
Abstract
The decline of sewage purification efficiency in winter is a frequent problem in sub-plateau municipal sewage treatment plants (MSTPs). Understanding the links between activated sludge (AS) bacterial community and sewage purification is crucial for exploring the cause of this problem. In this study, Illumina high-throughput sequencing technology was applied to investigate the seasonal changes of AS bacterial community in sub-plateau MSTPs. The sequencing result indicates that the bacterial community OTU number, diversity, and relative abundance in winter are significantly lower than that in summer samples. The discriminant linear effect size analysis (LEfSe) reveals that Proteobacteria and Chloroflexi members were enriched in summer AS, while Actinobacteria and Firmicutes were enriched in winter AS. The results indicate that different core bacterial community assembly was developed in summer and winter, respectively. The changes in bacterial community may be the reasons for the lower sewage purification efficiency in winter. Furthermore, redundancy analysis (RDA) shows that temperature and dissolved oxygen (DO) are the principal factors that drive the seasonal changes in the core bacterial community diversity, richness and structure in sub-plateau MSTPs. Thus, the sub-plateau AS selects for a unique community assembly pattern and shapes the particular AS ecosystem. These results expand previous understanding and provide insight into the relationship between bacterial community and performance of sub-plateau MSTPs.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The sub-plateau region is defined as the transition zone from the Tibet plateau to the loess plateau in northwest China, where the average altitude ranges from 1000–2500 m and environmental ecosystems are fragile. Therefore, the local municipal sewage treatment plants play an important role in protecting the aquatic ecological system, especially as the headstream of Yangtze river and Yellow river. However, the efficiency of sewage purification in sub-plateau municipal sewage treatment plants (MSTPs) is clearly lower than that in plain MSTPs, and obviously decreased in winter season [13, 28].
Recent studies showed that microbial community structure and diversity were major factors related to nutriment removal and process stability in engineered microbial systems [8, 10, 21, 26]. While activated sludge (AS) bacterial community structure in different regions has significant geographical difference [9, 29]. It may be assumed that the local environmental variables such as the influent microbiota, temperature, altitude, dissolved oxygen (DO), and operational processes had remarkable effect on the AS microbial community structure [19, 21]. Previous study has shown that influent wastewater was a source of AS bacterial community populations, but there was a significant difference between influent and AS [25]. In addition, other research indicated that the environmental and operational factors play a greater role in shaping AS community than influent [16]. For example, DO concentration significantly affected the microbial community structure and the efficiency of nutrients removal [14, 23], temperature also evidently affected the microbial community structure, activity, diversity, and process performances [5], and altitude was an important factor influencing denitrifying bacteria community [19]. Like that, we can be supposed that the unique AS microbial community presents in sub-plateau MSTPs and the efficiency of sewage purification links with them. And now, no relevant literature focused on AS microbial community structure and functions in sub-plateau MSTPs. This study attempts to clarify the AS bacterial community structures and seasonal changes in sub-plateau MSTPs, which would be beneficial to explore the reasons for the poor performance in winter, and guide to develop the appropriate operation strategy for sub-plateau MSTPs.
In this study, 24 AS samples were collected from four full-scale sub-plateau MSTPs. Bacterial community diversity, structure, and relative abundance between summer and winter AS samples were measured by Illumina MiSeq sequencing. The objectives of this study were (1) to reveal the core AS bacterial community in sub-plateau MSTPs, (2) to compare the differences in bacterial community diversity, richness, and structure between summer and winter AS samples, (3) to unravel the correlation between bacterial community and sewage purification efficiency.
Materials and methods
MSTP introduction and sample collection
AS samples were collected from the aeration tanks of the four full-scale MSTPs, which basic information is listed in Table 1. During the sampling period, the MSTPs were in good condition and working in a steady state, though a slight sludge foaming was observed in the aeration tanks in winter. Triplicate samples for each tank were collected in summer and winter, respectively. Each AS sample was collected using a 500-mL sterilized polyethylene bottle, then centrifuged at 5000 rpm for 15 min and the sediments stored at − 80 ℃ before DNA extraction. At the same time, influent and effluent quality indices, including chemical oxygen demand (COD), ammonium (NH4+-N), and total phosphorus (TP), were measured by total carbon and nitrogen analyzer (Multi, N/C2100, Analytik Jena) and water quality analyzer (DR3900, Hach, American). The data are shown in Table 2.
