
Abstract
Spinocerebellar ataxia type 6 (SCA6), an autosomal dominant triplet repeat disease, predominantly affects the cerebellum with a late onset and generally good prognosis. Dysphagia is commonly associated with the outcomes of neurodegenerative diseases such as SCA6. Although the characteristics of dysphagia have been rarely reported in SCA6, our previous study indicated that dysphagia is generally milder in SCA6 than in SCA3, another inherited ataxia with multisystem involvement. However, abnormalities in the pharyngeal phase in SCA6 were indistinguishable from those in SCA3, with no explainable reason. To determine the reason, we repeatedly performed videofluoroscopic examinations (VF) in 14 patients with SCA6. The results showed that the gross progression of dysphagia was apparently slow, but four patients had progressive dysphagia at an early disease stage; dysphagia began within 10 years from the onset of ataxia and rapidly progressed. A common clinical feature of the four patients was a significantly older age at the onset of ataxia (74.0 vs. 60.3 years), associated with significantly shorter triplet repeats. This finding surprisingly indicated that patients who had shorter repeats and thereby later onset and potentially better prognoses were at risk for dysphagia-associated problems. Ischemic changes, homozygous mutation, and diabetes mellitus as well as aging might have contributed to the observed progressive dysphagia. We found that conventionally monitored somatosensory evoked potentials at least partly reflected progressive dysphagia. Despite the small study group, our findings suggest that clinicians should carefully monitor dysphagia in patients with SCA6 who are older at disease onset (>60 years).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxias (SCAs) are autosomal dominant disorders. SCA type 6 (SCA6), referred to as “pure” cerebellar ataxia, typically has a late-onset (>50 years) and a good prognosis [1]. This disease is a polyglutamine disease caused by CAG triple expansions in the CACNA1A gene and is the commonest subtype of autosomal dominant ataxia worldwide [2, 3]. The progression of ataxia in patients with SCA6 is slower than that in patients with other common types of spinocerebellar ataxia, such as SCA1, SCA2, and SCA3 [2], all of which are characterized by multiple system involvement, generally including the cerebellar, autonomic, pyramidal, and extrapyramidal systems [4].
Dysphagia leads to aspiration pneumonia and suffocation, and is therefore a major cause of death in neurodegenerative disorders such as SCAs. However, dysphagia in SCA6 is incompletely understood. Only a few studies have reported swallowing dysfunction in SCA6 [5, 6]. Dysphagia has been estimated to occur in 8–50% of patients with SCA6 [5]. We previously showed that patients with SCA3 as well as those with SCA6 have considerable dysphagia on videofluoroscopic examination (VF), with generally milder involvement in SCA6 [6]. Our results also demonstrated that patients with SCA6 have appreciable dysfunctions during the pharyngeal phase, even in patients with early-stage disease or mildly disturbed activities of daily living (ADL). We thought that such considerable dysphagia in SCA6 was attributed to pathological lesions in the brainstem, especially in the facial nuclei, trigeminal nuclei, solitary nuclei, and reticular nuclei [7], which are also affected in SCA3 and other neurodegenerative diseases [7, 8]. However, the precise underlying reasons remain unclear.
We aimed to clarify the characteristics and progression of dysphagia in SCA6 by repeatedly performing VF. We evaluated dysphagia using the scale established by the Japanese Society of Dysphagia rehabilitation, which can separately evaluate the oral and pharyngeal phases [9–11], as well as the dysphagia outcome and severity scale (DOSS), which is used internationally [12]. The DOSS globally evaluates swallowing abnormalities, but cannot separately evaluate abnormalities during the oral phase and the pharyngeal phase.
Methods
Patients
We retrospectively studied dysphagia in 14 patients with SCA6 (4 men and 10 women; age at the first VF examination, 64.2 ± 12.0 years, mean ± SD). All patients visited our hospital directly or were referred by their regular physicians because of ataxia. SCA was clinically diagnosed by board-certified neurologists. Some patients had undergone genetic testing before presentation at our hospital. The others underwent genetic testing in our hospital, as described previously [6]. The numbers of cytosine, adenine, and guanine (CAG) repeats in the affected gene ranged from 21 to 27 (n = 12). The number of repeats was inversely correlated with the age at the onset of ataxia (Fig. 1), as reported previously [3]. The duration of disease was 11.1 ± 6.9 years. The Barthel index, which was used to evaluate activities of daily living, was 78.9 ± 23.7 (range 20–100).

