
Abstract
The cellular stress response decreases cellular injury, either via primary induction of cytoresistance or by secondary enhancement of cellular repair mechanisms. The most frequently studied and best understood effectors of the cellular stress response are the heat shock proteins (HSP). HSP are among the oldest tools in the cellular protein machinery, demonstrating extremely high conservation of the genetic code since bacteria. Molecular chaperons, with the HSP-70 being the prototype, cooperate in transport and folding of proteins, preventing aggregation, and even resolubilizing injured proteins. Increasing evidence supports a role for HSP during the recovery from renal ischemia, in particular in cellular salvage from apoptotic cell death and cytoskeletal restoration. Recent studies also report the potential for biomolecular profiling of newborns for the risk of acute renal failure. In peritoneal dialysis novel data suggest the use of HSP expression for biocompatibility testing. More importantly, HSP are prime therapeutic candidates for clinical situations associated with predictable insults, such as organ procurement in transplant medicine and repetitive exposure to hyperosmolar and acidotic peritoneal dialysis fluids. The next challenge will be to define the regulatory pathways of the cellular stress response in these models to introduce novel therapeutic interventions, such as new pharmaceutics enhancing the HSP expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although considerable progress has been made in understanding the pathogenesis of many chronic diseases such as chronic renal failure, the progression of tissue damage frequently occurs even when the primary disease process has been adequately treated and appears to have become inactive. The development of new therapeutic strategies to counteract this self-perpetuating organ damage is still of vital importance. At the cellular level the cellular response to stress may offer a therapeutic target to induce cytoprotection.
The cellular stress response produces a “switch” of the cellular machinery from housekeeping activities towards reaction against stressors [1]. This corresponds to a homeostatic reaction initiated upon recognition of a “stressful” condition. The result is a decrease in cellular injury, either by primary induction of cytoresistance that prevents injury or by secondary enhancement of cellular repair, counteracting ongoing damage. By these means the cellular stress response increases the cell’s chance of survival. The most frequently studied and best understood effectors of the cellular stress response are heat shock proteins (HSP) [2]. This review first provides a historical overview, then focuses on HSP-70 in renal ischemia including clinical data, and finally reviews experimental research on HSP in peritoneal dialysis. The reader should always keep in mind that production and activation of HSP are not the only cellular response to stressful stimuli.
Historical overview
Modern stress research began 40 years ago with Ritossa’s [3] description of specific heat-induced alterations in Drosophila melanogaster. In these fruit flies the connection of several thousand DNA strands to a polytene chromosome made it possible to identify sites of active transcription (“puffs”) by simple light microscopy. Hyperthermic stress resulted in loss of puffs that were usually present under control conditions, whereas new puffs appeared reproducibly at specific sites. Ritossa’s work was the first description of the reprogramming of the cellular gene expression machinery from “routine” to the “cellular stress response.”
Ten years later these observations were extended to the protein level [4]. When cells were exposed for a given period to radioactively labeled amino acids, proteins could be identified that were synthesized at the time of interest. During such an autoradiography the protein distribution pattern under control conditions showed a wide range. Under stressful conditions, however, there was decreased marking of the routinely expressed proteins but the appearance of “stress induced” protein bands. This technique impressively demonstrated the reduction in overall protein synthesis in favor of massive induction of a special class of proteins, which were called “heat-shock proteins” based on the classical experimental stress, heat-shock, and grouped according to their molecular weight. This review focuses on the best described HSP family in the human system, the 70-kDa HSP (HSP-70).
Mechanisms of HSP cytoprotection
HSP have subsequently been shown to be rapidly induced by a variety of cellular stressors, such as heat, UV light, and cytotoxic agents [5]. All these inducers of HSP have in common that they are proteotoxic or proteochaotic, resulting in increased cellular levels of denatured proteins [6]. The most widely accepted hypothesis regarding the regulation of HSP is currently that heat shock factor (HSF), a constitutive transcription factor, is inhibited by constitutive HSP-70 under control conditions. Increased levels of disrupted and denatured proteins compete for the binding of HSP and thereby initiate the activation of HSF, resulting in increased transcription of HSP genes [7, 8, 9].
