
Abstract
The potential effects of macrophage migration inhibitory factor (MIF) on the natural immune response are due to the inhibition of immune cell activation, which is regulated by glucocorticoids. In this study, we investigated MIF −173G/C genotype and C allele frequency in 214 patients with idiopathic nephrotic syndrome (INS) and 103 healthy volunteers. We found significant increases in GC genotype (OR=3, p=0.0009) and C allele frequency (OR=2.5, p=0.0007) in INS. Upon classifying patients as steroid responsive (n=137) or resistant (n=77), a 20-fold over-expression of the CC-genotype was found in the steroid-resistant group (OR=20, p=0.0002). Moreover, a significant increase in C allele frequency in patients with focal segmental glomerulosclerosis (FSGS) has also been noted when compared with other histopathological groups (OR=3.2, p=0.0017). Furthermore, significant increases in the CC genotype (15.6% vs 3.3%) and C allele (75% vs 32%) frequencies have been found in patients with permanent renal function failure (p=0.013 and p=0.0002, respectively). Patients with the CC genotype were found to be at considerably increased risk of permanent renal failure (OR=5.43, p=0.013) and end-stage renal disease (OR=5.53, p=0.020). Additionally, there was a correlation between age of detection of proteinuria and CC genotype. We found an earlier age of onset of proteinuria in patients with the CC genotype (1.9±1.7 years) than in patients who were GC-heterozygous (3.7±3.1 years) and GG-homozygous (3.6±2.9 years, p=0.88). In summary, our results indicate that the MIF −173 C allele confers an increased risk of susceptibility to INS and plays a crucial role in glucocorticoid responsiveness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Macrophage migration inhibitory factor (MIF) is a pleiotropic lymphocyte and macrophage cytokine; it is likely to play an important role in natural immunity. MIF is produced by T cells but it is also expressed by many cell types, including monocytes/macrophages and vascular endothelia. In contrast to other cytokines, MIF is constitutively expressed in a variety of immune and non-immune cells, and its tissue distribution is almost ubiquitous [1]. In kidney, MIF is constitutively expressed in some renal epithelial cells, including tubular epithelial cells and glomerular parietal and visceral epithelial cells (podocytes) [2, 3]. Most of the critical functions of MIF encompass the regulation of macrophage function [4], lymphocyte immunity [5] and endocrine functions [6, 7]. Because of its broad regulatory properties, MIF is a critical mediator of a number of immune and inflammatory diseases, including septic shock [8], juvenile idiopathic arthritis (JIA) [9, 10], ulcerative colitis [11], psoriasis [12], inflammatory lung diseases [13], and cancer [14, 15].
When present at low levels, glucocorticoids up-regulate MIF expression while down-regulating it at higher glucocorticoid concentrations, due to a counter-regulatory mechanism [16]. This characteristic of MIF has considerable potential applications to glucocorticoid-sensitive immunopathological diseases such a rheumatic arthritis (RA) [17, 18]. Glucocorticoid therapy represents a common anti-inflammatory treatment in RA. On the other hand, it is well known that a proportion of patients fail to respond to prescribed glucocorticoids. Santos et al demonstrated that dexamethasone treatment inhibits antigen-induced arthritis, and MIF treatment reverses the effects of the administered steroids [17].
The pathologic role of MIF in kidney disease has been extensively studied in experimental and human glomerulonephritis [2, 3, 19, 20, 21]. In a rat model of immunologically-induced crescentic antiglomerular basement membrane glomerulonephritis, Lan and colleagues [20] demonstrated that MIF plays a key regulatory role in the pathogenesis of immunologically-induced kidney disease. Treatment with anti-MIF mABs resulted in a marked inhibition of interleukine-1α (IL-1α) expression by both intrinsic kidney cells and macrophages, and inhibits inducible nitric oxide synthase expression in glomerular interstitial and tubular cells [19]. Furthermore, renal MIF expression was found to be significantly up-regulated in severe proliferative forms of human GN [3].
Moreover, since most of the mechanistic aspects of MIF action are not yet fully elucidated, potent novel MIF-based therapeutic tools may arise for several pathophysiologic, inflammatory and immune conditions. In this study, we studied the −173G/C polymorphism of the MIF gene to address the question of whether MIF is crucial to glucocorticoid resistance in idiopathic nephrotic syndrome (INS).
