
Abstract
A protease producing marine bacterium, Bacillus halodurans CAS6 isolated from marine sediments, was found to produce higher enzyme by utilizing shrimp shell powder. Optimum culture conditions for protease production were 50 °C, pH 9.0, 30 % NaCl and 1 % shrimp shell powder (SSP) and the protease purified with a specific activity of 509.84 U/mg. The enzyme retained 100 % of its original activity even at 70 °C, pH 10.0 and 30 % NaCl for 1 h. The purified protease exhibited higher stability when treated with ionic, non-ionic (72–94 %) and commercial detergents (76–88 %), and organic solvents (88–126 %). Significant blood stain removal activity was found with the enzyme in washing experiments. The culture supernatant supplemented with 1 % SSP showed 93.67 ± 2.52 % scavenging activity and FT-IR analysis of the reaction mixture confirmed the presence of antioxidants such as cyclohexane and cyclic depsipeptide with aliphatic amino groups. These remarkable qualities found with this enzyme produced by Bacillus halodurans CAS6 could make this as an ideal candidate to develop the industrial process for bioconversion of marine wastes and antioxidant synthesis.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Proteases are one among the most important groups of industrial enzymes, accounting for more than 65 % of the total industrial enzyme market [1] and find applications in detergents, feather processes, food processing, silk gumming, pharmaceuticals, bioremediation, biosynthesis and biotransformation [2–4]. Recently, the application of proteases in synthesis of oligopeptides has received great attention as a viable alternate to chemical approach [5, 6].
Marine industry generates significant amount of shellfish wastes that can serve as a renewable resource with rich protein and chitin contents [7]. Disposal of marine wastes presents a major problem due to objectionable odour, high nutrient content and disposal regulations. So far marine wastes have been used for metalloproteases, chitinases, antifungal hydrolytic enzymes, carotenoids, amino acids and lipases production [8, 9]. As a prominent source of protein and chitin, marine shellfish wastes can be used for the extraction of peptides and chitin oligomers for antioxidant formulations [10].
Bioconversion of marine waste materials have been proposed as an alternative to waste treatment. Furthermore, many studies have been recently reported on bioconversion of shellfish wastes for production of proteases and chitinases [4, 11]. To further enhance the utilization of marine crustacean wastes, in this study an attempt was made to (1) optimize the culture conditions for protease production by B. halodurans CAS6, (2) evaluate the thermo and pH stability of isolated protease and (3) apply purified protease as a cleansing additive in blood stain removal and antioxidant activity.
Materials and methods
Materials
Marine wastes such as shrimp shell powder (SSP), crab shell powder (CSP) and squid pen powder (SPP) used in this study were prepared as described earlier by Annamalai et al. [11]. Shrimp and crab shells and squid pens were purchased from local fish processing unit and washed thoroughly with tap water and sun dried. The dried shells were milled and sieved (100 μm) to get fine powder and used as the carbon source for protease production. Bovine Serum Albumin (BSA), reagents for protein estimation and SDS-PAGE, DEAE-Cellulose and Sephadex G-50 were purchased from Hi-Media, Mumbai, India. Silica gel TLC plates (0.25 mm), trichloroacetic acid (TCA), casein and other analytical grade chemicals were purchased from Merck, Mumbai, India.
Microorganism
The protease producing bacterial strain was isolated from marine sediments of Parangipettai coast, Tamilnadu, India using casein milk agar medium (pH 9.0) and identified by morphological, biochemical identification schemes and confirmed by 16S rRNA gene sequencing. In brief, the DNA was isolated by phenol chloroform method [12]. The primer sequences were selected from the conserved regions as previously reported for the bacterial 16S rRNA gene [13]. Sequencing was done using forward primer (5′-CAGGCCTAACACATGCAAGTC-3′) and reverse primer (5′-GGGCGGTGTGTACAAGGC-3′). PCRs were performed with following conditions: 35 cycles consisting of 95 °C for 1 min and 72 °C for 5 min, followed by final extension of 5 min at 72 °C. The 16S rRNA gene sequences were obtained by an automated DNA Sequencer (Megabace, GE) and homology of the isolated gene with sequences in the GenBank database was analyzed.
