
Abstract
The up-regulation of secondary metabolic pathways following herbivore attack and the subsequent reduction in herbivore performance have been identified in numerous woody plant species. Eucalypts constitutively express many secondary metabolites in the leaves, including terpenes and formylated phloroglucinol compounds (FPCs). We used clonal ramets from six clones of Eucalyptus grandis and two clones of E. grandis × camaldulensis to determine if methyl jasmonate (MeJA) treatment could induce changes in the foliar concentrations of either of these groups of compounds. We also used bioassays to determine if any changes in the performance of larvae of Paropsis atomaria, a chrysomelid leaf beetle, could be detected in treated ramets versus the untreated controls, thus indicating whether MeJA induced the up-regulation of defences other than terpenes or FPCs. We found no significant effects of MeJA treatment on either the foliar concentrations of terpenes and FPCs or on herbivore performance. We did, however, detect dramatic differences in larval performance between Eucalyptus clones, thereby demonstrating large variations in the levels of constitutive defence. Larval feeding on clones resistant to P. atomaria resulted in high first instar mortality and disruption of normal gregarious feeding behaviour in surviving larvae. Histological examination of larvae feeding on a resistant clone revealed damage to the midgut consistent with the action of a toxin. These findings concur with mounting evidence that most evergreen perennial plants lack foliar-induced defences and suggest that constitutively expressed secondary metabolites other than those commonly examined in studies of interactions between insect herbivores and Eucalyptus may be important in plant defence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Much of the theory on the evolution of plant defence assumes that defences are costly (Coley et al. 1985; Feeny 1976; Herms and Mattson 1992; Rhoades and Cates 1976). Given the limited resources available to plants for defence, researchers suggest that trade-offs between allocation to different types of defences within species should occur and that such a trade-off should exist between inducible and constitutive defence strategies (Herms and Mattson 1992; Karban and Myers 1989; Mattson et al. 1988). A meta-analysis of defence strategies (Koricheva et al. 2004) did find a negative correlation between inducible and constitutive defence strategies but, overall, the presence of an induced defence strategy is difficult to predict a priori because plants can possess many complementary defensive strategies (Nykanen and Koricheva 2004). For example, some conifers demonstrate induced oleoresin production as a defence against bark beetles (Byun-McKay et al. 2006; Litvak and Monson 1998; Martin et al. 2002; Raffa and Smalley 1995), indicating that species with high concentrations of constitutive defensive secondary metabolites can still evolve induced defensive strategies if these strategies result in a fitness advantage.
Increased resistance to herbivores following defoliation is often derived from qualitative and quantitative changes in plant biochemistry. Typically, there is up-regulation of biochemical pathways producing defensive secondary metabolites or increased expression of proteinase inhibitors or polyphenol oxidases that inhibit digestion (reviewed by Haukioja 1990; Karban and Baldwin 1997). Such defences may already be constitutively expressed in the plant at a lower concentration. Many studies investigating the effects of prior leaf damage on subsequent insect performance, however, demonstrate minor or no additional effects on herbivore performance relative to those of constitutively expressed defences (Nykanen and Koricheva 2004). For example, the variation in the concentration of phenolic glycosides, the most important secondary metabolite determining the performance of lepidopteran defoliators on quaking aspen (Hwang and Lindroth 1998; Osier et al. 2000), is primarily determined by genotype and is unaffected by defoliation (Osier and Lindroth 2001). In contrast, quantitative changes in condensed tannins occur after defoliation in this species, but these do not contribute to significant changes in herbivore performance [Osier and Lindroth 2001; but see Donaldson and Lindroth (2004) for the effects of tannins on a specialist beetle]. Consequently, the general importance of induced responses to insect population dynamics and how much they contribute to relative levels of defoliation between host genotypes remains unknown.
The foliage of species in the genus Eucalyptus (Myrtaceae) contains relatively high concentrations of secondary metabolites, including tannins, other phenolics, terpenes and another group of defensive secondary metabolites, formylated phloroglucinol compounds (FPCs) (Eschler et al. 2000). While eucalypts possess significant constitutive defences, the existence of induced responses in Eucalyptus has not been demonstrated, although a single study on a lepidopteran defoliator (Thyrinteina arnobia) of plantation Eucalyptus in Brazil did show a small increase in larval mortality on previously damaged leaves (Holtz et al. 2003). In eucalypts, terpenes and FPCs are constitutively expressed in leaves, and intraspecific variation in concentrations are largely due to genetic differences between individual trees (Andrew et al. 2005, 2007; Barton et al. 1991). Large variations in foliar concentrations of terpenes and FPCs within and between Eucalyptus species suggest that a concomitant variability in resistance to herbivores could be anticipated. Several studies have shown that FPCs deter feeding by vertebrate herbivores (Lawler et al. 2000; Marsh et al. 2003; Moore et al. 2005; Wallis et al. 2002) and confer a cross resistance to Christmas beetles (Andrew et al. 2007), but their effect on other insect taxa remains unknown. Some field studies (Edwards et al. 1993; Stone and Bacon 1994) suggest terpene concentration or composition may limit herbivory by some insects on Eucalyptus, but other studies on paropsine chrysomelids (a group of specialist eucalypt herbivores) have failed to detect any negative relationship between terpenes and insect performance (Lawler et al. 1997; Morrow and Fox 1980; Patterson et al. 1996). This is likely due to the ability of paropsine chrysomelids to metabolise most of the terpenes ingested (Ohmart and Larsson 1989).