Materials and methods
Study patients
All of the patients (127 boys and 87 girls, 3.5±2.9 years old; follow-up time 4.7±3.9 years) with INS admitted to our department between January 2002 and December 2003 were included in this study, according to ISKDC (International Study of Kidney Disease in Children) criteria. 103 healthy controls (77 girls and 28 boys) were recruited from the Department of Pediatrics and randomly enrolled in the study. Their medical histories were taken and blood was sampled during the first routine visit. Written informed consent was obtained from the patients or parents. The procedures were in accordance with the ethical standard for human experimentation established by the Declaration of Helsinki of 1975, revised 1983.
Glomerular filtration rate (GFR) was estimated using the Schwartz formula. Permanent renal failure was defined as GFR below 80 mL/min per 1.73 m2, and ESRF (end-stage renal failure) was defined as GFR below 10 mL/min per 1.73 m2 or the need for any renal replacement therapy. Frequent relapse was defined as 2 relapses within 6 months after cessation of the first steroid treatment or 3 or more relapses within any 12-month period in an initially steroid-responsive patient. Steroid dependency was defined as 2 consecutive relapses under steroid treatment or occurring within 14 days after steroid withdrawal. Steroid resistance was defined as no achievement of remission despite treatment with prednisolone at 2 mg/kg per day for 4 weeks. Positive family history was associated with NS only in 26 patients (12.1%), hematuria in 32 patients (15%), hypertension in 23 patients (10.7%), renal failure in 42 patients (19.6%), and end-stage renal disease in 23 patients (10.7%). Steroid resistance was present in 34% (77/214) of the patients.
The following were accepted as indicators for renal biopsy indication: nephrotic syndrome under one years old or above 12 years old, macroscopic hematuria, microscopic hematuria and hypertension, hypocomplementemia, renal functional failure unrelated to hypovolemia, steroid-resistant nephrotic syndrome, and relapses in steroid-dependent patients with greater than 0.5 mg/kg per day prednisolone dose. Out of a total of 135 performed renal biopsies, minimal change nephrotic syndrome was in 29 patients (29.5%), diffuse mesangial sclerosis in 18 patients (13.3%), IgM nephropathy in 21 patients (15.6%) and focal segmental glomerulosclerosis in 67 patients (49.6%) (Table 1). A higher frequency of FSGS was observed than expected in our study group, because our center is a third-line reference center for pediatric nephrology patients in the Ege region of Turkey, and generally problematic and resistant patients are referred to our center.
MIF genotyping
Germline DNA was extracted from peripheral blood using Qiagen MiniBlood Purification System kits (Qiagen, Ontaria, Canada). The PCR–restriction fragment length polymorphism (PCR-RFLP) method of Donn R et al [22] was used to genotype the −173G/C polymorphism of MIF. The presence of a C at −173 of MIF creates an Alu I restriction enzyme site. The PCR primers were designed to amplify a 366-bp fragment that contained both the polymorphic and a nonpolymorphic Alu I site. The MIF-173 forward PCR-RFLP primer was 5′-ACT-AAG-AAA-GAC-CCGAGGC-3′, and the MIF-173 reverse PCR-RFLP primer was 5′-GGG-GCA-CGT-TGG-TGT-TTA-C-3′.
Amplification was carried out on a GeneAmp PCR System 9700 (PE Applied Biosystems, Foster City, CA, USA) in a 25 μl reaction mixture in 0.2 ml thin-wall PCR strip tubes (Axygen Scientific, Inc., Union City, CA, USA) containing 1 μl genomic DNA solution, GeneAmp Gold Buffer (15 mmol/L Tris-HCl, pH 8.0, 50 mmol/L KCl; PE Applied Biosystems), 1.5 mmol MgCl2, 50 μmol/l each of the dGTP, dATP, dTTP and dCTP (Promega, Madison, WI, USA), 25 pmol each of the forward and reverse primers, and 1.0 U AmpliTaq Gold polymerase (PE Applied Biosystems). The cycling conditions comprised a hot start at 95 °C for 10 min, followed by 35 amplification cycles at 95 °C for 45 s, 60 °C for 45 s, and 72 °C for 45 s, and a final extension at 72 °C for 7 min.