Optimization of culture conditions and carbon source for protease production
Optimization of culture conditions for protease production was performed in 500-mL Erlenmeyer flasks with 100 mL of basal medium containing 0.1 % K2HPO4 and 0.05 % MgSO4·7H2O (pH 9.0) and 0.1–2 % (w/v) of various carbon sources to be investigated such as SSP, CSP, SPP and shrimp and crab shell powder (SCSP) at 1:1, 1:3 and 3:1 ratio, w/w). Flasks with sterilized culture medium were inoculated with 24 h grown bacterial culture (1 % inoculum (v/v), ~105 cells/ml) and incubated in a shaker incubator (150 rpm) at various temperature (30–70 °C), pH (6.0–12.0) and NaCl (0–30 %, w/v) concentration for 60 h. The flasks were maintained at 55 °C, pH 9.0 and 25 % NaCl concentration while optimizing other parameters. The culture broths were centrifuged (12,000×g, 4 °C, for 20 min) and the cell free supernatants were used for further protease analysis.
Enzyme production and purification
Based on results obtained from optimization experiments, B. halodurans CAS6 was grown in 100 mL basal medium containing 1 % SSP, 0.1 % K2HPO4, and 0.05 % MgSO4·7H2O (pH 9.0), and seeded with 1 % inoculum (~105 cells/mL) and incubated in shaking incubator (150 rpm) for 60 h at 55 °C. After incubation, culture broth was centrifuged (4 °C and 12,000×g for 20 min), and the supernatant was used as crude enzyme.
To the centrifuged culture broth obtained from above enzyme production experiment, ammonium sulphate was added with constant stirring for 30 min to obtain a 60 % saturation (w/v) and allowed to stand overnight at 4 °C. The precipitates were collected by centrifugation at 6,000×g for 15 min, dissolved in 50 mM Tris–HCl buffer (pH 9.0) and dialyzed against the same buffer (4 °C) for 12 h. The dialysed protein was loaded onto DEAE-Cellulose column (5 × 25 cm) and eluted with linear gradient of NaCl (0–1 M) at a flow rate of 0.5 mL/min. Fractions were collected and analyzed for enzyme activity, and fractions with enzyme activity were pooled together and concentrated by ammonium sulphate (60 % saturation, w/v) precipitation. The resulting precipitate was collected by centrifugation and dissolved in 50 mM Tris–HCl buffer (pH 9.0). The concentrated precipitate was loaded onto a Sephadex G-50 column (2.5 × 25 cm) equilibrated with 50 mM Tris–HCl buffer (pH 9.0) and eluted with the same buffer at a flow rate of 15 mL/h. Fractions exhibiting protease activity were pooled together and used as a purified enzyme for further study.
Protein estimation
Protein content of the enzyme during various purification steps was determined by the method of Lowry et al. [14] using bovine serum albumin as the standard.
Enzyme assay
Protease activity was measured with a diluted enzyme solution (0.2 mL) mixed with 1.25 mL of 1.25 % casein (w/v) in 50 mM Tris–HCl buffer (pH 9.0) and incubated for 30 min at 37 °C. The reaction was terminated by adding 5 mL of 0.19 M trichloroacetic acid. The reaction mixture was centrifuged and the soluble peptides in the supernatant were measured by the method of Todd [15] with tyrosine as the reference compound. One unit of enzyme activity is defined as the amount of enzyme required to liberate 1 μmol of tyrosine per min under the defined assay conditions.
Molecular weight determination of the purified protease
Molecular weight of the purified protease was determined by SDS-PAGE with 10 % polyacrylamide gel following the method described by Laemmli [16]. After electrophoresis, protein bands were stained with Coomassie Brilliant Blue R-250 prepared with methanol–acetic acid–water (5:1:5, v/v), and the stained gels were decolorized with 7 % acetic acid. The standard reference proteins used for comparison were phosphorylase b (97.4 kDa), albumin (66.2 kDa), ovalbumin (45 kDa), carbonic anhydrase (29 kDa), trypsin inhibitor (20.1 kDa) and α-lactalbumin (14.3 kDa). Molecular weight of the protease isolated in this study was estimated by comparing the mobility with standard proteins.
Effect of temperature, pH and NaCl on activity and stability of purified protease
Effect of temperature, pH and NaCl concentrations on enzyme activity was evaluated by incubating the reaction mixture (enzyme + substrate) at different temperatures (30–95 °C), pH (4.0–12.0) and various concentrations of NaCl (0–35 %, w/v). To check the stability of the purified protease, the enzyme was preincubated for 1 h at above mentioned reaction conditions and the residual activity (%) was measured as described in enzyme assay. Buffers used in experiments were 50 mM of sodium citrate (pH 3.0–6.0), 50 mM sodium phosphate (pH 6.0–8.0), 50 mM glycine–NaOH (9.0–11.0) and 50 mM dilute NaOH for pH 12.0–13.0.