Correlation of numbers of repeats with ages at onset of ataxia. The section sign indicates a homozygous patient. Linear regression analysis without the data for this homozygous patient showed a significant inverse correlation (p < 0.01 and r = −0.84). The solid symbols indicate patients with progressive dysphagia at an early disease stage (Patients 1–4)
To clarify the progression pattern of dysphagia, we performed a second VF examination at least 6 months after the first VF examination in each patient. All patients underwent VF at various time points; some patients were evaluated 3 times or more.
One patient had a 13-year history of diabetes mellitus, which was recently relatively well controlled (HbA1c = 6.0%) by 120 mg/day gliclazide and 0.9 mg/day voglibose. No patient had other factors related to dysphagia, such as structural abnormalities, spinal osteophytosis, radiation therapy, cardiac failure, a history of major surgical intervention, or neuroleptic intake. All patients provided informed consent to undergo VF.
Gene Analyses
We analyzed the SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, SCA31, and DRPLA genes in some of the patients with SCA6 [6, 10]. The remaining patients with SCA6 underwent genetic diagnosis in other hospitals. This study was approved by the Institutional Review Board of Kindai University. The patients or their guardians provided written informed consent.
VF Procedures and Evaluations
VF was performed as described previously, with slight modifications [11]. Briefly, VF was carried out with the subject in a seated position. Video images were recorded on DVDs in two viewing planes (lateral and anteroposterior). A diluted solution of barium (5 ml) was swallowed twice. If the swallowing problem was not very severe as indicated by the procedure and rating scales described below, a concentrated solution of barium was then swallowed twice; the amount was not restricted, and the subject was requested to swallow as usual to detect swallowing problems encountered in daily life. Worse scores according to the scales described below were obtained on each swallow. Afterwards, 6 g of barium mixed with jelly was swallowed.
The VF results were evaluated according to a scale established by the Japanese Society of Dysphagia Rehabilitation (total score, 21 = normal and 7 = severely affected), as well as according to the DOSS.
The following VF variables were assessed using the Japanese scale: lip closure, bolus formation, bolus transport during the oral phase and pharynx constriction, larynx elevation, bolus stasis at the vallecular and pyriform sinus, and aspiration during the pharyngeal phase. A 3-point scale was used to semi-quantify each variable in a VF series: 3 (normal), 2 (disturbed), and 1 (severely disturbed). One speech language pathologist and one neurologist who were blinded to all clinical details independently scored the results for each patient. Both had more than 10 years of experience in swallowing evaluation. More than 90% of the point scores were matched between the initial evaluators. If there was any discrepancy, three additional neurologists who were also swallowing experts decided which assigned score was appropriate.
Swallowing scores and the numbers of CAG repeats, durations of disease, and degrees of disturbed ADL were analyzed by linear regression analysis.
MRI Findings
MRI was performed with MAGNETOM Symphony 1.5T (SIEMENS, Germany). Some patients underwent MRI several times.
Somatosensory Evoked Potentials (SEP) and Auditory Evoked Potentials (AEP)
Somatosensory evoked potentials (SEP) elicited by median nerve stimulation were monitored in nine patients (three with progressive dysphagia at an early stage and six of the remaining patients). The other patients did not undergo evaluation of SEP, and most were no longer followed up. Auditory evoked potentials (AEP) were performed in 11 patients (3 with progressive dysphagia at an early stage and 8 of the remaining patients).
Statistical Analyses
Statistical analyses were performed with the use of SPSS software (version 22). The methods used for analyses are described in the text or figure legends. p values of less than 0.05 were considered to indicate statistical significance.
Results
VF Evaluation
Total VF scores in the patients with SCA6 varied considerably (Fig. 2). Only two patients (Patients 4 and 5) had total score of 14 points or below (the point at which we usually consider to perform percutaneous endoscopic gastrostomy) at the ages of 74 and 72 years, respectively. Four patients (two men and two women) had progressive dysphagia at an early disease stage (<10 years). They were 70.5 ± 11 years old at the onset of ataxia (range 61–87 years), which was significantly older than the other patients (Table 1). The four patients were 74 ± 10 years old at the first VF. The number of CAG repeats ranged from 21 to 22, including one patient with a homozygous mutation. The mean Barthel index was 90 (75–100) in the four patients. Their details are summarized in Table 1. Two of them had silent aspiration.