Amplified PCR product (3 μl) was digested in a 10 μl final reaction volume using 1 μl of reaction buffer 2 and four units of Alu I restriction enzyme (New England Biolabs, Beverly, MA), at 37 °C overnight. Controls of known genotype were included for every set of digestions carried out. The PCR products for the MIF gene were analyzed on a 3.0% agarose gel pre-stained with ethidium bromide (0.5 μg/ml) for visualization under ultraviolet light. Gels (20×20 cm) were run at 100 mV in 1xTBE buffer for 60 min.
To validate the accuracy and reproducibility of the results we randomly ran 100 samples taken from both patients and control subjects using the ddNTP primer extension method and capillary electrophoresis using SnaPshot kit (PE Applied Biosystems) [9]. Briefly, the −173 single-nucleotide polymorphism was genotyped using 20 ng of genomic DNA, and was amplified in a 10 μL final PCR reaction volume containing 5 pmol of each primer, 0.08 nmol of dNTPs, 1 μM KCl buffer, and 0.6 units of AmpliTaq polymerase (PE Applied Biosystems, Foster City, CA, USA). After amplification, the PCR product was incubated with 1 unit each of shrimp alkaline phosphatase (Amersham, Buckinghamshire, UK) and Exo I (New England Biolabs, Beverly, MA, USA) at 37 °C for 1 hour and at 72 °C for 15 minutes. An extension reaction was carried out for 25 cycles of 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 30 s. 6 μL of this extension product was incubated with 1 unit of calf intestine alkaline phosphatase (Amersham). 1 μL of this product was then pooled with 5 μL of deionized formamide and electrophoresed on a 310 ABI Genetic Analyzer (PE Applied Biosystems). Results were analyzed using GeneScan analysis and Genotyper version 3.6 software (PE Applied Biosystems).
Statistical analyses
The Hardy–Weinberg equilibrium (HWE) assumption was assessed for case and control groups by comparing the observed numbers of different genotypes with those expected under HWE for the estimated allele frequency (Table 2). The distributions of the MIF genotypes among cases versus steroid response and histology were assessed with the Mann-Whitney U-test. Fisher’s exact test was used to estimate odds ratios (ORs) and 95% confidence intervals (CIs) to gauge the relationship of the MIF genotype to the risk of nephrotic syndrome. GraphPad PRISM program (version 4.0 for Windows, GraphPad Software, San Diego, CA, USA) was used for statistical analyses.
Results
Spectrum of MIF −173G/C gene polymorphism
Using blood samples from Turkish INS patients and controls, we tested for the association of INS with G/C polymorphism in MIF gene. The findings are summarized in Table 3. Results show a significant increase of the GC and a non-significant increase of the CC genotype (OR=3, 95% CI 1.5–5.8, p=0.0009; OR=1.8, 95% CI 0.49–6.6, p=0.561, respectively), as well as a significant decrease of the GG genotype (OR=0.34, 95% CI 0.19–0.64, p=0.004) in INS patients. An increase in the C allele frequency was also significant (Table 3). The strength of the observed associations did not differ significantly between the sexes.
Genotype-renal function correlation
When we subclassified our data into steroid-responsive (n=137) and -resistant (n=77) patient groups, we found a 20-fold over-expression of the CC genotype in the steroid-resistant patients (OR=20, 95% CI 2.5–160, p=0.0002) (Table 4). Parallel significant increases were found in the GC genotype (OR=2.3, 95% CI=1.2–4.2, p=0.0109) and in the C allele frequency (OR=3.6, 95% CI 2.2–6.0, p<0.0001).
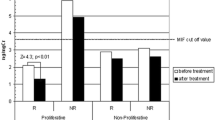
Moreover, the histopathology yielded significant evidence for an association between the C allele and distinct histological variants of primary INS. We found a significant increase in the C allele frequency in patients with FSGS and IgM nephropathy when compared with other histopathological groups (OR=3.2, 95% CI 1.5–6.8, p=0.0017; OR=3, 95% CI 1.2–7, 7, p=0.0372, respectively) (Fig. 1 and Table 5).

MIF gene “C” allele frequency for different histologic groups with nephrotic syndrome. Mean values and 95% confidence intervals are shown. The frequency was significantly higher in IgM and FSGS (see Table 5)
In order to investigate the above trend, we also calculated the relative risks associated with the phenotypes characteristic in INS patients. Using this, we found a significant increase in the CC genotype (15.6% vs 3.3%) frequency in the patients with permanent renal function failure compared to those with normal renal function (OR=5.43, 95% CI 1.55–19.04, p=0.013). CC polymorphism was also related to an increased risk of end-stage renal disease (OR=5.53, 95% CI 1.48–20.64, p=0.020).