Substrate specificity
The substrate specificity of the purified protease was assayed with different substrates such as Casein, Azocasein, BSA, Egg albumin, Gelatin, Haemoglobin and BSA. The reaction mixture containing 200 μl of enzyme and 200 μl of substrate (1 mg/ml) was incubated at 50 °C for 30 min and the relative activity was estimated by standard protease assay.
Effect of metal ions and detergents (ionic, non-ionic and commercial) on enzyme activity
Effect of metal ions on enzyme activity was investigated using Mn2+, Fe2+, Co2+, Mg2+, Ca2+, Hg2+ and EDTA at 0.1 and 1 mM concentration, respectively. The influence of protease inhibitors such as phenylmethylsulfonyl fluoride (PMSF), ethylenediaminetetraacetic acid (EDTA), β-mercaptoethanol, dithio-bis-nitrobenzoic acid (DTNB) and soybean tryptin inhibitor (SBTI) on protease activity was investigated. Effect of ionic and non-ionic detergents on enzyme activity was evaluated using SDS, Triton X-100, Tween 80 and sodium deoxycholate at a final concentration of 1 % (v/v). Effect of commercial detergents on protease activity was studied with 1 % (w/v) solution of commercial detergents such as Ariel, Rin, Henko and Surf. The purified protease was preincubated with aforementioned metal ions, detergents and commercial detergents at 50 °C for 1 h and then the residual activity was assayed. The activity of the purified protease without addition of any metals and detergents was considered as control and activity was taken as 100 %.
Effect of organic solvents on enzyme activity
The activity of the purified protease in the presence of organic solvents was evaluated by incubating with various organic solvents such as xylene, isopropanol, hexane and benzene (0.5 % v/v) for 4 h at 50 °C and then assayed for residual enzyme activity.
Blood stain removal activity of purified protease
The blood stain removal property of purified protease obtained from the present study was evaluated using white cotton cloth pieces (10 × 10 cm) stained with blood and dried at 95–100 °C for 5 min. Four sets of washing experiments were carried out as described below:
Set 1: distilled water (100 mL) + blood-stained cloth (control).
Set 2: distilled water (100 mL) + blood-stained cloth + 1 mL of commercial detergent (Surf excel 5 mg/mL, w/v).
Set 3: distilled water (100 mL) + blood-stained cloth + 1 mL of commercial detergent (Surf excel 5 mg/mL) + purified enzyme (2 mL, 500 U/mL).
Set 4: distilled water (100 mL) + blood-stained cloth + purified enzyme (2 mL, 500 U/mL).
All four experimental sets were incubated at 50 °C for 30 min and the cloth pieces were rinsed with water for every 5 min, dried and visually examined for stain removal activity. Untreated cloth pieces stained with blood and washed with distilled water were taken as control.
Antioxidant activity of the culture supernatant of B. halodurans CAS6 fermented with marine wastes
Antioxidant activity of the culture supernatants of B. halodurans CAS6 fermented with marine wastes such as SSP, CSP and SPP (150 μl) were assayed by mixing 37.5 μl of methanol solution containing 0.75-mM 2,2-diphenyl-1-picrylhydrazyl (DPPH) (Hi-Media, India). The reaction mixture was shaken vigorously and allowed to stand for 30 min in dark, and the optical density (OD) of the reaction mixture was measured at 517 nm against a blank (control) without culture broth [17]. The scavenging activity was calculated as follows:
Characterization of antioxidant materials
Antioxidant materials in the culture supernatants were analyzed by silica gel thin-layer chromatography (TLC) using n-butanol–methanol–16 % aqueous ammonia (5:4:3, v/v) as the mobile phase [18]. After developing the TLC plates, the compounds were visualized by spraying ethanol containing 0.5 % (w/v) ninhydrin. The antioxidant materials were identified based on their R f value in comparison with the standard oligopeptides (Ala-Cys-Ala-His-Asp-Lys-Val, Leu-Leu-Gly-Pro-Gly-Leu-Thr-Asn-His-Ala, Thr-Arg-Asn-Tyr-Tyr-Val-Arg-Ala-Val-Leu-OH and Asp-Leu-Gly-Leu-Gly-Leu-Pro-Gly-Ala-His). Functional groups which exhibiting antioxidant activity was identified by FT-IR spectrum recorded in a FT-IR spectrometer (Biorad-40 model, USA) using KBr pellets as background [10].