Terpenoids are a diverse group of compounds, many of which are involved in plant defence and exhibit toxic and/or inhibitory effects on herbivores (Gershenzon and Croteau 1991; Hummelbrunner and Isman 2001; Langenheim 1994). The treatment of conifers with methyl jasmonate (MeJA) mirrors the effects of bark beetle activity both histologically and by stimulating terpene (oleoresin) production (Franceschi et al. 2002; Hudgins et al. 2004; Martin et al. 2002). In addition, recent work has shown that terpenes accumulate in Norway spruce (Picea abies) needles and monoterpene emissions increase after MeJA treatment (Martin et al. 2003). Monoterpene synthase activities and monoterpene emissions from the foliage of conifers following simulated and real insect damage have also been reported (Litvak and Monson 1998). In conifers and many angiosperms, the jasmonate (octodecanoid) signal pathway (Farmer and Ryan 1992; Liechti and Farmer 2002; Reymond and Farmer 1998) thus appears to be closely linked to the increased expression of terpene synthases and, in some cases, an increase in accumulated terpenes.
In this study we sought to determine if exposure to MeJA would induce accumulation or qualitative changes in foliar terpenes and FPCs in clonal ramets of E. grandis and two E. grandis hybrids. We also conducted bioassays to try to detect any induced responses in foliage via changes in the growth and development of the first three larval instars of Paropsis atomaria (Coleoptera: Chrysomelidae). These bioassays demonstrated an unexpectedly large variation in resistance to P. atomaria between clones of E. grandis that was independent of MeJA treatment. Consequently, we also carried out additional analyses aimed at determining the underlying cause of this variability.
Materials and methods
Plant material
Plant material was grown under shade cloth at the plant culture facility, School of Botany and Zoology, The Australian National University, Canberra, ACT, Australia. Experimental plants consisted of seven Eucalyptus grandis and two E. grandis × camaldulensis hybrid clones with 8–12 ramets per clone. The ramets were fertilised with slow release Osmocote® fertiliser and were approximately 15 months old and >1 m tall at the time of the experiment.
Experimental design
Half the ramets in each clonal trial were exposed to MeJA in a chamber (1 × 1 × 1.2 m) for 24 h, with the remaining ramets housed in an identical chamber as a control. Each chamber consisted of a frame welded from 10 mm2 hollow steel rod covered with clear plastic and sealed with adhesive clear plastic tape. Ramets of five of the nine clonal lines were exposed to MeJA vapour using a modified version of the method of Farmer and Ryan (1990). In brief, a solution of 10% MeJA in ethanol was applied at a rate of 1 μl of solution per litre of air (120 μl MeJA total) in the chamber by pipetting small volumes onto cotton wool-tipped bamboo skewers inserted upright in the potting medium. Control ramets were treated with ethanol only. Rather than risk exposing the chambers to sunlight in the summer where there was a chance of overheating, the sealed chambers were placed under artificial light for 24 h. Although we chose to expose the plants to MeJA vapour, many other researchers have instead sprayed variable quantities of the compound directly onto foliage. To determine if the concentration of methyl MeJA in the chambers was too low and/or the method of application ineffective, the second set of four clones were treated with MeJA by spraying the leaves of each set of treated ramets with a solution containing 250 μl of MeJA in 40 ml of 10% ethanol. We applied this solution with an atomiser and then immediately sealed the chamber. Control plants were similarly treated with ethanol, and all ramets were then incubated in the chambers as previously described. All clones exposed to MeJA via one of the two application methods were treated within 7 days of each other. The rate of MeJA application per plant in both methods was comparable to that used in other studies on woody plants, such as Norway spruce (Martin et al. 2003).
Bioassay experiments
After 24 h exposure to MeJA or ethanol only, treated and control ramets were kept separate to avoid any potential induction of defence chemistry by endogenous elicitors generated by treated plants. Eight days after treatment with MeJA, all ramets were transferred to a pair of climate-controlled glasshouses with natural light and a 30/18°C day/night temperature regime. As treated and control ramets could not be housed together, the locations of the treated and control ramets for the first set of five and the second set of four clones were switched between glasshouses to counter glasshouse-specific effects.
Paropsis atomaria colonies of locally collected adults were cultured in the laboratory. Adults were housed in timber and mesh cages (0.4 × 0.4 × 0.5 m) and supplied regularly with fresh E.grandis foliage. We collected egg batches daily, and when several egg masses hatched simultaneously, they were placed in containers until the neonates started wandering in search of food. The larvae were effectively randomised between trees by the inherent mixing of neonates within the hatching container.
Four of the seven E. grandis clones plus the two hybrid clones were used for bioassays, which consisted of depositing 20 neonates onto a small slip of paper and clipping this paper to approximately the same position of an expanding shoot tip on each ramet. Neonates were then free to select appropriate expanding leaves to feed upon. Each cohort was then monitored to determine the time taken to reach moult (measured as time for 50% of remaining larvae in a cohort to moult) and mortality over each instar stage. The experiment was terminated when a cohort had completed third instar development (of a total four instars) or, more rarely, when all larvae in a cohort had died. In the former scenario, the larvae were collected, weighed and dried at 50°C to a constant mass (or for 48 h). For a subset of the clones, mortality within the first cohort was unusually high, prompting us to repeat the bioassay with a second larval cohort placed on these clones 4–5 days after the first to assess if this result was repeatable.