DNA extraction and Illumina MiSeq sequencing
A total of 24 samples were collected, and about 5 g of each AS sample was used to extract genomic DNA by the Fast DNA® SPIN Kit for Soil (MP Biomedicals, CA, USA). After performing the assay for concentration and purity DNA, the V3–V4 regions (~465 nucleotides) of bacterial 16S rRNA gene were amplified with primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) [6]. Finally, the purified PCR amplicons were paired-end sequenced on the Illumina MiSeq PE300 platform (Illumina, USA) in Majorbio Co., Ltd (Shanghai, China). All 16S rDNA sequences have been deposited into the NCBI Sequence Read Archive (SRA) database with Bioproject accession number PRJNA551365.
16S gene sequence analysis
Raw sequences were quality filtered and clustered into operational taxonomic units (OTUs) by USEARCH 7.0 (https://drive5.com/uparse/) [3, 7]. Then the OTUs were classified into exact bacterial taxonomy against the SILVA database (https://www.arb-silva.de) and Ribosomal Database Project (RDP, https://rdp.cme.msu.edu/) by Bayesian classifier at 70% threshold. Based on the OTUs information, estimators of community diversity (Shannon and Simpson) and community richness (Chao and Ace), principal co-ordinates analysis (PCoA), and redundancy analysis (RDA) were performed using the online Majorbio I-Sanger Cloud Platform (www.i-sanger.com). Partial least squares discriminant analysis (PLS-DA) was used to find resultant variables between sample groups. LEfSe method uses the Kruskal–Wallis test to identify features with significant differences between winter and summer samples, and LDA to evaluate the effect of size on each feature [9, 20]. An LDA threshold score of 4.0 and a significant α of 0.05 was used to detect biomarkers.
Statistical analysis
All statistical analyses were performed on SPSS 23.0 (SPSS Inc., Chicago, USA). The p value < 0.05 was statistically significant. Student's t test was used to compare the differences in bacterial diversity and functional genera between winter and summer samples. One-way analysis of variance (ANOVA) and Tukey's HSD comparisons were used to determine statistically significant differences among different groups. Correlations among core bacterial community richness, diversity, and environmental variables were considered as statistically significant if p value < 0.05 and the Spearman's rank correlation coefficients (SRCCs) ≥ 0.5 or ≤ − 0.5. All data are presented as the means of triplicate samples (n = 6) and expressed as the mean ± SE. The relevant column charts reflecting community structures were drawn by Microsoft Excel 2016.
Results and discussion
The core AS bacterial communities in sub-plateau MSTPs
The qualified 16S rRNA gene sequences were aligned against the SILVA database and affiliated into different taxonomic levels at 97% similarity. A total of 46 phyla were identified from the AS samples. The dominated bacterial phyla (relative abundance > 1%) account for 93.98–99.25% of the total community abundance (Fig. 1a). Among them, Proteobacteria was the most abundant members (26.10–34.62%), followed by Actinobacteria (13.74–27.78%), Bacteroidetes (18.72–21.04%), Chloroflexi (7.67%-18.52%), and Firmicutes (4.06–6.88%) in summer samples. But in winter samples, Actinobacteria was the most abundant members (31.39–41.59%), followed by Proteobacteria (18.23–22.94%), Bacteroidetes (11.14–27.80%), Firmicutes (7.64–11.83%), and Chloroflexi (3.92–8.82%). The rest of the 35 phyla (relative abundance < 1% in each sample) were defined as “other phyla” accounting for 0.75–6.02%. It is clear that the relative abundance of bacterial phylum community varied greatly between the summer and winter samples, but the core bacterial phylum community structure remained stable to some extent. Furthermore, the core bacterial phylum community structure was shared with various geographical AS samples from the plain, plateau regions and even in the Polar Arctic Circle.[1, 9, 12, 17, 30]. These research studies further support the judgment that the core AS bacterial phylum community structure shows no latitudinal gradient and ecological coherence.

