tanaka W, Holst JJ, Deacon CF, Gottesdiener KM, Wagner JA (2006) Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 91:4612–4619
Scott R, Wu M, Sanchez M, Stein P (2007) Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract 61:171–180
Chan JC, Scott R, Arjona Ferreira JC, Sheng D, Gonzalez E, Davies MJ, Stein PP, Kaufman KD, Amatruda JM, Williams-Herman D (2008) Safety and efficacy of sitagliptin in patients with type 2 diabetes and chronic renal insufficiency. Diabetes Obes Metab 10:545–555
Ahren B (2008) Novel combination treatment of type 2 diabetes DPP-4 inhibition + metformin. Vasc Health Risk Manag 4:383–394
Ahren B, Foley JE (2008) The islet enhancer vildagliptin: mechanisms of improved glucose metabolism. Int J Clin Pract Suppl 175:8–14
Pratley RE, Rosenstock J, Pi-Sunyer FX, Banerji MA, Schweizer A, Couturier A, Dejager S (2007) Management of type 2 diabetes in treatment-naive elderly patients: benefits and risks of vildagliptin monotherapy. Diabetes Care 30:3017–3022
Rosenstock J, Sankoh S, List JF (2008) Glucose-lowering activity of the dipeptidyl peptidase-4 inhibitor saxagliptin in drug-naive patients with type 2 diabetes. Diabetes Obes Metab 10:376–386
Rosenstock J, Aguilar-Salinas C, Klein E, Nepal S, List J, Chen R (2009) Effect of saxagliptin monotherapy in treatment-naive patients with type 2 diabetes. Curr Med Res Opin 25:2401–2411
Deacon CF, Holst JJ (2009) Saxagliptin: a new dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes. Adv Ther 26:488–499
Tahrani AA, Piya MK, Barnett AH (2009) Saxagliptin: a new DPP-4 inhibitor for the treatment of type 2 diabetes mellitus. Adv Ther 26:249–262
Chacra AR, Tan GH, Apanovitch A, Ravichandran S, List J, Chen R (2009) Saxagliptin added to a submaximal dose of sulphonylurea improves glycaemic control compared with uptitration of sulphonylurea in patients with type 2 diabetes: a randomised controlled trial. Int J Clin Pract 63:1395–1406
DeFronzo RA, Hissa MN, Garber AJ, Luiz Gross J, Yuyan Duan R, Ravichandran S, Chen RS (2009) The efficacy and safety of saxagliptin when added to metformin therapy in patients with inadequately controlled type 2 diabetes with metformin alone. Diabetes Care 32:1649–1655
Wolf R, Frederich R, Fiedorek FT, Donovan M, Xu Z, Harris S, Chen R (2009) Evaluation of CV risk in the saxagliptin clinical trials. Diabetes 59(Suppl 1):8, LB
Matthaei S, Bierwirth R, Fritsche A, Gallwitz B, Häring HU, Joost HG, Kellerer M, Kloos C, Kunt T, Nauck MA, Schernthaner G, Siegel E, Thienel F (2009) Medicinal antihyperglycemic therapy of type 2 diabetes. Guidelines of the German Diabetes Association. Exp Clin Endocrinol Diabetes 117:522–557
National Institute for Health and Clinical Excellence (2009) Available at http://guidance.nice.org.uk/CG87
Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP Jr, Selby JV (2009) Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 301:1565–1572
Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP (2007) Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 9:194–205
Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH Jr, Probstfield JL, Simons-Morton DG, Friedewald WT (2008) Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 358:2545–2559
Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, Zinman B (2009) Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 32:193–203
Vilsboll T, Rosenstock J, Yki-Jarvinen H, Cefalu WT, Chen Y, Ling Y, Meehan AG, Katz L, Engel SS, Kaufman KD, Amatruda JM (2009) Sitagliptin, a selective DPP-4 inhibitor, improves glycemic control when added to insulin, with or without metformin, in patients with type 2. Diabetes 58(Suppl 1):588, P
Fonseca V, Baron M, Shao Q, Dejager S (2008) Sustained efficacy and reduced hypoglycemia during one year of treatment with vildagliptin added to insulin in patients with type 2 diabetes mellitus. Horm Metab Res 40:427–430
Perry T, Holloway HW, Weerasuriya A, Mouton PR, Duffy K, Mattison JA, Greig NH (2007) Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp Neurol 203:293–301
Müssig K, Oncu A, Lindauer P, Heininger A, Aebert H, Unertl K, Ziemer G, Häring HU, Holst JJ, Gallwitz B (2008) Effects of intravenous glucagon-like peptide-1 on glucose control and hemodynamics after coronary artery bypass surgery in patients with type 2 diabetes. Am J Cardiol 102:646–647
Nikolaidis LA, Elahi D, Hentosz T, Doverspike A, Huerbin R, Zourelias L, Stolarski C, Shen YT, Shannon RP (2004) Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation 110:955–961
Nikolaidis LA, Doverspike A, Hentosz T, Zourelias L, Shen YT, Elahi D, Shannon RP (2005) Glucagon-like peptide-1 limits myocardial stunning following brief coronary occlusion and reperfusion in conscious canines. J Pharmacol Exp Ther 312:303–308
Nikolaidis LA, Elahi D, Shen YT, Shannon RP (2005) Active metabolite of GLP-1 mediates myocardial glucose uptake and improves left ventricular performance in conscious dogs with dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 289:H2401–H2408
Carraro-Lacroix LR, Malnic G, Girardi AC (2009) Regulation of Na+/H+ exchanger NHE3 by the glucagon-like peptide 1 receptor agonist exendin-4 in renal proximal tubule cells. Am J Physiol Renal Physiol 297:F1647–F1655
Schlatter P, Beglinger C, Drewe J, Gutmann H (2007) Glucagon-like peptide 1 receptor expression in primary porcine proximal tubular cells. Regul Pept 141:120–128
Hirata K, Kume S, Araki S, Sakaguchi M, Chin-Kanasaki M, Isshiki K, Sugimoto T, Nishiyama A, Koya D, Haneda M, Kashiwagi A, Uzu T (2009) Exendin-4 has an anti-hypertensive effect in salt-sensitive mice model. Biochem Biophys Res Commun 380:44–49
Meier JJ, Nauck MA, Kranz D, Holst JJ, Deacon CF, Gaeckler D, Schmidt WE, Gallwitz B (2004) Secretion, degradation, and elimination of glucagon-like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes 53:654–662
Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132:2131–2457
Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, Bertolotto C, Di Mario U, Harlan DM, Perfetti R (2003) Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 144:5149–5158
Waldron-Lynch F, von Herrath M, Herold KC (2008) Towards a curative therapy in type 1 diabetes: remission of autoimmunity, maintenance and augmentation of beta cell mass. Novartis Found Symp 292:146–155
Raman VS, Heptulla RA (2009) New potential adjuncts to treatment of children with type 1 diabetes mellitus. Pediatr Res 65:370–374
Creutzfeldt WO, Kleine N, Willms B, Orskov C, Holst JJ, Nauck MA (1996) Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon-like peptide I(7–36) amide in type I diabetic patients. Diabetes Care 19:580–586
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gallwitz, B. The evolving place of incretin-based therapies in type 2 diabetes. Pediatr Nephrol 25, 1207–1217 (2010). https://doi.org/10.1007/s00467-009-1435-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-009-1435-z