Terpene analysis
Immediately before each bioassay, we collected and stored on ice approximately 5 g of fully expanded new leaves (approximately upwards of ten leaves) from each ramet for later quantification of secondary metabolites. Recently expanded leaves were used to avoid confounding variation associated with rapid changes in terpene concentration in expanding leaves. Terpene extraction methods and gas chromatography-mass spectroscopy (GC-MS) analysis were based on the method of Ammon et al. (1985). Briefly, approximately 1.5 g of fresh leaf was added to a weighed volume (15 ml) of ethanol containing the internal standard, n-tridecane (0.25 g/l). The extraction was then left for at least 14 days before GC analysis. The remaining leaf sample was weighed and then freeze-dried (for FPC analysis) and weighed again to determine moisture content. We used an Agilent 6890 N GC (Agilent Technologies, Santa Clara, CA) with helium as the carrier gas at a flow rate of 1 ml/min, split injector with a split ratio of 25:1 (injection temperature 250°C, 1 μl injection volume) and a 60 m × 0.25 mm × 0.25 μm Heliflex AT-35 column. Terpenes were separated using a 25-min programme with an initial temperature of 100°C held for 6 min, an increase of 20°C/min until 200°C, then 5°C/min until 250°C, which was held for 4 min. The coupled MS was an Agilent 5973 with a quadrupole mass selective detector, transfer line temperature 230°C, source temperature 230°C, quadrupole temperature 150°C, ionisation potential 70 eV and a scan range of 40–350 amu. Compounds were identified by comparing mass spectra to reference spectra in the Wiley and National Institute of Standards and Technology libraries. Because we were interested in concentration changes for individual peaks rather than changes in ratios of concentrations of terpenes, we calculated concentrations based on the ratio of peak area to the internal standard peak area (milligram internal standard equivalents per gram dry leaf).
FPC analysis
We extracted and analysed the FPCs in each leaf sample using the method of Wallis and Foley (2005) with about 50 mg of freeze-dried ground leaf. The specific FPC compounds separated by high pressure liquid chromatography (HPLC) were quantified using authentic standards purified in the laboratory.
Protease inhibition assays
To test for the presence of protease inhibitors, we prepared protein fractions from expanding leaf tissue by two different methods. The second method, sequential fractionation, was used in order to minimise the probablility that a null finding using our initial approach was due to failure of column separation to isolate any inhibitors present. All of the subsequent steps described herein were carried out in refrigerated centrifuges or in a cold room (4°C) to minimise oxidation and protein precipitation by phenolic compounds in the extract. The protein concentrations of the final eluent/fractions were then determined using the method of Bradford (1976), with bovine serum albumin as a standard.
Solid-phase silica-bonded filtration
Approximately 0.5 g fresh weight of expanding leaf tissue from each clonal ramet was frozen in liquid nitrogen and ground with a small amount of glass powder using a mortar and pestle. This sample was split between two microcentifuge tubes and thawed on ice before the addition of 1 ml extraction buffer containing 2% (w/v) polyvinylpolypyrrolidone (PVPP), 0.1 M Tris–Cl, pH 7.6 and 0.05 M Na2EDTA to each tube. The tubes were mixed on a vortex and centrifuged at 12,000g for 10 min before decanting the supernatant and repeating the process. The pooled supernatant was then centrifuged in a Beckman ultracentrifuge (Beckman Coulter, Fullerton, CA) at 44,000g for 45 min. Protein was separated from this crude extract using a solid phase separation column with a silica-bonded trifunctional octadecyl 18 carbon hydrocarbon chain (Bond Elut C18-EWP; Varian, Palo Alto, CA) coupled to a 5-ml syringe. After the column had been primed with 1 ml methanol, it was flushed with 2 ml extraction buffer (without PVPP). The supernatant was then loaded onto the column, followed by washing with a further 4 ml of buffer. The column was then eluted with 2 ml 50% aqueous MeOH. The methanol was removed, and the sample concentrated to about 50% of the initial volume by lyophilisation.
PEG fractionation of crude extract
The preparation of the crude extract was the same as described above but approximately 1.2 g fresh weight of expanding leaf tissue was used, divided between 4 × 2-ml microfuge tubes containing 1.5 ml extraction buffer [1% (w/v) polyethylene glycol (PEG) (8000 MW)] per tube. Four fractions were then obtained from the pooled crude extract by the sequential addition of solid PEG (followed by mixing until dissolved) to achieve 10, 20 and 30% solutions and a final fraction precipitated by lowering the pH to 5 using 10% acetic acid. The sample was centrifuged at 30,000g for 15 min between PEG additions to pellet the protein fractions, which were resuspended in 400 μl buffer containing 0.1 M MOPS pH 6.8, 0.15 M NaCl and 0.005 M MgCl2 to test their activity.
Inhibition assays
As our interest initially lay in determining relative differences in protease inhibition activity, all assays were conducted in pairs to directly compare the most resistant and the most susceptible clones. Assays consisted of combining 50 μl of insect enzyme extract (see below) with up to 100 μl of plant protein extract in a microcentrifuge tube. We adjusted the volume of plant extract per assay to use an equivalent amount of protein from all fractions from the same plant and also equivalent amounts from the paired comparison clone. We then added MOPS buffer so that each tube contained 650 μl. The mixture was then allowed to stand at room temperature for 10 min before the addition of 500 μl of 1% azocasein solution. All assays were completed in triplicate with positive controls (larval extract without plant extract) and negative controls (no added extracts). After mixing, 100 μl was immediately pipetted into 500 μl of ice-cold 10% TCA and allowed to stand at 4°C for at least 30 min. The tubes were then centrifuged at 14,000g for 5 min, and the absorbance of the supernatant was measured at 340 nm. The assays were incubated at 28°C, and 100 μl subsamples were removed for spectrophotometry at approximately 4.5-h intervals until the absorbance of the positive control had at least doubled.