Bacterial community composition of core bacterial phyla (> 1%) a classified genera (top 30). b Note: ANS—samples from AN MSTP in summer, XGS—samples from XG MSTP in summer, ZTS—samples from ZT MSTP in summer, XNS—samples from XN MSTP in summer, ANW—samples from AN MSTP in winter, XGW—samples from XG MSTP in winter, ZTW—samples from ZT MSTP in winter, XNW—samples from XN MSTP in winter, refer to Table 2 in detail
At the genus level, AS bacterial community profiles were displayed by a hierarchically clustered heatmap based on Bray–Curtis similarity index, see in Fig. 1b, which shows that all samples were divided into two clades in accordance with seasonal changes. Although the relative abundance of core genera among different AS samples changed dramatically, these genera has some overlap across the winter and summer samples, including members of Candidatus M. parvicella, Saprospiraceae, Caldilineaceae, Trichococcus, Saccharibacteria (TM7), Terrimonas, among which Candidatus Microthrix and Trichococcus in winter were significantly richer than that in summer samples. This change is consistent with a phenomenon that filamentous foaming frequently appeared in winter at the surface of the aeration tank. So it maybe concluded that these two genera be linked to the sludge foaming and bulking. At all, if the core community was identified by the abundance and occurrence frequency of OTUs, the core community in AS from the sub-plateau region is clearly distinguished from other geographical regions, suggesting that the sub-plateau AS selects for a unique core community and shapes the specific AS ecology.
The changes in bacterial community diversity and abundance
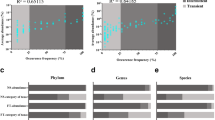
The abundance-based coverage of clean reads was observed to be higher than 98% in all AS samples, indicating that the obtained sequence could well cover the bacterial communities. After clustering and alignment, a total of 3094 OTUs was identified at 97% similarity across all samples with a range of 988–1738 OTUs in individual AS samples. The OTU number in the summer samples (1421 ± 304) was significantly higher than that in the winter samples (1202 ± 159) (Table S1), while 46.1% OTUs (n = 605) was shared between them. Moreover, Chao richness index among different group samples indicates that the abundance of bacterial community in the summer samples is higher than that in the winter samples (Fig. 2a), excluding the XN–MSTPs samples because of its exclusive process. Shannon′s diversity index values were 4.49 ± 0.20 and 5.61 ± 0.43 in the winter and summer samples, respectively. Bacterial diversity was positively linked with sewage temperature (R2 = 0.9139, p = 0.005) (Fig. 2c, d). The changes of the bacterial community were further assessed by principal co-ordinates analysis (PCoA), represented as PC1 and PC2 at 33.46% and 23.65% of the variance in all community structure, respectively. AS samples collected from the same MSTP were distinctly separated into two groups in accordance with winter and summer samples, and the bacterial community in Xining MSTP winter samples were distinctly different from other samples (Fig. 2b), probably due to the sewage temperature was lowest in Xining MSTP. These results indicate that temperature is a crucial environmental factor driving the dynamics of bacterial community diversity and abundance, these changes could be attributed to the variation in sensitivity and resistance of microorganisms under different temperatures [5, 22].