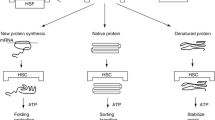
Whereas the role of HSP-70 during cellular repair processes remains to be fully elucidated, constitutively expressed HSP-70 is known to bind to newly translated immature proteins and prevent premature and improper binding and folding [10]. The amino acid sequence of the nascent polypeptide chain provides all of the information needed to generate the final product, and physicochemical forces result in folding of a well formed three-dimensional protein. For many years it was thought that these processes were passive and spontaneous. The detection of HSP changed that concept. This has been particularly well demonstrated in Escherichia coli, where HSP are necessary to allow the assembly of the complex protein structure of a bacteriophage [11]. In the human system similar “protective transport” has been observed in mitochondria, where HSP-70 molecules were shown to associate with immature polypeptide chains, preventing inadequate binding and folding of that structure. This function of HSP is called “molecular chaperoning.”
Today we know that there is a continuous balance between counteracting cellular proteotoxic and proteoprotective conditions (Fig. 1). Following this concept it is only logical that conditions that result in protein denaturation, and thus in loss of protein structure integrity, will also result in increased production of “renaturing tools” within the cell protein machinery, such as HSP [12, 13]. Molecular chaperons, with HSP-70 being the prototype, cooperate in transport and folding of proteins, without alteration in their own structure, by binding to hydrophobic, normally hidden domains of disrupted or immature proteins. They may prevent aggregation or even resolubilize injured proteins. In addition, the HSP-70 family, as well as the other HSP families, are involved in multiple cellular functions, such as protein degradation and cellular signaling mechanisms. Thus a major function of HSP is “homoeostasis” of cellular functions, a task that positively affects the survival of each cell—and of the whole organism under stressful conditions.

There is a continuous balance between decreases and increases in protein structure complexity. Under proteotoxic or proteochaotic stressful conditions a “perfect protein” (upper right) is disrupted and may either aggregate and be discarded (lower left) or be preserved by HSP-70 as a molecular chaperon (lower right). Upon activation of HSP-70 function (e.g., by ATP-hydrolysis) such reversibly disrupted and intermediate proteins again improve their structural and functional integrity
Taken together, HSP are among the oldest tools in the cellular protein machinery. HSP-70 has been extremely well conserved since bacteria, which leads to two important conclusions. First, HSP-70 is critical for cellular survival, a hypothesis that has been repeatedly verified by the finding that HSP-70 knockout animals are not viable. Second, it is unlikely that HSP-70 will ever prove to be a magic “silver bullet” in any given specific pathogenetic mechanism since the more than 80% of the genetic code of HSP-70 is much older than any of the complex molecular mechanisms that have evolved in eucaryotic and multicellular organisms since bacteria. High levels of HSP rather represent a setting of a “positive environment” during and, following stressful conditions, providing improved chances for effective recovery.
HSP in renal ischemia
Renal ischemia is a particularly well studied, clinically relevant stressor. There is increasing evidence for the importance of HSP during the recovery from ischemia [14, 15, 16, 17, 18, 19, 20]. Renal ischemia at the cellular basis results in remarkably heterogeneous injury ranging from cellular dysfunction in nonlethally injured cells to frank cell death [21]. Whereas necrosis is generally a rapidly occurring “catastrophic” form of cell death, apoptosis (=programmed cell death) is a carefully regulated active process [22, 23]. In both experimental and clinical acute renal injury apoptosis is the major form of cell death [21, 24, 25]. With regards to therapeutic approaches the nonlethally injured cells are the most important targets for interventions. Depending on the balance between cellular injury, repair, and death pathways, each renal epithelial cell may either recover (and survive) or undergo apoptosis (and die) (Fig. 2).

Scheme of stress-induced outcome at the cellular level. Any given cell may initially die in a necrotic way, remain unharmed, or be sublethally injured, depending on the dose and duration of stress exposure. Injured cells are stressed cells. With regards to therapeutic approaches these vital but stressed cells present the most important targets. In contrast to the two extreme outcomes (“necrotic” or “unharmed”) the outcome of these stressed cells (“recovery” vs. “apoptosis”) depends on active cellular mechanism and is thus likely to be particularly amenable to specific cytoprotective interventions
In contrast to receptor mediated apoptosis, the signal that leads to stress-induced apoptosis somehow “detects” that the damage can no longer be contained by the cellular repair mechanisms [26]. In renal tubular cells energy depletion induces apoptosis through specific molecular pathways that regulate this balance between recovery and apoptosis, based on protein-protein interactions [25, 26, 27]. HSPs, which can influence the assembly, transport, and folding of other proteins, are therefore prime candidates to affect the execution or inhibition of apoptotic signaling pathways [28, 29]. New experimental data indeed show that HSP confer cytoprotection against apoptosis [30, 31]. In energy-depleted renal epithelial cells both heat pretreatment and the selective overexpression of HSP-70 have been shown to decrease apoptosis in vitro [32]. HSP-70 inhibit stress-induced release of cytochrome c from mitochondria as well as the release and nuclear translocation of apoptosis-inducing factor, resulting in attenuated triggering of the caspase cascade [33, 34]. At present there are no studies extending these findings into the in vivo system.