Results and discussion
Microorganism
In the present study, a protease producing strain CAS6 was isolated from marine sediments of Parangipettai coast, Tamilnadu, India. Morphological and biochemical characteristics of the strain revealed that it is a gram-positive, endospore-forming bacillus with catalase enzyme activity (Table 1). 16S rRNA gene sequence analysis confirmed the identity of the strain CAS6 as B. halodurans with 99 % similarity with reference sequences (Table 2, GenBank accession no. HQ116805).
Optimization of culture conditions and carbon source for protease production
To study the effect of carbon sources on protease production, fermentation was carried out with basal medium containing various concentrations of (0.1–2 % w/v) SSP, CSP, SPP and SCSP. Results from this study revealed that 1 % SSP (3,413 IU/mL) and 3:1 % SCSP (3 parts of shrimp and 1 part of crab shell powder) (3,246 IU/mL) were found as suitable carbon sources for protease production than other marine wastes used such as CSP (2,572 IU/mL) and SPP (2,963 IU/mL) (Fig. 1). Eventhough the protein content of SPP was higher than that of SSP and SCSP, the resultant protease production was lesser due to the protein and chitin ratio (nearly 1:1) which is more suitable as an inducer for protease production [6]. Similar to results obtained in this study, SSP was found as the suitable substrate and as an inducer for the production of proteases by Chryseobacterium taeanense [4], Bacillus cereus TKU006 [6] and chitinases from Alcaligenes faecalis AU02 [11] and Bacillus amyloliquefaciens V656 [19].

Protease production with various carbon sources by B. halodurans CAS6. SSP shrimp shell powder, CSP crab shell powder, SPP squid pen powder, SCSP shrimp and crab shell powder. SCSP (shrimp:crab shell powder) ratio: SCSP 1:3, SCSP 1:1 and SCSP 3:1. B. halodurans CAS6 was incubated in a shaker incubator at 55 °C and pH 9.0 for 60 h with above substrate combinations
The optimization of culture conditions studies revealed that increase in cell growth and protease production was observed while increasing the temperature from 30 °C and reached maximum at 50 °C (Fig. 2 a, b). The effect of pH on cell growth and protease production showed that B. halodurans CAS6 was able to grow and produce protease in wide range of pH between 7.0 and 10.0 and optimum was found at pH 9.0 (Fig. 2c, d). The optimum pH for growth and protease production was found between 9.0 and 10.0 and it is a common characteristic feature of alkaliphilic and haloalkaliphilic organisms [6]. Regarding NaCl concentration, protease production was increased with increasing concentrations from 0–30 % and reached its maximum at 30 % (w/v) (Fig. 2e, f). There was a significant reduction in enzyme production found in the absence of NaCl (0 %). The strain B. halodurans CAS6 used in this study was isolated from marine sediments and that might be the reason for the higher protease production found at 30 % and lesser at 0 % NaCl.

Cell growth and protease production by B. halodurans CAS6 at various temperature (a, b), pH (c, d) and NaCl concentrations (e, f). B. halodurans CAS6 was incubated in a shaker incubator at various temperature (30–70 °C), pH (6.0–12.0) and NaCl (0–30 %, w/v) concentration for 60 h
Enzyme production and purification
Protease production was carried out in 500 mL Erlenmeyer flask containing 100 mL of culture medium with 1 % SSP. As shown in Fig. 3, cell growth and protease production reached their maximum at 36 h and gradually decreased after wards. Wang et al. [6] reported the protease (0.573 U/mL) and chitinase (0.089 U/mL) production by Bacillus cereus TKU006 using 2 % SSP after 48 h incubation. When compared with earlier reports, results obtained in this study clearly indicates the efficient utilization of SSP (1 %) by B. halodurans CAS6 (3,413 IU/mL) with less incubation time. Thus, B. halodurans CAS6 could be an ideal candidate for conversion marine wastes as cheaper sources into valuable product development process with less cost of production.