The difference of core bacterial community in diversity (c, d), relative abundance (a), and community structure in different MSTPs between summer and winter samples (b). Abbreviations as in Fig. 1 caption
The bacterial populations enriched in the summer and winter AS samples
To determine the classified bacterial taxa with significant abundance differences between the summer and winter AS samples, we performed biomarker analysis using the method of linear discriminant analysis (LDA) effect size (LEfSe) and T test (95% confidence intervals). The result showed that, of the 46 phyla, 22 bacterial phyla were significantly distinguished in relative abundance between summer and winter samples. As shown in Fig. 3a, Proteobacteria and Chloroflexi are richer in summer samples, Actinobacteria and Firmicutes are more abundant in winter samples. The difference of bacterial community richness between summer and winter samples is significant (p ≤ 0.05). In contrast, the other 24 bacterial phyla populations showed no significance differences (p ≥ 0.05), including a few frequent bacterial phyla, such as Bacteroidetes, Saccharibacteria, Verrucomicrobia. At genus level, the dominant bacterial community populations with significant abundance differences are shown in Fig. 3b, including Candidatus M. parvicella, Trichococcus, Ornithinibacter, Comamonadaceae, etc., Nevertheless, there was also about half of the community populations which showed no significant differences in relative abundance between summer and winter samples. These results indicate that different core bacterial community assembly was developed in summer and winter, respectively. Moreover, this assembly pattern is different from the other geographic MSTPs [15, 18, 29]. For example, Haliangium, Roseiflexus, Smithella, and Lachnospiraceae were detected in the plateau Wastewater Treatment Plants (WWTPs), but the Dokdonella, Nitrospira, Terrimonas, Chitinophagaceae, Saprospiraceae, and Haliangium were dominant in the plain WWTPs [9].

Student's t test for the relative abundance of the core bacterial community at phylum level a and at genus level b between the summer and winter samples. *0.01 < p ≤ 0.05, **0.001 < p ≤ 0.01, ***p ≤ 0.001
Furthermore, a significant difference was observed among the 21 bacterial clades present with an LDA threshold of 4.0, as shown in Fig. 4; it is unexpected that the more bacterial populations were significantly enriched in the winter samples, only six clades showed abundance advantage in summer samples. In detail, Proteobacteria (phylum), Chloroflexi (phylum), Deltaproteobacteria (class), Gammaproteobacteria (class), Myxococcales (order), and Betaproteobacteria (class) were enriched in summer samples. Actinobacteria (phylum and class), Acidimicrobiales incertae sedis (order), Candidatus M. parvicella (genus), Acidimicrobiales (family), and Firmicutes (phylum) were enriched in winter samples. Meanwhile, the efficiency of sewage purification in summer was clearly higher than that in winter (Table 2). Thus, we could assume that the AS bacterial community assembly enriched in the summer played more effective roles in removing organic pollutants and nutrients. This result agrees with the previous study that Proteobacteria could metabolize acetate, butyrate, glucose, and propionate by multiple metabolic mechanisms [11]. Chloroflexi populations play an important role in stabilizing flocs in AS and removing pollutants [9, 27]. In contrast, Actinobacteria members enriched in the winter AS, such as Candidatus M. parvicella, were associated with the foams or the AS bulking in the sub-plateau MSTPs [2, 24]. It could be deemed that the overgrowth of Actinobacteria inhibited other bacterial groups and results in the decline of sewage purification at low temperature. This result could be attributed to the decreasing of bacterial community diversity, relative abundance and the changes in core bacterial community structure.

LEfSe analysis of bacterial community populations enriched in winter and summer AS samples, respectively
The changes in bacterial community related to environmental and operational factors
To investigate the principal factors responsible for the changes of bacterial community in different seasons, RDA was performed to explore the relationship between the relative abundance of community and environmental/operational parameters. As shown in Fig. 5a, acute angles emerged among temperature (T) and DO, altitude (A), SRT, and HRT, respectively, indicating that these factors had a synergetic impact on the relative abundance of community. AS bacterial community was RDA1-affiliated, which corresponded more closely with temperature and DO. The altitude, SRT, and HRT had little influence on AS bacterial community. Proteobacteria and Chloroflexi were positively correlated with temperature and DO, but Actinobacteria, Firmicutes, and Bacteroidetes were negatively correlated with temperature and DO, and SRT and HRT were positively correlated with altitude (Fig. 5a). The most abundant bacterial genus Candidatus M. parvicella and Trichococcus were negatively correlated with temperature and DO, but positively correlated with SRT and HRT (Fig. 5b). These results were in agreement with the previous study that these two genera overgrew and caused sludge foaming under low temperature [24]. In addition, RDA also showed a weak correlation between the bacterial communities, and HRT and altitude. Pprevious studies also have demonstrated that the distinctive AS community abundance and structure are shaped by different operational parameters and environmental conditions [4, 26]. Among them, temperature and DO are the most important variables that shaped the particular AS ecosystem in sub-plateau MSTPs. For example, temperature exerts a profound impact on the biological hydrolysis performance and the microbial community structures such as the main protein or carbohydrate fermenting bacteria genera changed with temperature [4]. DO concentration also significantly affects the microbial community structure as well as the efficiency of sewage purification [23]. Thus, to some extent, an optimized microbial community structure would be established in a given environmental/operational parameters, and the performance of MSTP tight links with it.