With regard to nonlethal cellular injury following renal ischemia there is an increasing amount of both in vitro and in vivo data on the HSP-70 mediated mechanisms involved in cytoskeletal reorganization. Upon renal ischemia or energy depletion, disruption, and aggregation of the actin cytoskeleton is rapidly induced in renal tubule cells [35, 36]. Microvilli are fragmented, cell-cell and cell-substratum junctions disassociate, and cellular polarity is impaired. Cellular polarity, however, is of utmost importance for tubule cell function, such as vectorial substrate transport. Ischemia induces the rapid, duration-dependent translocation of apical and basolateral membrane-specific proteins and lipids into the alternate membrane domain. For example, upon energy depletion Na,K-ATPase transiently shifts from the basolateral into the apical membrane domain of proximal tubule cells where it remains functional but uses ATP to pump sodium back into the tubule lumen. Several clinical sequelae, such as renal sodium loss and reduced glomerular filtration due to tubuloglomerular feedback, can be explained by this loss of cellular polarity.
There is broad experimental evidence that HSP are involved in the restoration of the cytoskeletal integrity and cellular polarity. First, it was shown that following renal ischemia or energy depletion the cellular stress response and HSP are strongly induced, whereas expression of other proteins, such as Na,K-ATPase is downregulated, suggesting that dislocated cytoskeletal proteins are recycled [15, 37, 38, 39, 40]. The HSP also show a distinct temporal pattern of intracellular distribution that coincides with the restoration of disrupted proteins during recovery of the proximal tubule cell from ischemia. Based on observations that HSP may reactivate injured and disrupted proteins, it was then demonstrated that HSP-70 is released and Na, K-ATPase stabilized within the cytoskeletal fraction of ischemic rat renal cortex [41, 42]. Based on these findings a new subcellular in vitro assay was designed based on effects of recombinant HSP or anti-HSP antibodies [43, 44]. Findings in this system were also consistent with “repair” of disrupted proteins involved in the cytoskeletal organization of the proximal tubule cell, mediated by the molecular chaperoning mechanisms of HSPs.
Pretreatment with a nonlethal dose of cellular stress has been shown to result in survival of a subsequent, usually lethal dose of the same or other injury [1, 2, 45]. Organisms and tissues overexpressing HSP are more resistant to cellular stress than those whose HSP expression is suppressed [46, 47]. This increased resistance to repeat cellular injury is called cytoprotection; the treatment resulting in this cytoprotection is termed conditioning [48]. Based on this approach it was shown that in vivo pretreatment with renal ischemia (ischemic conditioning) not only induces HSP but also prevents cytoskeletal disruption in rat renal cortex after repeat ischemia [49, 50]. This stabilization was abolished by blocking antibodies against HSP ex vivo. In another novel approach using gene silencing techniques the detachment of Na, K-ATPase from its cytoskeletal anchorage was markedly increased, with decreased HSP-70 induction in energy depleted renal tubule cells, demonstrating that inhibition of stress response results in worsening injury [51]. Comparison of susceptibility for acute renal failure between different strains of rats also suggested that HSP expression affects the severity of injury following ischemic insult [52]. These data confirmed and extended those of several other studies that also reported cytoprotection upon overexpression of HSP in models of renal ischemia [53, 54, 55].
In the immature kidney high levels of HSP-70 have also been found to be correlated with protection from hypoxic or ischemic injury. Under experimental conditions HSF was more activated and HSP-70 expression higher in immature than in mature tubule cells. This was associated with tolerance of the immature tubules to anoxia [56]. More recently cytoprotection has also been shown to result in enhanced cytoskeletal stabilization in ischemic immature renal tubule cells [57]. This innate renal overexpression of HSP-70 does not require preconditioning and may explain the clinical observation that immature neonates appear relatively resistant to acute renal failure. Interestingly, this concept found strong support in a recent clinical study demonstrating that neonates with an HSP-70 polymorphism that is less inducible have a greater risk of acute renal failure than age-matched and clinically matched infants with a different allelic variant of HSP-70 [58]. Biomolecular profiling of patients may therefore allow identification of those patients who are more susceptible to injury.