Additionally, there was a correlation between age for detection of proteinuria and genotype (Fig. 2 compares nephrotic syndrome diagnosis ages (mean±SD) in NS patients with different MIF −173G/C genotypes). We found an earlier age of onset of proteinuria in patients with the CC genotype (1.9±1.7 years) than for the patients who were GC heterozygotes (3.7±3.1 years) and GG homozygotes (3.6±2.9 years, p=0.88). However, no correlation was found between C allele frequency and proteinuria detection age (r=0.018, p=0.74). MIF polymorphism was also tested for association with hematuria. An observed increase in hematuria for CC homozygous patients (54.5% vs 13.3% for GC and 12.6% for GG) was attributed to the strong association of MIF polymorphism and INS.

Comparison of nephrotic syndrome diagnosis ages (mean±SD) in NS patients with different MIF −173G/C genotypes
Discussion
Nephrotic syndrome is a common chronic illness in childhood. Idiopathic primary nephrotic syndrome (INS) has a reported incidence of 2–7 cases per 100,000 children and a prevalence of nearly 16 cases per 100,000 [23]. Childhood nephrotic syndromes are most commonly caused by one of two idiopathic diseases: MCNS and FSGS. The heterogeneity of the clinical course of INS is in stark contrast to the homogeneity of initial clinical manifestations and pathophysiology. The most important prognostic indicator in INS is steroid responsiveness. Almost 95% of the patients with MCNS and 20% of those with FSGS achieve remission after an eight-week course of prednisone [23]. Unfortunately, roughly 60% of the steroid-responsive patients experience 5 or more relapses. New therapeutic approaches are needed for steroid-responsive and resistant patients. To assess the functional and prognostic relevance of the −173 single-nucleotide G-to-C polymorphism of the MIF gene in patients with INS, it is useful to evaluate its association with glucocorticoid responsiveness.
Recent reports have suggested that MIF can be produced by intrinsic renal cells and that it plays a significant pathophysiologic role in kidney disease. Sasaki et al recently showed that renal expression of MIF was up-regulated in the kidneys of Tg mice, and the glomerular pathology consisted of progressive mesangial sclerosis with increased collagen IV accumulation without a significant increase in glomerular cells. Immunohistochemical and in situ hybridization in neonatal Tg kidneys demonstrated that MIF expression was up-regulated in almost all podocytes, in developing glomeruli and in some tubular epithelial cells [19].
Functional interaction of polymorphism on poor outcome of systemic inflammation has also been shown previously. Brown et al showed that there was a significant correlation between the urine MIF concentration and MIF expression within the kidney [24]. Thus, urine MIF reflects MIF expression within the injured kidney. Benedetti et al showed that the MIF-173 C allele is a predictor of poor outcome in systemic-onset juvenile idiopathic arthritis, and that patients carrying a MIF-173 C allele had serum and synovial fluid levels of MIF that were significantly higher than those in patients with the GG genotype [25]. A C allele at the position of the −173 MIF gene increases MIF expression through the creation of an activator protein 4 response element in the MIF promoter. It is possible that the CATT repeat element may influence some aspects of the MIF gene [9]. In this study, we found that patients with CC polymorphism exhibit a considerably increased risk of permanent renal failure and end-stage renal disease. The frequency of the C allele (8.11%) in our control population was also lower than that reported in Japan (19%) [11], but very similar to the frequencies reported for Caucasian Europeans in the UK (11.9%) [9]. Furthermore, the histopathology yielded significant evidence for an association between the CC genotype of MIF polymorphism and distinct histological variants of primary idiopathic nephrotic syndrome. We also found a significant increase in the CC genotype frequency in patients with FSGS. In conjunction with these studies we note that that podocyte-expressed MIF can affect podocyte functionality. This effect might aid the acceleration of chronic podocyte injury, with resulting progressive glomerulosclerosis.
Additional interest in MIF as a potential therapeutic target stems from its unique relationship to glucocorticoids. Combining the clinical findings and genotype relationship in INS patients yielded significant evidence for an association between the CC genotype of the MIF −173G/C polymorphism and clinical findings such as proteinuria, haematuria, renal function failure, histopathology, and most notably steroid responsiveness. This was even stronger when considering only the C allele frequency. Determination of MIF genotype in nephrotic syndrome may predict responsiveness to steroids and it may be useful for guiding clinicians when choosing an appropriate therapy.