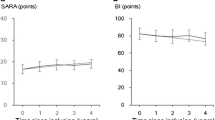
Progression of dysphagia and disturbed activities of daily living in patients with SCA6. a Total score of swallowing function, b oral phase, c pharyngeal phase, d) dysphagia outcome and severity scale (DOSS), and e Barthel index of activities of daily living. Solid symbols indicate patients with progressive dysphagia at an early stage of disease (Patients 1–4). The symbols are the same as those used in Fig. 1
Results of Linear Regression Analyses Assessing the Relations of the Swallowing Scores to the Numbers of CAG Repeats, Durations of Disease, and Degrees of Disturbed ADL
The total scores of swallowing abnormalities in all patients with SCA6 did not correlate with the number of CAG repeats, durations of disease, or degrees of disturbed ADL (Supplemental Fig. 2). However, the total scores of swallowing abnormalities in patients with later onset of dysphagia paralleled the degrees of disturbed ADL.
MRI Changes
Patient 1 had T2-high lesions probably caused by ischemic changes (Fig. 3). The lesions were found in periventricular white matter in the frontal, parietal, and occipital lobes, with predominance of the occipital lobes. Some lesions extended to the subcortical white matter corresponding to the motor cortex for the oral area. Patient 5 also had ischemic changes in the white matter and brainstem. Interestingly, the MRI findings after progression of the ataxia showed more ischemic changes in the white matter and in the brainstem, including pontocerebellar fibers and/or pontine nuclei in the pons, and the corticospinal tract in the middle brain, according to an atlas describing anatomical orientations in MRI images [13]. Cerebellar atrophy did not apparently differ between patients with progressive dysphagia at an early stage and the other patients.

MRI of patients with SCA6. a MRI of a patient with progressive dysphagia at an early disease stage (Patient 1). MRI showed diffuse cerebral atrophy (arrows) and typical cerebellar atrophy. T2-high signal lesions probably due to ischemic changes (arrowheads) were present in the basal ganglia (panel 3), deep white matter (panel 4), and subcortical white matter (panel 5). b MRI of a patient with late-onset dysphagia and then rapid progression (Patient 5). MRI before rapid progression already showed T2-high signals in the cerebral white matter and brainstem (arrowheads). Such signals apparently increased after rapid progression in the pontine base (white arrows, panels 1–2), in the middle brain (white arrows, panels 3–4), and in the deep white matter (white arrows, panel 5), and extended to the subcortical white matter (black arrows)
Results of SEP and AEP Evaluation
The cortical potential (N20) of SEP elicited by stimulation of the right (rt) median nerve in the patients who had progressive dysphagia at an early disease stage (19.4 ± 1.0 ms) was longer than that in the rest of the patients (18.0 ± 0.8 ms, Student’s t test, Fig. 4). The left (lt) median nerve SEP in the patients with progressive dysphagia (19.2 ± 1.1 ms) was slightly but not significantly longer than that in the rest of the patients (18.3 ± 0.8 ms). However, one of the patients with progressive dysphagia (Patient 4), who had silent aspiration, had apparently longer latency on both sides (rt 20.4 and lt 20.4) than the control ranges (17.5–19.9 ms). Other measures, including N9 (the brachial plexus point), N13 (the cervical point), or N20–N13 (the central conduction velocity), did not differ significantly between the two groups. The amplitudes of N20 and the results of AEP did not differ significantly between the two groups.

Results of the somatosensory potentials (SEP) elicited by right median nerve stimulation. The patients with late-onset dysphagia (Late) had shorter latency of cortical potential (N20) than those with progressive dysphagia at an early disease stage (Early, p < 0.05, Student’s t-test). Symbols are the same as those used in Fig. 1
Patient Outcomes
One patient (7.7%), Patient 5, who had two episodes of suffocation, underwent percutaneous endoscopic gastrostomy during this study. At the time of this writing, no patient with SCA6 had aspiration pneumonia or bronchitis.