HSP in renal transplantation
The setting of renal transplantation shares many features of models of renal ischemia and is particularly attractive to extend the basic principles of HSP-70 mediated cytoprotection. There are only limited data on HSP in ischemic renal injury in the human system; however, upregulation of HSP-70 expression, mostly upon heat pretreatment, has been shown to confer resistance against subsequent injury in experimental models of ischemia and transplantation [48, 59, 60]. A recent clinical study compared the renal tubular expression of HSP-70 in donor kidneys to the subsequent clinical course [61]. Interestingly, HSP-70 expression was low regardless of pretransplant insults and posttransplant outcome [61, 62]. After engraftment HSP-70 was detectable in the first urine produced by renal grafts in the immediate postoperative period [63]. As HSP-70 was expressed only basally, even in biopsy specimens of the subset of donor kidneys that subsequently developed complications, one may speculate that therapeutic interventions based on HSP-70 upregulation would at least be feasible. However, such nonspecific conditioning treatment may also activate or induce unwanted cellular processes, such as increasing immunogenicity [64].
Taken together, HSP present prime candidates for clinical situations associated with acute predictable insults, such as organ procurement in transplant medicine, protocol toxic drug exposure in oncology, and repetitive exposure to hyperosmolar and acidotic peritoneal dialysis fluids.
HSP in peritoneal dialysis
Although HSP have long been known for their cytoprotective role, the essential step from bench to bedside for HSP-mediated cytoprotection has not yet been successful [65, 66]. Obviously its clinical application is seriously hampered because of the need for planned pretreatment and subsequent acute (repeat) insult. Clinically relevant injuries, however, are generally complex and occur rather unpredictably with regard to both time course and dose.
Peritoneal dialysis may offer a unique opportunity to apply cytoprotection as an innovative therapeutic tool. The clinical setting of peritoneal dialysis consists of repeated, timed predictable exposure of peritoneal mesothelial cells to acute and uniform cellular insults upon peritoneal dialysis fluids (PDF) exposure; the final part of this review is therefore dedicated to this topic (Fig. 3). In vitro, several studies have demonstrated marked cytotoxic effects on human mesothelial cells of acute PDF exposure, due to their low pH, hyperosmolarity, and high concentrations of lactate, glucose, and glucose degradation products [67]. Experimentally and clinically, prolonged treatment frequently results in severe chronic damage to the integrity of the peritoneal membrane [68, 69]. Children appear particularly prone to the vicious circle of high dependence on peritoneal membrane function (substantially higher need of ultrafiltration than adults) and high risk of membrane failure (increased exposure to glucose) [70].

Scheme of the potential role of the cellular stress response in peritoneal dialysis. Toxic physicochemical properties of PDF cause injury in mesothelial cells. Although some cells die while some proliferate, the majority of mesothelial cells are likely to be stressed and undergo cellular repair. Therefore both decreased toxicity of PDF and enhanced cellular repair result in intact mesothelial cells
Although new and improved formulations have recently shown to be less toxic in several in vitro and in vivo experimental and clinical studies, the main working principle of PDF requires use of a hypertonic solution for mediation of solute and water removal [71]. PDF therefore is never likely to offer a physiologically inert or completely biocompatible fluid, and thus repeated filling and drainage of the abdominal cavity will always produce some acute stress. Based on the experiences in the model of renal ischemia one expects that stressors such as PDF will result not only in mesothelial cell injury but also in induction of HSP. Indeed, specific HSP isoforms are upregulated in mesothelial cells upon exposure to PDF with and without modification of its physicochemical properties (such as pH and GDP content) in in vitro and in vivo systems [72, 73, 74, 75, 76]. Whereas these first studies focused on the upregulation of HSP expression as a marker of biocompatibility of PDF, new in vitro results confirm that nonlethal pretreatment also confers cytoprotective effects in experimental peritoneal dialysis [77] (Fig. 4). Human mesothelial cells respond to nonlethal PDF exposure or heat stress with the development of acquired resistance against PDF (a specific finding termed “cross tolerance”). The specific role of HSP-70 was confirmed by overexpression of HSP-70 using transient gene transfection [77]. These studies showed effective protection of mesothelial cells against exposure to PDF. These data are in good agreement with those of other recent studies that also demonstrate evidence for HSP-70 mediated cytoprotection, at conditions which are known to result in similar ATP depletion as reported after PDF exposure. These in vitro data are also well in accord with the previously reported finding of increased staining for HSP-70 in peritoneal biopsy specimens of patients on complicated peritoneal dialysis [78].