Protease production at optimized culture concisions by B. halodurans CAS6. Culture medium: 100 mL basal medium (0.1 % K2HPO4, 0.05 % MgSO4·7H2O and 1 % SSP). Culture conditions: 55 °C, pH 9.0 and incubation time 66 h. Data are the average of three experiments
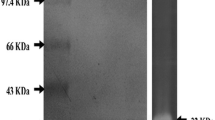
A summary of the enzyme purification steps were presented in Table 3. Purification of protease with ammonium sulphate followed by DEAE-cellulose and Sephadex G-50 column chromatography yielded 51.93, 17.93 and 12.54 % protein recovery, respectively. The overall purification fold was about 7.96 with the specific activity of 509.84 U/mg. The homogeneity of the purified protease was confirmed by the single band in SDS-PAGE analysis. Molecular weight of the purified protease from B. halodurans CAS6 was about 28 kDa (Fig. 4) which is similar to the metalloprotease from B. cereus (28 kDa) [20] and different from other reported proteases such as B. cereus (45.6 kDa) [21] and B. cereus TKU006 (39 kDa) [6].

SDS-PAGE analysis of crude and purified protease from B. halodurans CAS6. Lane 1 molecular markers and their molecular weight—lysozyme (14.3 kDa), trypsin inhibitor (20.1 kDa), carbonic anhydrase (29 kDa), ovalbumin (43 kDa), bovine serum albumin (66 kDa) and phosphorylase ‘B’ (97.4 kDa); Lane 2 crude enzyme; and Lane 3 purified protease
Effect of temperature, pH and NaCl on enzyme activity and stability
Effect of temperature, pH and NaCl on enzyme activity and stability of the purified protease was studied with casein as substrate. The optimum temperature for enzyme activity of purified protease of B. halodurans CAS6 was 50 °C. Thermo stability studies revealed that the enzyme was 100 % stable up to 70 °C and retained 83 % of its original activity at 80 °C and 40 % at 95 °C (Fig. 5a). Thus, the purified protease from Bacillus halodurans CAS6 was considerably more stable than the proteases of Bacillus sp. B001 [22] and Alcaligens faecalis [23].

a Effect of temperature, b pH and c NaCl concentration on enzyme activity and stability of purified protease from B. halodurans CAS6. Effect of temperature, pH and NaCl concentrations on enzyme activity was evaluated at different temperatures (30–95 °C), pH (4.0–12.0) and with various concentrations of NaCl (0–35 %, w/v). Data are the average of three experiments
The purified protease of B. halodurans CAS6 was active over a wide range of pH between 6.0 and 12.0 and optimum was found at pH 9.0 (Fig. 5b). Regarding pH stability, the purified protease was 100 % stable at pH 10.0 and retained 82 % of its original activity at pH 11.0. The pH stability of the protease of B. halodurans CAS6 was comparatively higher than the previously reported proteases of B. cereus (6.0–10.0) [21], A. faecalis AU01 (7.0–10.0) [23] which indicates its possible industrial applications. In general, proteases with an optimal temperature of 50–70 °C and pH of 9.0–12.0 are desirable for applications in detergents and tanning processes [24].
The effect of NaCl on activity and stability of purified protease revealed that the enzyme activity was increased with increasing the concentration from 0–30 % and optimum was found at 30 %. The halostability studies indicated that purified protease was 100 % stable at 30 % NaCl concentration and it retained 80 % of its original activity at 35 % (Fig. 5c). The halostability of the protease of B.halodurans CAS6 was comparatively higher than the protease of Bacillus aquimaris strain VITP4 which was stable up to 2 M NaCl (i.e. 11.68 %) [25] and Salinivibrio sp. strain AF-2004 stable up to 4 M NaCl (i.e. 22.37 %) [26]. The high salt tolerance is characteristic feature of this enzyme which has number of application in any biotechnological process that depends on high salinity or osmotic pressures.
Substrate specificity
The substrate specificity studies revealed that the protease activity was higher when casein (100 %) was used as substrate followed by gelatine (90 %), Azocasein (72 %) and egg albumin (59 %) respectively, but there was no activity found with haemoglobin and BSA. Similarly, proteases of B. cereus TKU006 [6] and Bacillus laterosporus-AK1 [27] were also reported to produce higher protease activity when casein used as substrate.