Although we have not yet identified the mechanism, several distinct pathways for an interaction between glucocorticoids and MIF have been proposed on the basis of in vitro studies [26, 27]. Unlike other proinflammatory cytokines, MIF is biphasically regulated by glucocorticoids, with suppression at high concentrations of glucocorticoids and induction at lower concentrations [5, 6]. At the physiological stage, it is apparent that MIF is expressed constitutively in many tissues and in plasma, and that, in the absence of disease, both stress and glucocorticoid administration result in an increase in circulating MIF levels [28, 29, 30, 31]. Together, these studies support the notion that MIF is a potent immune modulator.
Two other major proinflammatory cytokines, interleukin-1β (IL-1β) and TNF-α, have been reported to be expressed at podocytes in several types of glomerulonephritis, as in the case of MIF [31]. Podocyte-expressed MIF and other cytokines can effect podocyte functionality and they participate in the acceleration of chronic podocyte injury, resulting in progressive glomerulosclerosis. Furthermore, overexpressed MIF derived from systemic circulation or through retrograde diffusion from podocyte could lead to systemic organ injuries through its proinflammatory effect.
In summary, we have found that the C allele of the MIF gene at position −173 is strongly related to steroid resistance in idiopathic nephritic syndrome. Furthermore, there is a positive correlation between C allele and histopathology for FSGS and IgM nephropathy. Our results indicate that the CC genotype plays a crucial role in glucocorticoid suppression during NS. These findings, which provide new insights into the antagonistic relationship between MIF and glucocorticoids, may aid the creation of therapeutic strategies such as anti-MIF antibodies and MIF antagonists for steroid-resistant nephrotic syndrome.
References
Calandra T, Roger T (2003) Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol 3:791–800.
Imamura K, Nishihira J, Suzuki M, Yasuda K, Sasaki S, Kusunoki Y, Tochimaru H, Takekoshi Y (1996) Identification and immunohistochemical localization of macrophage migration inhibitory factor in human glomerulonephritis. Biochem Mol Biol Int 40:1233–1242
Lan HY, Yang N, Nikolic-Paterson DJ, Yu XQ, Mu W, Isbel NM, Metz CN, Bucala R, Atkins RC (2000) Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int 57:499–509
Calandra T, Bernhagen J, Mitchell RA, Bucala R (1994) The macrophage is an important and previously unrecognized source of macrophage inhibitory factor. J Exp Med 179:1985–1992
Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa T, Donnelly T, Bucala R (1996) An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA 93:7849–7854
Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R (1995) MIF as a glucocorticoid-induced modulator of cytokine production. Nature 377:68–71
Donn RP, Ray DW (2004) Macrophage migration inhibitory factor: molecular, cellular and genetic aspects of a key neuroendocrine molecule. J Endocrinol 182:1–9
Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R (1993) MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365:756–759
Donn R, Alourfi Z, De Benedetti F, Meazza C, Zeggini E, Lunt M (2002) Mutation screening of the macrophage migration inhibitory factor gene: positive association of a functional polymorphism of macrophage migration inhibitory factor with juvenile idiopathic arthritis. Arthritis Rheum 46:2402–2409
Donn R, Alourfi Z, Zeggini E, Lamb R, Jury F, Lunt M, Meazza C, De Benedetti F, Thomson W, Ray D (2004) A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum 50:1604–1610
Nohara H, Okayama N, Inoue N, Koike Y, Fujimura K, Suehiro Y,Hamanaka Y, HigakiS, Yanai H, Yoshida T, Hibi T, Okita K, Hinoda Y (2004) Association of the −173G/C polymorphism of the macrophage migration inhibitory factor gene with ulcerative colitis. J Gastroenterol 39:242–246
Donn RP, Plant D, Jury F, Richards HL, Worthington J, Ray DW, Griffiths CE (2004) Macrophage migration inhibitory factor gene polymorphism is associated with psoriasis. Dermatol J Invest 123:484–487