RDA of the correlation between bacterial community and environmental/operational variables at phylum level (a) and genus level (b). Abbreviations as in Fig. 1 caption
Conclusions
There was significant difference of the core bacterial community between summer and winter AS sample in sub-plateau MSTPs. The OTU number, community diversity, and richness of bacterial community have decreased dramatically in winter. The different core bacterial community assembly was developed in summer and winter, respectively. As Proteobacteria and Chloroflexi are enriched in summer, Actinobacteria and Firmicutes are enriched in winter. These changes are closely associated with the performance of sewage purification. Additionally, the core bacterial community assembly in sub-plateau region is clearly distinguished from other geographical regions, suggesting that the sub-plateau AS selects for a unique community assembly pattern and shapes the particular AS ecosystem. Further analysis shows that temperature and DO are the principal factors that drive the dynamics of bacterial community diversity and relative abundance in sub-plateau MSTPs.
References
Alejandro G-M, Maija S, Barbara M-P, Alejandro R-S, Anna M, Riku V (2018) Microbial ecology of full-scale wastewater treatment systems in the Polar Arctic Circle: Archaea Bacteria and Fungi. Sci Rep-UK 8:1–11
Andreasen K, Nielsen PH (2000) Growth of Microthrix parvicella in nutrient removal activated sludge plants: studies of in situ physiology. Water Res 34:1559–1569
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fr aser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Chen H, Chang S (2017) Impact of temperatures on microbial community structures of sewage sludge biological hydrolysis. Bioresource Technol 245(Pt A):502–510
Chen Y, Lan S, Wang L, Dong S, Zhou H, Tan L, Li X (2017) A review: Driving factors and regulation strategies of microbial community structure and dynamics in wastewater treatment systems. Chemosphere 174:173–182
Chu Z, Wang K, Li X, Zhu M, Yang L, Zhang J (2015) Microbial characterization of aggregates within a one-stage nitritation–anammox system using high-throughput amplicon sequencing. Chem Eng J 262:41–48
Fan X, Gao J, Pan K, Li D, Dai H, Li X (2018) Functional genera, potential pathogens and predicted antibiotic resistance genes in 16 full-scale wastewater treatment plants treating different types of wastewater. Bioresource Technol 268:97–106
Fan X, Gao J, Pan K, Li D, Dai H (2017) Temporal dynamics of bacterial communities and predicted nitrogen metabolism genes in a full-scale wastewater treatment plant. RSC Adv 7(89):56317–56327
Fang D, Zhao G, Xu X (2018) Microbial community structures and functions of wastewater treatment systems in plateau and cold regions. Bioresource Technol 249:684–693
Gao J, Liu G, Li H, Xu L, Du L, Yang B (2016) Predictive functional profiling using marker gene sequences and community diversity analyses of microbes in full-scale anaerobic sludge digesters. Bioprocess Biosyst Eng 39(7):1115–1127
Guo J, Peng Y, Ni B, Han X, Fan L, Yuan Z (2015) Dissecting microbial community structure and methane-producing pathways of a full-scale anaerobic reactor digesting activated sludge from wastewater treatment by metagenomic sequencing. Microbe Cell Fact 14(1):1–11
Ibarbalz FM, Figuerola ELM, Erijman L (2013) Industrial activated sludge exhibits unique bacterial community composition at high taxonomic ranks. Water Res 47:3854–3864
Jin LY, Zhang GM, Tian HF (2014) Current state of sewage treatment in China. Water Res 66:85–98
Kang XH, Wu XK, Wang H, Zeng XY, Leng Y, Li SW (2018) Illumina sequencing reveals bacterial community shift and its role in a full-scale A2O sewage treatment process at low temperatures. Desalin Water Treat 109:193–204