Quantification of cytoprotection conferred by a conditioning protocol in human mesothelial cells. Left Staining for HSP-70 is shown for nonpretreated cultures (Con) and cultures pretreated by PDF for 60 min (PT) and allowed to recover for 24hours. Right Staining for Vital Dye (marker for mortality) is shown in cells exposed to sham treatment or PDF pretreatment before (Con, PT) and after usually lethal PDF exposure (LT, PT+LT). PDF pretreatment induced HSP-70 and protected cellular viability (see [77])
Taken together, recent findings in experimental peritoneal dialysis demonstrate that mesothelial HSP expression not only promises a new biocompatibility test but also confers marked cytoprotection against exposure to PDF. The therapeutic use of cytoprotection may offer attractive new alternatives to improve long-term outcome on peritoneal dialysis. However, as pretreatment protocols based on heat or transfection are not feasible in the clinical setting, the next challenge will be to further define the regulatory pathways of the cellular stress response in peritoneal dialysis. Manipulation of these pathways may then allow introduction of alternate therapeutic interventions, such as new pharmaceutics enhancing the HSP expression [79, 80].
References
Nover, L (1991) Heat shock response. CRC, Boca Raton
Morimoto RI, Tissieres A, Georgopoulos C (1994) The biology of heat shock proteins and molecular chaperons. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Ritossa FM (1962) A new puffing pattern induced by a temperature shock and DNP in Drosophila. Experimenta 18:571–573
Tissieres A, Mitchell HK, Tracy UM (1974) Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J Mol Biol 85:389–398
Morimoto RI (1993) Cells in stress: transcriptional activation of heat shock genes. Science 259:1409–1410
Hightower LE (1991) Heat shock, stress proteins, chaperons and proteotoxicity. Cell 66:191–197
Ananthan J, Goldberg AL, Voellmy R (1986) Abnormal proteins serve as eukaryotic stress signals and trigger the activation of heat shock genes. Science 232:522–524
Mifflin LC, Cohen RE (1994) Characterization of denatured protein inducers of the heat shock (stress) response in Xenopus laevis oocytes. J Biol Chem 269:15710–15717
Mifflin LC, Cohen RE (1994) HSC70 moderates the heat shock (stress) response in Xenopus laevis oocytes and binds to denatured protein inducers. J Biol Chem 269:15718–15723
Hartl FU, Hayer-Hartl M (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852–1858
Polissi A, Goffin L, Georgopoulos C (1995) The Escherichia coli heat shock response and bacteriophage lambda development. FEMS Microbiol Rev 17–:159–169
Pelham HRB (1984) Hsp-70 accelerates the recovery of nuclear morphology after heat shock. EMBO J 3:3095–3100
Pelham HRB (1986) Speculation on the functions of the major heat shock and glucose-related proteins. Cell 46:959–961
Emami A, Schwartz JH, Borkan SC (1991) Transient ischemia or heat stress induces a cytoprotectant protein in rat kidney. Am J Physiol 260:F479–F485
Van Why SK, Hildebrandt F, Ardito T, Mann AS, Siegel NJ, Kashgarian M (1992) Induction and intracellular localization of HSP-72 after renal ischemia. Am J Physiol 263:F769–F775
Van Why SK, Siegel NJ (1998) Heat shock proteins in renal injury and recovery. Curr Opin Nephrol Hypertens 7:407–412
Smoyer WE, Ransom R, Harris RC, Welsh MJ, Lutsch G, Benndorf R (2000) Ischemic acute renal failure induces differential expression of small heat shock proteins. J Am Soc Nephrol 11:211–221
Schober A, Muller E, Thurau K, Beck FX (1997) The response of heat shock proteins 25 and 72 to ischaemia in different kidney zones. Pflugers Arch 434:292–299