Makita H, Nishimura M, Miyamoto K, Nakano T, Tanino Y, Hirokawa J, Nishihira J, Kawakami Y (1998) Effect of anti-macrophage migration inhibitory factor antibody on lipopolysaccharide-induced pulmonary neutrophil accumulation. Am J Respir Crit Care Med 158:573–579
Takahashi N, Nishihira J, Sato Y, Kondo M, Ogawa H, Ohshima T, Une Y, Todo S (1998) Involvement of macrophage migration inhibitory factor (MIF) in the mechanism of tumor cell growth. Mol Med 4:707–714
Shimizu T, Abe R, Nakamura H, Ohkawara A, Suzuki M, Nishihira J (1999) High expression of macrophage migration inhibitory factor in human melanoma cells and its role in tumor cell growth and angiogenesis. Biochem Biophys Res Commun 264:751–758
Lolis E (2001) Glucocorticoid counter regulation: macrophage migration inhibitory factor as a target for drug discovery. Curr Opin Pharmacol 1:662–668
Santos L, Hall P, Metz C, Bucala R, Morand EF (2001) Role of macrophage migration inhibitory factor (MIF) in murine antigen-induced arthritis: interaction with glucocorticoids. Clin Exp Immunol 123:309–314
Barton A, Lamb R, Symmons D, Silman A, Thomson W, WorthingtonJ, et al (2003) Macrophage migration inhibitory factor genepolymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes Immun 4: 487–491
Sasaki S, Nishihira J, Ishibashi T, Yamasaki Y, Obikane K, Echigoya M, Sado Y, Ninomiya Y, Kobayashi K (2004) Transgene of MIF induces podocyte injury and progressive mesangial sclerosis in the mouse kidney. Kidney Int. 65:469–481
Lan HY, Bacher M, Yang N, Mu W, Nicolic-Paterson DJ, Metz C, Meinhardt A, Bucala R, Atkins RC (1997) The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med 185:1455–1465
Tesch CH, Nikolic-Paterson DJ, Metz CN, Mu W, Bacher M, Bucala R, Atkins RC, Lan HY (1998) Rat mesangial cells express macrophage migration inhibitory factor in vitro and in vivo. J Am Soc Nephrol 9:417–424
Donn RP, Shelley E, Ollier WE, Thomson W (2001) A novel 5′-flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum 44:1782–1785
Eddy AA, Symons JM (2003) Nephrotic syndrome in childhood. Lancet 362:629–639
Brown FG, Nikolic-Paterson DJ, Hill PA, Isbel NM, Dowling J, Metz CM, Atkins RC (2002) Urine macrophage migration inhibitory factor reflects the severity of renal injury in human glomerulonephritis. J Am Soc Nephrol 13:S7–S13
De Benedetti F, Meazza C, Vivarelli M, Rossi F, Pistorio A, Lamb R, Lunt M, Thomson W, Ravelli A, Donn R, Martini A (2003) Functional and prognostic relevance of the −173 polymorphism of the macrophage migration inhibitory factor gene in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum 48: 1398–407
Lue H, Kleemann R, Calandra T, Roger T, Bernhagen J (2002) Macrophage migration inhibitory factor (MIF): mechanisms of action and role in disease. Microbes Infect 4:449–460
Gregersen PK, Bucala R (2003) Macrophage migration inhibitory factor, macrophage migration inhibitory factor alleles, and the genetics of inflammatory disorders: incorporating disease outcome into the definition of phenotype. Arthritis Rheum 48:1171–1176
Baugh JA, Donnelly SC (2003) Macrophage migration inhibitory factor: a neuroendocrine modulator of chronic inflammation. J Endocrinol 179:15–23
Fingerle-Rowson G, Koch P, Bikoff R, Lin X, Metz CN, Dhabhar FS, Meinhardt A, Bucala R (2003) Regulation of macrophage migration inhibitory factor expression by glucocorticoids in vivo. Am J Pathol 162:47–56
Sampey AV, Hall PH, Mitchell RA, Metz CN, Morand EF (2001) Regulation of synoviocyte phospholipase A2 and cyclooxygenase 2 by macrophage migration inhibitory factor. Arthritis Rheum 44: 1273–1280
Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J (2000) Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 408:211–216
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Berdeli, A., Mir, S., Ozkayin, N. et al. Association of macrophage migration inhibitory factor −173C allele polymorphism with steroid resistance in children with nephrotic syndrome. Pediatr Nephrol 20, 1566–1571 (2005). https://doi.org/10.1007/s00467-005-1930-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-005-1930-9