Insect protease extracts
First instar larvae from the captive population were placed on an expanding shoot of E. grandis and allowed to feed for approximately 5 days. Protease extracts were made in a microfuge tube using a stainless steel pestle to homogenise 20 entire larvae in 400 μl of ice cold buffer containing 0.1 M MOPS pH 6.8, 0.15 M NaCl and 0.005 M MgCl2. The homogenate was then centrifuged for 10 min at 8000g (4°C) and kept on ice until required.
Condensed tannin assay
Tannin extraction
We used the radial diffusion method of Gedir et al. (2005) with minor modifications to identify relative differences in the condensed tannin content of expanding foliage of the E. grandis clones. Young foliage was harvested from clonal ramets and kept on ice. The leaf samples were then flash frozen in liquid nitrogen and freeze dried before being ground to a fine powder in an SDI Ultramat 2 ball mill (Henry Schein Regional, Rosebery, Australia). Approximately 500 mg of leaf powder was used per extraction, supplying 1.5 ml of acetone extract obtained by the method of Gedir et al. (2005); this was then reduced to approximately 75 μl in a rotary evaporator. The residue was resuspended by adding 250 μl of 50% methanol and sonicating before precipitating any nondissolved material by centrifugation at 2000g for 5 min. The assay was conducted by depositing 10 μl of this preparation into 4.5-mm diameter wells punched into gel plates (Hagerman 1987) and incubating the plates at 30°C for 120 h.
Diffusion ring measurement
The gel plates were scanned against a black background using a flat bed scanner. An occluding disc was drawn over the ring of the precipitated protein on the images to obtain a sharp boundary, the area of which was then measured (in mm2) using ImageJ digital imaging software [Rasband WS (1997–2006) ImageJ, http://rsb.info.nih.gov/ij/, U.S. National Institutes of Health, Bethesda, MD] This area was then divided by the dry weight of leaf extracted and used as a relative measure of tannin content.
Statistical analyses
Concentrations of foliar compounds were analysed using restricted maximum likelihood (REML) models to determine any differences between treated and untreated clonal ramets. Each compound was analysed separately with clone and MeJA treatment as fixed factors. To analyse data from the bioassays, we first examined the dependent variables of larval dry weight and development time using REML models with the same fixed factors but including a ramet-level random term to estimate variance within clones. If this was a minor component of total variance, and other data assumptions were satisfied, regression was then used to determine the explanatory power of the fixed effects clone, glasshouse location, MeJA treatment and cohort. The ability of these same fixed effects to explain mortality within instars was examined using binomial regression, but we included development time as an explanatory variable. Various terpenes and FPCs were also added as factors to this model to determine if their foliar concentrations could explain first instar survival.
Histological examination of first instar larvae
One E. grandis clone exhibited significant resistance to P. atomaria larvae (see Bioassay data section of Results for details). To determine an underlying cause, we sectioned first instar larvae in search of any histological changes that may indicate a toxicological response. Approximately 25 first instar larvae were placed on ramets of the most resistant and least resistant clones and allowed to feed for several days until larvae on the resistant clones started to exhibit high rates of mortality. Remaining larvae from two ramets per clone (total larvae per clone: approx. 7) were collected and prepared for sectioning as described below.
Sample preparation
The tissues were treated with Bouin’s fixative for 1 day before being washed with 70% ethanol to remove the fixative and begin dehydration by exposing them to an ethanol concentration series (70, 95 and 100% twice, then superdry 100% over a molecular sieve to remove any remaining water). Fixed tissue samples were mounted in paraffin wax blocks at 55°C.
Sectioning and staining
We used a rotary microtome to cut 5-μm sections that were fixed to glass slides. Tissues were deparaffinised in histolene for 10 min and re-hydrated in a series of decreasing ethanol concentrations before rinsing in phosphate buffered saline (PBS) to remove any remaining ethanol. Tissues were stained using Periodic acid–Schiff’s reagent to clarify the carbohydrate moieties within the digestive system.
Dehydration and clearing
After staining, the tissues were dehydrated with increasing concentrations of ethanol (70%, 95%, 2 × 100%) for 5 min each, and then histolene for 2 min to clear the tissues. After clearing the tissues were mounted permanently on the slide using Permount™ and a cover slip and allowed to dry on a slide warmer for 24 h.
Results
Chemical analyses
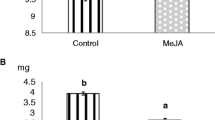
The variation in foliar concentrations of FPCs in the E. grandis clones was within the range we previously measured in this species (e.g. total sideroxylonal 0–17 mg/g dry leaf). Linear mixed models using REML showed there were consistent differences in foliar chemistry between clones for all compounds examined but that there were no significant effect of MeJA treatment on the foliar concentrations of either terpenes or FPCs (Table 1). The method of MeJA application also had no effect in any analysis; consequently, data were pooled for subsequent analyses.
Bioassay data
The estimated component of variance (ramet term) in the REML analyses indicated that random variation between ramets did not contribute significantly to variation in either mean dry weight of larvae or development time. Subsequent regression analyses demonstrated that the clone on which the larvae fed had a significant effect on their final weights and development rates while treatment with MeJA had no significant effect. Clonal differences did not account for much variation in larval weight (22.6%) but did explain around 70% of the variation in first instar development time (Table 2). Ranking the clones by final mean dry weight of larvae did not correspond to their ranking by any other parameters of larval performance measured.