Effect of metal ions and detergents (ionic, non-ionic and commercial) on enzyme activity
Effect of metal ions on activity of purified protease is summarized in Table 4. The enzyme activity was enhanced when treated with Ca2+, Mg2+, and Mn2+ at 0.1 and 1 mM concentrations. Similar to the present study, stimulatory effects of Ca2+ and Mg2+ have been reported previously [24]. There was no significant effect on enzyme activity was found with Fe2+, Co2+ and EDTA and strong inhibition was found with Hg2+ at 0.1 and 1 mM concentrations. These results were similar to the proteases from Bacillus sp. B001 [22] and Bacillus cereus TKU006 [6]. Regarding the effect of protease inhibitors on enzyme activity, there was no significant effects found with PMSF, β-mercaptoethanol, DTNB and SBTI. But, the metalloenzyme inhibitor (EDTA) significantly inhibited enzyme activity which clearly indicates that the protease purified from the present study belongs to metalloprotease.
The stability of enzyme with detergents revealed that the enzyme retained 94 and 72 % of its original activity in the presence of 1 % Tween 80 and Triton X-100 (non-ionic surfactant) and 82 and 85 % of activity with SDS and sodium deoxycholate (anionic surfactant), respectively (Table 5). Jellouli et al. [3] suggested that most of the proteases reported earlier were destabilized in the presence of ionic surfactants and oxidizing agent. The purified protease of B. halodurans CAS6 was stable in the presence of commercial detergents such as Rin (88 %), Ariel (79 %), Henko (85 %) and Tide (76 %) which was higher than the stability of protease from B. laterosporus-AK1 (75 % with Ariel, Henko 63 %, Surf excel 43 % and 38 % with Tide) [27]. The higher stability of the purified protease of B. halodurans CAS6 towards detergents will make this enzyme as an ideal candidate for application in detergent industry.
Effect of organic solvents on enzyme activity
The effect of organic solvents on stability of purified protease revealed that the activity was enhanced by the addition of xylene (118.27 %) and hexane (126.28 %), whereas there was a slight decrease in enzyme activity observed with isopropanol (88.16 %) and benzene (90.26 %) (Table 5). The solvent stability of the protease purified in this study was higher than the stability of alkaline protease from Chryseobacterium taeanense TKU001, which retained only 76 % of its activity in the presence of toluene, methanol, ethanol, ethyl ether, acetonitrile, isopropyl alcohol and isoamyl alcohol (25 % v/v) [4]. Enzymes are usually inactivated by the addition of organic solvents to the reaction solution. But stability of purified protease of B. halodurans CAS6 indicates that it could be useful for both type of reactions such as aqueous (buffer) and added solvents (co-solvents).
Application of purified protease on blood stain removal
The blood stain removal experiments conducted with purified protease revealed that the blood stains were removed completely within 15 min in set-3 where combination of detergent (5 mg/mL) and purified enzyme was used and it took 20 min when enzyme alone used (set-4) (Fig. 6). It clearly indicates that addition of enzyme with commercial detergent significantly enhanced the washing performances and blood stain removal. Banerjee et al. [28] reported blood stain removing activity of thermostable alkaline protease from Bacillus brevis removed blood stains in combination of 7 mg/mL + enzyme (2 mL) within 25 min. Thus, fast stain removal quality of the purified protease in this study suggested its possible commercial application as a detergent additive.

Blood stain removal activity of protease from B. halodurans CAS6. a Unstained cloth, b blood-stained cloth washed with distilled water, c blood-stained cloth washed with detergent and protease enzyme, and d blood-stained cloth washed with protease enzyme
Antioxidant activity of culture supernatant fermented with marine wastes
Most of the earlier studies suggested that peptides, chitin and chitosan have antioxidative [29] and anticarcinogenic properties [30]. To increase the utilization of marine wastes, culture supernatant of B. halodurans CAS6 fermented with marine wastes (SSP, CSP, SPP and SCSP) was evaluated for their antioxidant activity. Among the marine wastes tested, maximum antioxidant activity was found with the culture broth fermented for 42 h with 1 % SSP (93.67 ± 2.52 % scavenging activity), followed by SCSP 3:1 (90.33 ± 2.32 %), SCSP 1:1 (82.67 ± 2.64 %), SCSP 1:3 (75.67 ± 2.08 %), SPP (72.33 ± 2.20 %) and CSP (66.67 ± 2.05 %). However, the antioxidant activity of the culture supernatant of SSP was higher than the culture supernatant of B. cereus TKU006 supplemented with 1 % SPP (74 %) and incubated for 60 h [6]. Peptides present in the culture broth incubated with SSP may be responsible for the radical scavenging properties [31]. Shrimp waste also contains natural antioxidants, mainly phenolic compounds [32]. The antioxidant materials may contain oligopeptides that are electron donors which could able to react with free radicals to terminate the radical chain reaction [6].