Kurumi H, Masami M, Daisuke I, Michihiko I (2014) Bacterial community dynamics in a full-scale municipal wastewater treatment plant employing a conventional activated sludge process. J Biosci Bioeng 118(1):64–71
Lee SH, Kang HJ, Park HD (2015) Influence of influent wastewater communities on temporal variation of activated sludge communities. Water Res 73:132–144
Ma S, Ding L, Huang H, Geng J, Xu K, Zhang Y, Ren H (2016) Effects of DO levels on the surface force, cell membrane properties and microbial community dynamics of activated sludge. Bioresour Technol 214:645–652
Muszynski A, Tabernacka A, Milobedzka A (2015) Long-term dynamics of the microbial community in a full-scale wastewater treatment plant. Int Biodeterior Biodegrad 100:44–51
Niu L, Li Y, Wang P, Zhang W, Wang C, Cai W, Wang L (2016) Altitude-scale variation in nitrogen-removal bacterial communities from municipal wastewater treatment plants distributed along a 3600-m latitudinal gradient in China. Sci Total Environ 559:38–44
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett W.S, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12(6)
Seib MD, Berg KJ, Zitomer DH (2016) Influent wastewater microbiota and temperature influence anaerobic membrane bioreactor microbial community. Bioresource Technol 216:446–452
Sims A, Gajaraj S, Hu ZQ (2012) Seasonal population changes of ammonia-oxidizing organisms and their relationship to water quality in a constructed wetland. Ecol Eng 40:100–107
Stadler LB, Love NG (2016) Impact of microbial physiology and microbial community structure on pharmaceutical fate driven by dissolved oxygen concentration in nitrifying bioreactors. Water Res 104:189–199
Wang P, Yu Z, Zhao J, Zhang H (2016) Seasonal changes in bacterial communities cause foaming in a wastewater treatment plant. Microb Ecol 71(3):660–671
Wells GF, Wu CH, Piceno YM, Eggleston B, Brodie EL, DeSantis TZ, Andersen GL, Hazen TC, Francis CA, Criddle CS (2014) Microbial biogeography across a full-scale wastewater treatment plant transect: evidence for immigration between coupled processes. Appl Microbiol Biotechnol 98(10):4723–4736
Xia Y, Wen X, Zhang B, Yang Y (2018) Diversity and assembly patterns of activated sludge microbial communities: a review. Biotechnol Adv 36:1038–1047
Yoon DN, Park SJ, Kim SJ (2010) Isolation, characterization, and abundance of filamentous members of Caldilineae in activated sludge. J Microbiol 48:275–283
Zhang Q, Yang W, Ngo H, Guo W, Jin P, Dzakpasu M, Yang SJ, Wang Q, Wang X, Ao D (2016) Current status of urban wastewater treatment plants in China. Environ Int 92–93:11–22
Zhao D, Huang R, Zeng J, Yu Z, Liu P, Cheng S, Wu QL (2014) Pyrosequencing analysis of bacterial community and assembly in activated sludge samples from different geographic regions in China. Appl Microbiol Biotechnol 98:9119–9128
Zhou Z, Qiao W, Xing C, An Y, Shen X, Ren W, Jiang L, Wang L (2015) Microbial community structure of anoxic-oxic-settling-anaerobic sludge reduction process revealed by 454-pyrosequencing. Chem Eng J 266:249–257
Acknowledgements
This work was supported by the National Natural Science Foundation of China (3176110) and Natural Science Foundation of Gansu Province (148RJZA046).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kang, XH., Leng, Y., O, M.M. et al. The seasonal changes of core bacterial community decide sewage purification in sub-plateau municipal sewage treatment plants. Bioprocess Biosyst Eng 43, 1609–1617 (2020). https://doi.org/10.1007/s00449-020-02352-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-020-02352-2