The use of ‘Cohort’ as a fixed effect (denoting a second cohort of larvae on the same clone) did not account for any significant variation in larval growth and survival (Wald tests P = 0.950 and P = 0.799 for mean dry weight per larvae and first instar development time, respectively). Data from the first and second larval cohorts were then pooled in subsequent analyses to increase the sample size for those clones on which most larvae died. There was no effect of the location of each treatment within the two glasshouses, so this factor was excluded from models.
There was a marked effect of clonal host on development rate, with the larvae on Clone E attaining the second instar stage more than twofold faster than larvae on Clone F, on which development was significantly retarded (Fig. 1). Feeding on at least two of the clones C and F) appeared to impair the development of larvae, with a concomitant decrease in survival. First instar larvae raised on these latter clones suffered high mortality rates, which reached 100% in some cohorts, as did a second set of larval cohorts placed on the same ramets, suggesting that cohorts did not differ in their ability to develop on these clones. The feeding behaviour of surviving larvae on these clones was consistent between ramets as follows: neonates moved to the outermost expanding leaves and proceeded to initiate gregarious feeding normally; after 1–2 days, any remaining larvae ceased feeding on these expanding leaves and commenced wandering with ensuing disruption of gregarious feeding; survival to the second instar was associated with larvae moving to those leaves further down the stem that were still soft enough for first instars to consume but on which growth was slow.

Number of Paropsis atomaria larvae versus time during development on four Eucalyptus grandis clones and two E. grandis × camaldulensis clones. Data points are the number of surviving larvae at instar changes from the first to the start of the fourth instar (clonal mean ± SE). Clones (A–H) are as per Table 1. Filled square A, open square B, open triangle C, filled circle E, open circle F, filled inverted triangle H
The inclusion of ‘Development time’ as a variable in a generalised linear model for a binomial distribution with a logit link of survival across the first three instars confirmed the relationship between mortality and rate of development. This model demonstrated a significant increase in proportional survival in latter instar stages but also an overall significant negative effect on the probability of survival to the next instar with increasing development time (Table 3). This model became far more complex when clonal host plant was included as a factor, with multiple significant interactions between development time, instar and clone simultaneously influencing larval survival. This reflects the spectrum of responses to different clonal host plant composition during larval development shown in Fig. 1. Due to the higher mortality in first instars across all clones, we examined the effect of leaf secondary metabolites on first instar survival using generalised linear models as before. However, neither terpene nor FPC concentrations significantly influenced larval survival to the second instar (results not shown).
Protease inhibition and condensed tannin content assays
We tested the clones on which first instar larvae survived best (susceptible clones) and those on which survival was worst (resistant clones) to determine if differences were due to the activity of digestibility reducers, similar perhaps to a trypsin inhibitor found in seeds of E. urophylla (Tremacoldi and Pascholati 2002). Although the column separation successfully extracted leaf protein, extracts from resistant and susceptible clones did not differ in their ability to inhibit proteolytic activity. Either extract in sufficient quantities, however, did completely inhibit insect proteolytic activity in vitro (Fig. 2). Boiling the extract did not diminish its ability to inhibit enzyme activity, suggesting that inhibition did not involve proteinaceous inhibitors (data not shown).

The results from two paired assays demonstrating the dose-dependent decrease in protease activity with increasing volumes of column eluent from leaf protein extractions. Inhibition of larval protease activity was determined by a change in absorbance at 340 nm after 4 h relative to a positive control (+ve) which contained no leaf extract. The addition of 100 μl of extract totally suppressed protease activity as no change in absorbance was detected after an additional measurement at 8 h. Each graph represents one paired assay conducted in parallel with the same extract of larval proteinases and leaf protein extracts from one ramet of a resistant (filled circle) and non-resistant (open circle) clone
The protein fractions from PEG fractionation of the crude extract also did not differ in their inhibition of proteolytic activity and did not inhibit activity relative to positive controls. This was true for fractions from both resistant and susceptible clones (data not shown), providing further evidence that the inhibitory factor obtained by chromatography was not proteinaceous. Finally, although E. grandis clone extracts varied considerably in their ability to precipitate protein in the condensed tannin assay, resistant clones did not always contain higher concentrations of condensed tannins than susceptible clones.
Histological response to feeding on foliage from resistant clones
The midgut from larvae that fed on foliage from the resistant clonal ramets differed distinctly from those of larvae that ingested less resistant foliage (Fig. 3). The midguts from the latter larvae had intact peritrophic matrices adjacent to even brush borders of microvilli. In contrast, ingesting foliage from resistant clones appeared to disrupt the peritrophic matrix such that it was absent in the sections examined. In addition, the layer of microvilli appeared to be sparser and less even, possibly due to physical abrasion by ingested food particles after the loss of the peritrophic matrix (Tellam 1996).