Discussion
Our study revealed that the overall progression of dysphagia in patients with SCA6 was slow and mild, with development and progression continuing for longer than 10 years after the onset of ataxia; progression was limited in most patients. However, four patients in our study had progressive dysphagia at an early disease stage. Although the mechanism underlying the observed progressive dysphagia remains uncertain, a common clinical feature of the four patients was a significantly older age at the onset of ataxia (74.0 vs. 60.3 years), associated with significantly shorter triplet repeats. This finding surprisingly indicated that patients who had shorter repeats and thereby later onset and potentially better prognoses were at risk for dysphagia-associated problems. Thus, factors other than increased numbers of triplet repeats may be implicated in progressive dysphagia, as described below.
Ischemic changes, an age-dependent factor, have been associated with dysphagia [14]. However, only one of the four patients with progressive dysphagia at an early disease stage (Patient 1) had apparent ischemic changes in the cerebrum on MRI. Interestingly, one of the patients in the subgroup of patients with later onset of dysphagia (Patient 5) had relatively rapid progression of dysphagia later in her disease course, associated with the new appearance of probable ischemic changes on MRI. Among these MRI lesions, subcortical lesions may have significantly affected swallowing functions, since several reports demonstrated their relevance in dysphagia [15, 16]. By contrast, each of the small MRI lesions in the brainstem might not affect swallowing functions independently, since patients who had dysphagia with lesions in the midbrain or the pontine base usually had larger lesions [17, 18]. Nonetheless, speculation remains that the combinations of lesions in the brainstem together with those in the white matter may modify swallowing functions. Thus, ischemic changes may be at least partly implicated in the rapid progression of dysphagia.
Another factor potentially related to dysphagia is genetic background. One patient with progressive dysphagia at an early disease stage (Patient 2) was homozygous for SCA6 mutation. Homozygous patients were reported to have severer phenotypes in SCA6 [3, 19]. Such gene dosage effects are well known in experimental animals [20], but are not always obvious clinically [21]. Nonetheless, a previous study described a homozygote and a heterozygote in the same family (indicating similar environment factors and genetic backgrounds of other genes) in whom dysphagia was noted only in the homozygous patient in association with severer atrophy of the cerebellum on MRI [3].
Another patient with progressive dysphagia at an early disease stage (Patient 3) additionally had long-term (longer than 10 years) diabetes mellitus, a disease associated with dysphagia [22]. Diabetes mellitus causes peripheral sensory and motor neuropathy, with age-dependent severity [23]. Although the diabetes was recently relatively well controlled, we speculate that a long duration of mild diabetes might have contributed to the progressive dysphagia.
One of the four patients with progressive dysphagia at an early stage had no apparent reason for his severity of dysphagia. A previous study reported that aging directly disturbs pharyngeal function [24]. Thus, various factors in addition to the genetic background of SCA6 mutations might have worsened the dysphagia in association with advancing age.
We found that the median nerve SEP findings at least partly reflected the severity of dysphagia. Some of the patients with progressive dysphagia at an early disease stage had silent aspiration without its recognition, indicating that their sensory system was impaired. Although the tested regions were the upper extremities, their sensory involvement might reflect that in the swallowing systems. We understand that oropharyngeal sensory evoked potentials may be more closely related to dysphagia [25], but their evaluation requires special techniques in contrast to our conventional SEP. Although abnormalities were only noted unilaterally (the contralateral side showed a worsening tendency that did not reach statistical significance), patients with apparently abnormal SEP might be at risk for progressive dysphagia. In fact, the patient with the worst SEP results on either side (Patient 4) had silent aspiration.
The degree of disturbed ADL did not correlate with that of dysphagia in the total patients with SCA6, suggesting it is difficult to predict the likelihood of dysphagia on the basis of disturbed ADL. However, prediction of the risk of dysphagia might have been possible if patients with older ages at onset (>60 years) had been excluded. Despite the small number of the patients studied, our findings suggest that clinicians should carefully monitor dysphagia in patients with SCA6 who are older at disease onset. In patients with SCA6, optimal treatment of cardiovascular risk factors (hypertension, diabetes, etc.) is strongly recommended because ischemic brain lesions seem to worsen the natural course of SCA6 and predispose patients to dysphagia.