Antioxidant materials in the culture supernatant were analyzed using TLC and three distinct bands were observed when developed on the TLC plates. FT-IR spectral analysis suggested that the presence of aromatic ring stretch with secondary amine N–H stretch and carboxylic acid ester groups and an asymmetrical methylene with C–H bonding in the culture supernatant (1,501.37, 1,150.68, 1,545.21, 1,742.47, 2,921.76 and 2,853.02 cm−1) (Fig. 7). From the FT-IR spectrum analysis, it was clear that aromatic nitro compounds with an asymmetrical methylene linked C–H, cyclohexane ring with CH3–CH and cyclic depsipeptide with aliphatic amino groups were responsible for the antioxidant activity.

FT-IR spectrum of the antioxidant materials in the culture supernatant of halodurans CAS6 over wave number range of 4,000–400 cm−1
Conclusions
Considering the production cost and reutilization of bioresources, microbial protease production from marine wastes seems to provide a renewable carbon source. This waste-derived inexpensive protease production not only solves environmental problems but also promotes the economic values of the marine wastes. The interesting properties found with the purified protease in this study such as stability towards temperature, pH, NaCl, surfactants, blood stain removal and antioxidant activity will make this enzyme as a potential candidate for the development of waste based sustainable industrial process. Further research on mechanisms of enzyme action, amino acid sequencing and identification of genes responsible for enzyme production are in progress.
References
Banik RM, Prakash M (2004) Laundry detergent compatibility of the alkaline protease from Bacillus cereus. Microbiol Res 159:135–140
Bhaskar N, Sudeepa ES, Rashmi HN, Selvi AT (2007) Partial purification and characterization of protease of Bacillus proteolyticus CFR3001 isolated from fish processing waste and its antibacterial activities. Bioresour Technol 98:2758–2764
Jellouli K, Bougatef A, Manni L, Agrebi R, Siala R, Younes I, Nasri M (2009) Molecular and biochemical characterization of an extracellular serine-protease from Vibrio metschnikovii J1. J Ind Microbiol Biotechnol 36:939–948
Wang SL, Yang CH, Liang TW, Yen YH (2008) Optimization of conditions for protease production by Chryseobacterium taeanense TKU001. Bioresour Technol 99:3700–3707
Ma C, Ni X, Chi Z, Ma L, Gao L (2007) Purification and characterization of an alkaline protease from the marine yeast Aureobasidium pullulans for bioactive peptide production from different sources. Mar Biotechnol 9:343–351
Wang SL, Chao CH, Liang TW, Chen CC (2009) Purification and characterization of protease and chitinase from Bacillus cereus TKU006 and conversion of marine wastes by these enzymes. Mar Biotechnol 11:334–344
Viswanathan K, Omorebokhae R, Li G, Gross RA (2010) Protease-catalyzed oligomerization of hydrophobic amino acid ethyl esters in homogeneous reaction media using l-phenylalanine as a model system. Biomacromolecules 11:2152–2160
Chang WT, Chen YC, Jao CL (2007) Antifungal activity and enhancement of plant growth by Bacillus cereus grown on shellfish chitin wastes. Bioresour Technol 98:1224–1230
Sachindra NM, Bhaskar N (2008) In vitro antioxidant activity of liquor from fermented shrimp biowaste. Bioresour Technol 99:9013–9016
Nawani NN, Prakash D, Kapadnis BP (2010) Extraction, purification and characterization of an antioxidant from marine waste using protease and chitinase cocktail. World J Microbiol Biotechnol 26(8):1509–1517
Annamalai N, Rajeswari MV, Vijayalakshmi S, Balasubramanian T (2011) Purification and characterization of chitinase from Alcaligenes faecalis AU02 by utilizing marine wastes and its antioxidant activity. Ann Microbiol 61:801–807
Marmur J (1961) A procedure for the isolation of deoxyribonucleic acid from microorganisms. J Mol Biol 3:208–218
Marchesi J, Sato T, Andrew J, Martin T, Fry J, Hiom S (1998) Design and evaluation of useful bacterium specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol 64:795–799
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Todd EW (1949) Quantitative studies on the total plasmin and trypsin inhibitor of human blood serum. J Exp Med 39:295–308