Sections of midgut from two first instar P. atomaria larvae after feeding for several days on non-resistant (a) and resistant (b) E. grandis clones. a Midgut shows a dense, even layer of microvilli (MV) on midgut epithelial cells (EC), with a distinct peritrophic matrix (arrows) separating the brush border from the endoperitrophic matrix space (ENPS). b In larvae demonstrating a toxic response, the midgut lacks a peritrophic matrix and the layer of microvilli is greatly reduced in density and length. Scale bars: 0.1 mm
Discussion
In this study, the application of exogenous MeJA to E. grandis did not induce any changes in the foliar concentration of either terpenes or FPCs, two groups of compounds considered to be important defences in Eucalyptus. Our results do not exclude the possibility that simulated herbivory, real herbivory or treatment with MeJA increases terpene synthase activity because, as demonstrated by Litvak and Monson (1998) in needles from several conifer species, higher rates of terpene volatilisation associated with higher activity of terpene synthases can occur without necessarily increasing the pool of leaf terpenes. The release of volatile terpenes in responses to the application of MeJA or jasmonic acid occurs in many angiosperms (Arimura et al. 2004; Degenhardt and Lincoln 2006; Dicke et al. 1999; Halitschke et al. 2000; Hampel et al. 2005; Ozawa et al. 2000; Rodriguez-Saona et al. 2001). Changes in leaf volatiles induced by herbivory have been shown to be important signals for predators and parasitoids to locate their prey in some tritrophic interactions (reviewed by Dicke et al. 2003). It is unknown whether quantitative or qualitative induced changes in volatile emissions occur in Eucalyptus.
Another possibility is that induced changes only occur during some stages of leaf development. Rapid induced resistance that occurs in birches is absent in mature leaves (Neuvonen et al. 1988; Neuvonen and Haukioja 1991). In this study we restricted our sampling for terpenes and FPCs to new fully expanded leaves to minimise unwanted variation associated with leaf age as terpene concentration and composition changes rapidly during leaf expansion (Maarse and Kepner 1970; Simmons and Parsons 1987). It is possible that an induced response occurred in expanding tissues and was not detected in our study by direct measurement. It is worth noting, however, that first instar larvae (including P. atomaria) usually feed on expanding leaves and are generally the larval stage most sensitive to differences in diet quality. The fact that we were unable to detect induced effects in our bioassays suggests significant changes to defence chemistry did not occur.
Our results show that the application of exogenous MeJA did not induce any responses in ramets of E. grandis (or the two hybrid clones) that affected the performance of P. atomaria larvae relative to those on control ramets. Herbivory by P. atomaria also appeared to induce no systemic changes in any ramets, as indicated by the similar performance of cohorts of larvae on ramets previously damaged by the cohorts of larvae deposited on the plants several days before. Paropsis atomaria is a widely distributed species that uses a variety of eucalypt host species within its range (Carne 1966). Analysis of intraspecific genetic variation throughout the distribution of P. atomaria suggests there are no races restricted to either geographic areas or host plants in this species (Schutze et al. 2006). This implies that P. atomaria has evolved a digestive physiology and detoxification capacity with a high degree of plasticity to cope with a diet that varies greatly in the composition and concentration of plant secondary metabolites. Such plasticity has been demonstrated in species of both Coleoptera and Lepidoptera that alter the expression of digestive proteases to overcome protease inhibitors in their diet (Jongsma and Bolter 1997). Similarly, ingestion of the toxin nicotine by tobacco hornworms stimulates the activity of cytochrome P450 enzymes required for its detoxification (Snyder and Glendinning 1996). Consequently, we should have expected that, unless MeJA changed defensive chemistry dramatically in E. grandis, there would be only minor effects or no observable effect of induced changes on the development of a specialist insect herbivore such as P. atomaria.
Our results agree with other studies that failed to detect any change in defensive chemistry in response to damage or induction using other methods (Chapin et al. 1985; Delano-Frier et al. 2004). Comparisons of the extent and nature of foliar-induced responses across species and life history strategies suggest the existence of broad patterns. Both the review by Karban and Baldwin (1997) and the meta-analysis by Nykanen and Koricheva (2004) suggest that broadleaf deciduous trees commonly respond to herbivory by increasing their concentrations of foliar secondary metabolites. Evergreens, in contrast, are relatively unresponsive to treatments that otherwise induce secondary metabolites in broadleaf deciduous trees and instead counter herbivores by relying on constitutive defence and induced premature leaf abscission (Karban and Baldwin 1997; Nykanen and Koricheva 2004). The rapid responses to folivorous insects identified thus far in evergreen conifer foliage are best described as indirect defences because they rely on the production of volatiles that attract predators and parasitoids (Mumm and Hilker 2006). Eucalypts, with the exception of a few tropical species, are evergreens which retain their leaves for at least a year (Lowman and Heatwole 1987). The consistent and significant variation in larval performance on different clones suggests that constitutive defenses based on genetic differences explain differential insect performance on E. grandis hosts. These results support the typical differences in foliar defence strategies between evergreens and deciduous trees and suggest that variation in defences during leaf expansion may dictate differential rates of herbivory on eucalypt genotypes.
The results of this study may suggest that E. grandis is insensitive to exogenous MeJA; however, it is still probable that the jasmonate pathway mediates the general wounding response in eucalypts. Recent advances in the understanding of regulation of defence-related genes induced in response to herbivory or wounding have shown that jasmonic acid (and MeJA, its volatile counterpart) along with salicylic acid and ethylene regulate these genes (Reymond and Farmer 1998). Such regulatory mechanisms appear to be common to all plants examined to date, suggesting that jasmonate-regulated induced changes in metabolism would be detectable in many species even if they are not all involved in defence. This has been demonstrated by the complexity of transcriptional responses in studies investigating patterns of mRNA production induced in plants by wounding and/or herbivores (see reviews by Gatehouse 2002; Kessler and Baldwin 2002).