References
Schols L, Kruger R, Amoiridis G, Przuntek H, Epplen JT, Riess O. Spinocerebellar ataxia type 6: genotype and phenotype in German kindreds. J Neurol Neurosurg Psychiatry. 1998;64:67–73.
Ashizawa T, Figueroa KP, Perlman SL, Gomez CM, Wilmot GR, Schmahmann JD, Ying SH, Zesiewicz TA, Paulson HL, Shakkottai VG, Bushara KO, Kuo SH, Geschwind MD, Xia G, Mazzoni P, Krischer JP, Cuthbertson D, Holbert AR, Ferguson JH, Pulst SM, Subramony SH. Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J Rare Dis. 2013;8:177.
Matsumura R, Futamura N, Fujimoto Y, Yanagimoto S, Horikawa H, Suzumura A, Takayanagi T. Spinocerebellar ataxia type 6. Molecular and clinical features of 35 Japanese patients including one homozygous for the CAG repeat expansion. Neurology. 1997;49:1238–43.
Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124:1–21.
Takahashi H, Ishikawa K, Tsutsumi T, Fujigasaki H, Kawata A, Okiyama R, Fujita T, Yoshizawa K, Yamaguchi S, Tomiyasu H, Yoshii F, Mitani K, Shimizu N, Yamazaki M, Miyamoto T, Orimo T, Shoji S, Kitamura K, Mizusawa H. A clinical and genetic study in a large cohort of patients with spinocerebellar ataxia type 6. J Hum Genet. 2004;49:256–64.
Isono C, Hirano M, Sakamoto H, Ueno S, Kusunoki S, Nakamura Y. Differences in dysphagia between spinocerebellar ataxia type 3 and type 6. Dysphagia. 2013;28:413–8.
Rub U, Brunt ER, Petrasch-Parwez E, Schols L, Theegarten D, Auburger G, Seidel K, Schultz C, Gierga K, Paulson H, van Broeckhoven C, Deller T, de Vos RA. Degeneration of ingestion-related brainstem nuclei in spinocerebellar ataxia type 2, 3, 6 and 7. Neuropathol Appl Neurobiol. 2006;32:635–49.
Seidel K, Mahlke J, Siswanto S, Kruger R, Heinsen H, Auburger G, Bouzrou M, Grinberg LT, Wicht H, Korf HW, den Dunnen W, Rub U. The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol. 2015;25:121–35.
Higo R, Nito T, Tayama N. Swallowing function in patients with multiple-system atrophy with a clinical predominance of cerebellar symptoms (MSA-C). Eur Arch Otorhinolaryngol. 2005;262:646–50.
Isono C, Hirano M, Sakamoto H, Ueno S, Kusunoki S, Nakamura Y. differential progression of dysphagia in heredity and sporadic ataxias involving multiple systems. Eur Neurol. 2015;74:237–42.
Hirano M, Isono C, Sakamoto H, Ueno S, Kusunoki S, Nakamura Y. Rotigotine transdermal patch improves swallowing in dysphagic patients with Parkinson’s disease. Dysphagia. 2015;30:452–6.
O’Neil KH, Purdy M, Falk J, Gallo L. The dysphagia outcome and severity scale. Dysphagia. 1999;14:139–45.
Haines DE. Neuroanatomy in clinical context. 9th ed. Baltimore: Wolters Kluwer; 2015.
Loeb C, Gandolfo C, Caponnetto C, Del Sette M. Pseudobulbar palsy: a clinical computed tomography study. Eur Neurol. 1990;30:42–6.
Cola MG, Daniels SK, Corey DM, Lemen LC, Romero M, Foundas AL. Relevance of subcortical stroke in dysphagia. Stroke. 2010;41:482–6.
Wan P, Chen X, Zhu L, Xu S, Huang L, Li X, Ye Q, Ding R. Dysphagia post subcortical and supratentorial stroke. J Stroke Cerebrovasc Dis. 2016;25:74–82.
Schmahmann JD, Ko R, MacMore J. The human basis pontis: motor syndromes and topographic organization. Brain. 2004;127:1269–91.
Kim JS, Kim J. Pure midbrain infarction: clinical, radiologic, and pathophysiologic findings. Neurology. 2005;64:1227–32.