Morita K, Wakui H, Komatsuda A, Ohtani H, Miura AB, Itoh H, Tashima Y (1995) Induction of heat-shock proteins HSP73 and HSP90 in rat kidneys after ischemia. Ren Fail 17:405–419
Aufricht C, Ardito T, Thulin G, Kashgarian M, Siegel NJ, Van Why SK (1998) Heat-shock protein 25 induction and redistribution during actin reorganization after renal ischemia. Am J Physiol 274:F215–F222
Bonegio R, Lieberthal W (2002) Role of apoptosis in the pathogenesis of acute renal failure. Curr Opin Nephrol Hypertens 11:301–308
Raffray M, Cohen GM (1997) Apoptosis and necrosis in toxicology: a continuum or distinct modes of cell death? Pharmacol Ther 75:153–177
Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P (1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 185:1481–1486
Oberbauer R, Rohrmoser M, Regele H, Muhlbacher F, Mayer G (1999) Apoptosis of tubular epithelial cells in donor kidney biopsies predicts early renal allograft function. J Am Soc Nephrol 10:2006–2013
Feldenberg LR, Thevananther S, del Rio M, de Leon M, Devarajan P (1999) Partial ATP depletion induces Fas- and caspase-mediated apoptosis in MDCK cells. Am J Physiol 276:F837–F846
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Lieberthal W, Levine JS (1996) Mechanisms of apoptosis and its potential role in renal tubular epithelial cell injury. Am J Physiol 271:F477–F488
Beere HM, Green DR (2001) Stress management—heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol 11:6–10
Mosser DD, Caron AW, Bourget L, Meriin AB, Sherman MY, Morimoto RI, Massie B (2000) The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol 20:7146–7159
Garrido C, Gurbuxani S, Ravagnan L, Kroemer G (2001) Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem Biophys Res Commun 286:433–442
Jäättelä M (1999) Heat shock proteins as cellular lifeguards. Ann Med 31:261–271
Wang Y, Knowlton AA, Christensen TG, Shih T, Borkan SC (1999) Prior heat stress inhibits apoptosis in adenosine triphosphate-depleted renal tubular cells. Kidney Int 55:2224–2235
Ruchalski K, Mao H, Singh SK, Wang Y, Mosser DD, Li F, Schwartz JH, Borkan SC (2003) HSP72 inhibits apoptosis-inducing factor release in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol 285:C1483–C1493
Li F, Mao HP, Ruchalski KL, Wang YH, Choy W, Schwartz JH, Borkan SC (2002) Heat stress prevents mitochondrial injury in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol 283:C917–C926
Molitoris BA, Dahl R, Geerdes A (1992) Cytoskeletal disruption and apical redistribution of proximal tubule Na-KATPase during ischemia. Am J Physiol 263:F488–F495
Molitoris BA, Geerdes A, McIntosh JR (1991) Dissociation and redistribution of Na (+),K (+)-ATPase from its surface membrane actin cytoskeletal complex during cellular ATP depletion. J Clin Invest 88:462–469
Van Why SK, Mann AS, Ardito T, Siegel NJ, Kashgarian M (1994) Expression and molecular regulation of Na(+)-K(+)-ATPase after renal ischemia. Am J Physiol 267:F75–F85
Van Why SK, Mann AS, Thulin G, Zhu XH, Kashgarian M, Siegel NJ (1994) Activation of heat-shock transcription factor by graded reductions in renal ATP, in vivo, in the rat. J Clin Invest 94:1518–1523
Van Why SK, Kim S, Geibel J, Seebach FA, Kashgarian M, Siegel NJ (1999) Thresholds for cellular disruption and activation of the stress response in renal epithelia. Am J Physiol 277:F227–F234
Eickelberg O, Seebach F, Riordan M, Thulin G, Mann A, Reidy KH, Van Why SK, Kashgarian M, Siegel N (2002) Functional activation of heat shock factor and hypoxia-inducible factor in the kidney. J Am Soc Nephrol 13:2094–2101
Aufricht C, Lu E, Thulin G, Kashgarian M, Siegel NJ, Van Why SK (1998) ATP releases HSP-72 from protein aggregates after renal ischemia. Am J Physiol 274:F268–F274