Although we restricted our histological examination of the larval midgut to a single contrast between larvae raised on either a resistant or on a susceptible E. grandis clone, the effect was dramatic. The loss of the peritrophic matrix and digestive function associated with ingesting resistant foliage indicates toxicity rather than a difference in digestibility of leaf tissue. In contrast to this clear difference and consistent with previous studies (Lawler et al. 1997; Morrow and Fox 1980; Patterson et al. 1996), we did not detect any differences in foliar chemistry, including protease inhibitors, that would explain the resistance of some clones to paropsine chrysomelid larval feeding. No terpene or FPC compound had any bearing on which clones appeared to resist P. atomaria larvae and which were susceptible although, admittedly, we sampled much older leaves than those on which the first two instars feed. Other research in our laboratory found a similar lack of correlation between FPC concentrations and defoliation of E. grandis by P. atomaria in trial plantations (Henery et al. 2008).
Likewise, the lack of correlation between the concentration of condensed tannins in expanding leaves and P. atomaria performance on individual clones suggests that these compounds do not cause first instar mortality, which agrees with the results of previous work on this eucalypt herbivore (Fox and Macauley 1977). Recent findings, however, suggest that the tannin assay used in this study is too general to differentiate between tannins with potentially differing activities against herbivores (Barbehenn et al. 2006a, b). In an attempt to explain the high larval mortality linked to some tannin bioassays, Ayres et al. (1997) suggested that reactions in the insect midgut converted certain tannins to other, more biologically active products that were toxic. It is now known that some tannins have high oxidative activities, suggesting that plants containing these compounds have active oxidative defenses against herbivores (Barbehenn et al. 2006a). Thus, we cannot discount the possibility that phenolic compounds damaged the insect midgut in P. atomaria larvae, which would explain the corresponding high mortality we observed.
Studies using other host eucalypt species may confirm whether the toxicological symptoms in P. atomaria larvae are peculiar to larvae feeding on E. grandis or whether they also occur in larvae feeding on other eucalypts. Identification of the causative agent, however, is likely to be difficult for the following reasons: (1) compounds belonging to an unidentified chemical group may be involved; (2) several compounds acting synergistically or being converted to a toxic product within the insect may be responsible; (3) any differences in leaf chemistry may be subtle because the toxicity primarily affects first instar larvae, which is the larval stage typically most sensitive to variations in plant defences.
References
Ammon DG, Barton AFM, Clarke DA, Tjandra J (1985) Rapid and accurate determination of terpenes in the leaves of Eucalyptus species. Analyst 110:921–924
Andrew RL, Peakall R, Wallis IR, Wood JT, Knight EJ, Foley WJ (2005) Marker-based quantitative genetics in the wild? The heritability and genetic correlation of chemical defenses in Eucalyptus. Genetics 171:1989–1998
Andrew RL, Wallis IR, Harwood CE, Henson M, Foley WJ (2007) Heritable variation in the foliar secondary metabolite sideroxylonal confers cross-resistance in Eucalyptus. Oecologia 153:891–901
Arimura G, Huber DPW, Bohlmann J (2004) Forest tent caterpillars (Malacosoma disstria) induce local and systemic diurnal emissions of terpenoid volatiles in hybrid poplar (Populus trichocarpa × deltoides): cDNA cloning, functional characterization, and patterns of gene expression of (−)-germacrene d synthase, PtdTPS1. Plant J 37:603–616
Ayres MP, Clausen TP, MacLean SF, Redman AM, Reichardt PB (1997) Diversity of structure and antiherbivore activity in condensed tannins. Ecology 78:1696
Barbehenn RV, Jones CP, Hagerman AE, Karonen M, Salminen JP (2006a) Ellagitannins have greater oxidative activities than condensed tannins and galloyl glucoses at high pH: potential impact on caterpillars. J Chem Ecol 32:2253
Barbehenn RV, Jones CP, Karonen M, Salminen JP (2006b) Tannin composition affects the oxidative activities of tree leaves. J Chem Ecol 32:2235
Barton AFM, Cotterill PP, Brooker MIH (1991) Short note: heritability of cineole yield in Eucalyptus kochii. Silvae Genet 40:37–38
Bradford MM (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem 72:248–254
Byun-McKay A, Godard KA, Toudefallah M, Martin DM, Alfaro R, King J, Bohlmann J, Plant AL (2006) Wound-induced terpene synthase gene expression in Sitka spruce that exhibit resistance or susceptibility to attack by the white pine weevil. Plant Physiol 140:1009–1021
Carne PB (1966) Ecological characteristics of the eucalypt-defoliating chrysomelid Paropsis atomaria OL. Aust J Zool 14:647–672
Chapin III FS, Bryant JP, Fox JF (1985) Lack of induced chemical defense in juvenile Alaskan (USA) woody plants in response to simulated browsing. Oecologia 67:457–459
Coley PD, Bryant JP, Chapin FS (1985) Resource availability and plant antiherbivore defense. Science 230:895–899