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Shimada K, Fujikawa K, Yahara K, Nakamura T (1992) Antioxidative properties of xanthan on the autoxidation of soybean oil in cyclodextrin emulsion. J Agric Food Chem 40:945–948
Kadokura K, Rokutani A, Yamamoto M, Ikegami T, Sugita H, Itoi S, Hakamata W, Oku T, Nishio T (2007) Purification and characterization of Vibrio parahaemolyticus extracellular chitinase and chitin oligosaccharide deacetylase involved in the production of heterodisaccharide from chitin. Appl Microbiol Biotechnol 75:357–365
Wang SL, Shih IL, Liang TW, Wang CH (2002) Purification and characterization of two antifungal chitinases extracellularly produced by Bacillus amyloliquefaciens V656 in a shrimp and crab shell powder medium. J Agric Food Chem 50:2241–2248
Doddapaneni KK, Tatineni R, Vellanki RN, Rachcha S, Anabrolu N, Narakuti V, Mangamoori N (2008) Purification and characterization of a solvent and detergent-stable novel protease from Bacillus cereus. Microbiol Res 164:383–390
Sousa F, Jus S, Erbel A, Kokol V, Cavaco-Paulo A, Gubitz GM (2007) A novel metalloprotease from Bacillus cereus for protein fibre processing. Enzyme Microb Technol 40:1772–1781
Deng A, Wu J, Zhang Y, Zhang G, Wen T (2010) Purification and characterization of a surfactant-stable high-alkaline protease from Bacillus sp. B001. Bioresour Technol 101:7100–7106
Annamalai N, Aunkumar, Saravanakumar A, Vijayalakshmi S, Balasubramanian T (2011) Characterization of protease from Alcaligens faecalis and its antibacterial activity on fish pathogens. J Environ Biol 32:781–786
Haddar A, Agrebi R, Bougatef A, Hmidet N, Sellami-Kamoun A, Nasri M (2009) Two detergent stable alkaline serine-proteases from Bacillus mojavensis A21: purification, characterization and potential application as a laundry detergent additive. Bioresour Technol 100:3366–3373
Shivanand P, Jayaraman G (2009) Production of extracellular protease from halotolerant bacterium, Bacillus aquimaris strain VITP4 isolated from Kumta coast. Process Biochem 44:1088–1094
Karbalaei-Heidari HR, Ziaee A, Schaller J, Amoozegar MA (2007) Purification and characterization of an extracellular haloalkaline protease produced by the moderately halophilic bacterium, Salinivibrio sp. strain AF-2004. Enzyme Microb Technol 40:266–272
Arulmani M, Aparanjini K, Vasanthi K, Arumugam P, Arivuchelvi M, Kalaichelvan T (2006) Purification and partial characterization of serine protease from thermostable alkalophilic Bacillus laterosporus-AK1. World J Microbiol Biotechnol 23:475–481
Banerjee UC, Sani RK, Azmi W, Soni R (1999) Thermostable alkaline protease from Bacillus brevis and its characterization as a laundry detergent additive. Process Biochem 35:213–219
He H, Chen X, Sun C, Zhang Y, Gao P (2006) Preparation and functional evaluation of oligopeptide-enriched hydrolysate from shrimp (Acetes chinensis) treated with crude protease from Bacillus sp. SM98011. Bioresour Technol 97:385–390
Liang TW, Chen YJ, Yen YH, Wang SL (2007) The antitumor activity of the hydrolysates of chitinous materials hydrolyzed by crude enzyme from Bacillus amyloliquefaciens V656. Process Biochem 42:527–534
Binsan W, Benjakul S, Visessanguan W, Roytrakul S, Tanaka M, Kishimura H (2008) Antioxidative activity of mungoong, an extract paste, from the cephalothorax of white shrimp (Litopenaeus vannamei). Food Chem 106:185–193
Seymour TA, Li SJ, Morrissey MT (1996) Characterisation of a natural antioxidant from shrimp shell waste. J Agric Food Chem 44:682–685
Acknowledgments
We thank the authorities of Annamalai University for providing facilities and Ministry of Earth Sciences—Coastal Ocean Monitoring and Prediction System (MoES-COMAPS), Government of India for providing financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Annamalai, N., Rajeswari, M.V., Thavasi, R. et al. Optimization, purification and characterization of novel thermostable, haloalkaline, solvent stable protease from Bacillus halodurans CAS6 using marine shellfish wastes: a potential additive for detergent and antioxidant synthesis. Bioprocess Biosyst Eng 36, 873–883 (2013). https://doi.org/10.1007/s00449-012-0820-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-012-0820-3