Skowyra D, Georgopoulos C, Zylicz M (1990) The E-coli DNA K gene product, the HSP 70 homolog, can reactivate heat-inactivated RNA polymerase in an ATP hydrolysis-dependent manner. Cell 62:939–944
Bidmon B, Endemann M, Muller T, Arbeiter K, Herkner K, Aufricht C (2000) Heat shock protein-70 repairs proximal tubule structure after renal ischemia. Kidney Int 58:2400–2407
Bidmon B, Endemann M, Muller T, Arbeiter K, Herkner K, Aufricht C (2002) HSP-25 and HSP-90 stabilize Na,K-ATPase in cytoskeletal fractions of ischemic rat renal cortex. Kidney Int 62:1620–1627
Minowada G, Welch WJ (1995) Clinical implications of the stress response. J Clin Invest 95:3–12
Neuhofer W, Muller E, Burger-Kentischer A, Fraek ML, Thurau K, Beck FX (1999) Inhibition of NaCl-induced heat shock protein 72 expression renders MDCK cells susceptible to high urea concentrations. Pflugers Arch 437:611–639
Barbe MF, Tytell M, Gower DJ, Welch WJ (1988) Hyperthermia protects against light damage in the rat retina. Science 241:1817–1820
Perdrizet GA, Shapiro DS, Rewinski MJ (1999) Surgical stress and the heat shock response: in vivo models of stress conditioning. Ann N Y Acad Sci 874:320–325
Zager RA, Baltes LA, Sharma HM, Jurkowitz MS (1984) Responses of the ischemic acute renal failure kidney to additional ischemic events. Kidney Int 26:689–700
Aufricht C, Bidmon B, Ruffingshofer D, Regele H, Herkner K, Siegel NJ, Kashgarian M, Van Why SK (2002) Ischemic conditioning prevents Na,K-ATPase dissociation from the cytoskeletal cellular fraction after repeat renal ischemia in rats. Pediatr Res 51:722–727
Riordan M, Garg V, Thulin G, Kashgarian M, Siegel J (2004) Differential inhibition of HSP72 and HSP25 produces profound impairment of cellular integrity. J Am Soc Nephrol 15:1557–1566
Basile DP, Donohoe D, Cao X, Van Why SK (2004) Resistance to ischemic acute renal failure in the Brown Norway rat: a new model to study cytoprotection. Kidney Int 65:2201–2211
Kelly KJ, Baird NR, Greene AL (2001) Induction of stress response proteins and experimental renal ischemia/reperfusion. Kidney Int 59:1798–1802
Park KM, Chen A, Bonventre JV (2001) Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem 276:11870–11876
Meldrum KK, Burnett AL, Meng X, Misseri R, Shaw MB, Gearhart JP, Meldrum DR (2003) Liposomal delivery of heat shock protein 72 into renal tubular cells blocks nuclear factor-kappaB activation, tumor necrosis factor-alpha production, and subsequent ischemia-induced apoptosis. Circ Res 92:293–299
Gaudio KM, Thulin G, Mann A, Kashgarian M, Siegel NJ (1998) Role of heat stress response in the tolerance of immature renal tubules to anoxia. Am J Physiol 274:F1029–F1036
Vicencio A, Bidmon B, Ryu J, Reidy K, Thulin G, Mann A, Gaudio KM, Kashgarian M, Siegel NJ (2003) Developmental expression of HSP-72 and ischemic tolerance of the immature kidney. Pediatr Nephrol 18:85–91
Fekete A, Treszl A, Toth-Heyn P, Vannay A, Tordai A, Tulassay T, Vasarhelyi B (2003) Association between heat shock protein 72 gene polymorphism and acute renal failure in premature neonates. Pediatr Res 54:452–455
Trieb K, Dirnhofer S, Krumbock N, Blahovec H, Sgonc R, Margreiter R, Feichtinger H (2001) Heat shock protein expression in the transplanted human kidney. Transpl Int 14:281–286
Redaelli CA, Wagner M, Kulli C, Tian YH, Kubulus D, Mazzucchelli L, Wagner AC, Schilling MK (2001) Hyperthermia-induced HSP expression correlates with improved rat renal isograft viability and survival in kidneys harvested from non-heart-beating donors. Transpl Int 14:351–360
Mueller T, Regele H, Posch M, Marszalek M, Schwarz C, Pichlhoefer B, Arbeiter K, Aufricht C (2004) HSP-72 expression in pre-transplant donor kidney biopsies and post-transplant outcome. Transplantation 78:292–295