Ikeuchi T, Takano H, Koide R, Horikawa Y, Honma Y, Onishi Y, Igarashi S, Tanaka H, Nakao N, Sahashi K, Tsukagoshi H, Inoue K, Takahashi H, Tsuji S. Spinocerebellar ataxia type 6: CAG repeat expansion in alpha1A voltage-dependent calcium channel gene and clinical variations in Japanese population. Ann Neurol. 1997;42:879–84.
Unno T, Wakamori M, Koike M, Uchiyama Y, Ishikawa K, Kubota H, Yoshida T, Sasakawa H, Peters C, Mizusawa H, Watase K. Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc Natl Acad Sci USA. 2012;109:17693–8.
Matsuyama Z, Kawakami H, Maruyama H, Izumi Y, Komure O, Udaka F, Kameyama M, Nishio T, Kuroda Y, Nishimura M, Nakamura S. Molecular features of the CAG repeats of spinocerebellar ataxia 6 (SCA6). Hum Mol Genet. 1997;6:1283–7.
Restivo DA, Marchese-Ragona R, Lauria G, Squatrito S, Gullo D, Vigneri R. Botulinum toxin treatment for oropharyngeal dysphagia associated with diabetic neuropathy. Diabetes Care. 2006;29:2650–3.
Valensi P, Giroux C, Seeboth-Ghalayini B, Attali JR. Diabetic peripheral neuropathy: effects of age, duration of diabetes, glycemic control, and vascular factors. J Diabetes Complicat. 1997;11:27–34.
Frederick MG, Ott DJ, Grishaw EK, Gelfand DW, Chen MY. Functional abnormalities of the pharynx: a prospective analysis of radiographic abnormalities relative to age and symptoms. Am J Roentgenol. 1996;166:353–7.
Pitts T, Hegland KW, Sapienza CM, Bolser DC, Davenport PW. Alterations in oropharyngeal sensory evoked potentials (PSEP) with Parkinson’s disease. Respir Physiol Neurobiol. 2016;229:11–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest that could potentially bias this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
455_2016_9771_MOESM2_ESM.tif
Relations between the total scores of dysphagia and the scores of activities of daily living according to the Barthel index score. Open circles, patients with late onset of dysphagia; crosses, patients with progressive dysphagia at an early stage of disease. (A) The total scores did not correlate with the Barthel index scores in the patients with SCA6 enrolled in this study. (B) The total scores and Barthel index scores were significantly related in patients with later onset of dysphagia. (C) The total scores did not correlate with the Barthel index scores in patients with progressive dysphagia at an early stage of disease. Supplementary material 2 (TIFF 9576 kb)
455_2016_9771_MOESM3_ESM.tif
The amplitudes of N20 elicited by the right median nerve stimulation. The difference did not reach statistical significance (N.S.) between patients with late-onset dysphagia (Late) and those with progressive dysphagia at an early disease stage (Early). Symbols are the same as those used in Fig. 1. Supplementary material 3 (TIFF 10072 kb)
455_2016_9771_MOESM4_ESM.tif
Auditory evoked potentials on left ear stimulation. The difference did not reach statistical significance (N.S.) between the patients with late-onset dysphagia (Late) and those with progressive dysphagia at an early disease stage (Early). Symbols are the same as those used in Fig. 1. Supplementary material 4 (TIFF 14032 kb)
455_2016_9771_MOESM5_ESM.mp4
VF results in a 74-year-old man who had SCA6 with progressive dysphagia at an early disease stage (Patient 4). The first video at age 70 shows moderate dysphagia, and the second shows dysphagia progression after 4 years. The patient’s evaluations are summarized in Supplemental Fig. 1. Supplementary material 5 (MP4 1882 kb)
455_2016_9771_MOESM6_ESM.mp4
VF results in a 74-year-old man who had SCA6 with progressive dysphagia at an early disease stage (Patient 4). The first video at age 70 shows moderate dysphagia, and the second shows dysphagia progression after 4 years. The patient’s evaluations are summarized in Supplemental Fig. 1. Supplementary material 6 (MP4 2965 kb)
Rights and permissions
About this article

Cite this article
Isono, C., Hirano, M., Sakamoto, H. et al. Progression of Dysphagia in Spinocerebellar Ataxia Type 6. Dysphagia 32, 420–426 (2017). https://doi.org/10.1007/s00455-016-9771-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00455-016-9771-1