Bellemare S, Vigneault N, Madore F, Raymond MA, Cailhier JF, Hebert MJ (2002) Enhanced development of caspase-independent cortical cell death during cold storage in kidneys of non-heart-beating donors. Transplantation 73:1742–1751
Mueller T, Bidmon B, Pichler P, Arbeiter K, Ruffingshofer D, VanWhy SK, Aufricht C (2003) Urinary heat shock protein-72 excretion in clinical and experimental renal ischemia. Pediatr Nephrol 18:97–99
Pockley AG (2001) Heat shock proteins, anti-heat shock protein reactivity and allograft rejection. Transplantation 71:1503–1507
Aufricht C (2004) HSP: helper, suppressor, protector. Kidney Int 65:739–740
Molitoris BA (2003) Transitioning to therapy in ischemic acute renal failure. J Am Soc Nephrol 14:265–267
Jorres A, Topley N, Gahl GM (1992) Biocompatibility of peritoneal dialysis fluids (editorial). Int J Artif Organs 15:79–83
Gotloib L, Shostack A, Bar Sella P, Cohen R (1987) Continuous mesothelial injury and regeneration during longterm peritoneal dialysis. Perit Dial Bull 7:148–155
Di Paolo N, Sacchi G (2000) Atlas of peritoneal histology. Perit Dial Int 20 [Suppl 3]:S5–S96
Arbeiter K, Vecsei A, Mueller T, Sanz C, Balzar E, Aufricht C (2003) Chronic peritoneal dialysis in children. Results of the Vienna Pediatric Dialysis Department (in German). Wien Klin Wochenschr 115:660–664
Breborowicz A, Oreopoulos DG (1996) Biocompatibility of peritoneal dialysis solutions. Am J Kidney Dis 27:738–743
Aufricht C, Endemann M, Bidmon B, Arbeiter K, Mueller T, Regele H, Herkner K, Eickelberg O (2001) Peritoneal dialysis fluids induce the stress response in human mesothelial cells. Perit Dial Int 21:85–88
Arbeiter K, Bidmon B, Endemann M, Bender TO, Eickelberg O, Ruffingshofer D, Mueller T, Regele H, Herkner K, Aufricht C (2001) Peritoneal dialysate fluid composition determines heat shock protein expression patterns in human mesothelial cells. Kidney Int 60:1930–1937
Ruffingshofer D, Endemann M, Arbeiter K, Bidmon B, Mueller T, Herkner K, Aufricht C (2003) Induction of heat shock protein 72 in mesothelial cells exposed to peritoneal dialysate effluent. Perit Dial Int 23:74–77
Mosser DD, Kotzbauer PT, Sarge KD, Morimoto RI (1990) In vitro activation of heat shock transcription factor DNA-binding by calcium and biochemical conditions that affect protein conformation. Proc Natl Acad Sci USA 87:3748–3752
Arbeiter K, Bidmon B, Endemann M, Ruffingshofer D, Mueller T, Regele H, Eickelberg O, Aufricht C (2003) Induction of mesothelial HSP-72 upon in vivo exposure to peritoneal dialysis fluid. Perit Dial Int 23:499–501
Bidmon B, Endemann M, Arbeiter K, Ruffingshofer D, Regele H, Eickelberg O, Aufricht C (2004) Overexpression of HSP-72 confers cytoprotection in experimental peritoneal dialysis. Kidney Int 66:2300–2307
Shioshita K, Miyazaki M, Ozono Y, Abe K, Taura K, Harada T, Koji T, Taguchi T, Kohno S (2000) Expression of heat shock proteins 47 and 70 in the peritoneum of patients on continuous ambulatory peritoneal dialysis. Kidney Int 57:619–631
Ishihara K, Horiguchi K, Yamagishi N, Hatayama T (2003) Identification of sodium salicylate as an hsp inducer using a simple screening system for stress response modulators in mammalian cells. Eur J Biochem 270:3461–3468
Morimoto RI, Santoro MG (1998) Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. Nat Biotechnol 16:833–838
Acknowledgements
The author thanks Larry Greenbaum, Scott K. Van Why, and Norm J. Siegel for critical review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aufricht, C. Heat-shock protein 70: molecular supertool?. Pediatr Nephrol 20, 707–713 (2005). https://doi.org/10.1007/s00467-004-1812-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-004-1812-6