Degenhardt DC, Lincoln DE (2006) Volatile emissions from an odorous plant in response to herbivory and methyl jasmonate exposure. J Chem Ecol 32:725–743
Delano-Frier JP, Martinez-Gallardo NA, Martinez-de la Verga O, Salas-Araiza MD, Barbosa-Jaramillo ER, Torres A et al (2004) The effect of exogenous jasmonic acid on induced resistance and productivity in amaranth (Amaranthus hypochondriacus) is influenced by environmental conditions. J Chem Ecol 30:1001–1034
Dicke M, Gols R, Ludeking D, Posthumus MA (1999) Jasmonic acid and herbivory differentially induce carnivore-attracting plant volatiles in lima bean plants. J Chem Ecol 25:1907–1922
Dicke M, van Poecke RMP, de Boer JG (2003) Inducible indirect defence of plants: from mechanisms to ecological functions. Basic Appl Ecol 4:27–42
Donaldson JR, Lindroth RL (2004) Cottonwood leaf beetle (Coleoptera: Chrysomelidae) performance in relation to variable phytochemistry in juvenile aspen (Populus tremuloides Michx.). Environ Entomol 33:1505–1511
Edwards PB, Wanjura WJ, Brown WV (1993) Selective herbivory by Christmas beetles in response to intraspecific variation in Eucalyptus terpenoids. Oecologia 95:551–557
Eschler BM, Pass DM, Willis R, Foley WJ (2000) Distribution of foliar formylated phloroglucinol derivatives amongst Eucalyptus species. Biochem Syst Ecol 28:813–824
Farmer EE, Ryan CA (1990) Interplant communication: airborne methyl jasmonate induces synthesis of proteinase inhibitors in plant leaves. Proc Natl Acad Sci USA 87:7713–7716
Farmer EE, Ryan CA (1992) Octadecanoid precursors of jasmonic acid activate the synthesis of wound inducible proteinase-inhibitors. Plant Cell 4:129–134
Feeny PP (1976) Plant apparency and chemical defense. In: Wallace JW, Mansell RL (eds) Biochemical interactions between plants and insects, vol 10. Plenum, New York, pp 1–40
Fox LR, Macauley BJ (1977) Insect grazing on Eucalyptus in response to variation in leaf tannins and nitrogen. Oecologia 29:145–162
Franceschi VR, Krekling T, Christiansen E (2002) Application of methyl jasmonate on Picea abies (Pinaceae) stems induces defense-related responses in phloem and xylem. Am J Bot 89:578–586
Gatehouse JA (2002) Plant resistance towards insect herbivores: a dynamic interaction. New Phytol 156:145–169
Gedir JV, Sporns P, Hudson RJ (2005) Extraction of condensed tannins from cervid feed and feces and quantification using a radial diffusion assay. J Chem Ecol 31:2761–2773
Gershenzon J, Croteau R (1991) Terpenoids. In: Rosenthal GA, Berenbaum MR (eds) Herbivores: their interactions with secondary metabolites. Academic, San Diego, pp 165–219
Hagerman AE (1987) Radial diffusion method for determining tannin in plant-extracts. J Chem Ecol 13:437–449
Halitschke R, Kessler A, Kahl J, Lorenz A, Baldwin IT (2000) Ecophysiological comparison of direct and indirect defenses in Nicotiana attenuata. Oecologia 124:408–417
Hampel D, Mosandl A, Wust M (2005) Induction of de novo volatile terpene biosynthesis via cytosolic and plastidial pathways by methyl jasmonate in foliage of Vitis vinifera L. J Agric Food Chem 53:2652–2657
Haukioja E (1990) Induction of defenses in trees. Annu Rev Entomol 36:25–42
Henery ML, Henson M, Wallis IR, Stone C, Foley WJ (2008) Predicting crown damage to Eucalyptus grandis by Paropsis atomaria with direct and indirect measures of leaf composition. For Ecol Manage. doi:10.1016/j.foreco.2008.03.003
Herms DA, Mattson WJ (1992) The dilemma of plants: to grow or defend. Q Rev Biol 67:283–335
Holtz AM, de Oliveira HG, Pallini A, Marinho JS, Zanuncio JC, Oliveira CL (2003) Adaptation of Thyrinteina arnobia to a new host and herbivore induced defense in eucalyptus. Pesqu Agropecu Bras 38:453–458
Hudgins JW, Christiansen E, Franceschi VR (2004) Induction of anatomically based defense responses in stems of diverse conifers by methyl jasmonate: a phylogenetic perspective. Tree Physiol 24:251–264
Hummelbrunner LA, Isman MB (2001) Acute, sublethal, antifeedant, and synergistic effects of monoterpenoid essential oil compounds on the tobacco cutworm, Spodoptera litura (Lep., Noctuidae). J Agric Food Chem 49:715–720
Hwang SY, Lindroth RL (1998) Consequences of clonal variation in aspen phytochemistry for late season folivores. Ecoscience 5:508–516
Jongsma MA, Bolter C (1997) The adaptation of insects to plant protease inhibitors. J Insect Physiol 43:885–895
Karban R, Baldwin IT (1997) Induced responses to herbivory. University of Chicago Press, Chicago
Karban R, Myers JH (1989) Induced plant responses to herbivory. Annu Rev Ecol Syst 20:331–348
Kessler A, Baldwin IT (2002) Plant responses to insect herbivory: the emerging molecular analysis. Ann Rev Plant Biol 53:299–328
Koricheva J, Nykanen H, Gianoli E (2004) Meta-analysis of trade-offs among plant antiherbivore defenses: are plants jacks-of-all-trades, masters of all? Am Nat 163:E64–E75
Langenheim JH (1994) Higher-plant terpenoids: a phytocentric overview of their ecological roles. J Chem Ecol 20:1223–1280
Lawler IR, Foley WJ, Woodrow IE, Cork SJ (1997) The effects of elevated CO2 atmospheres on the nutritional quality of Eucalyptus foliage and its interaction with soil nutrient and light availability. Oecologia 109:59–68
Lawler IR, Foley WJ, Eschler BM (2000) Foliar concentration of a single toxin creates habitat patchiness for a marsupial folivore. Ecology 81:1327–1338
Liechti R, Farmer EE (2002) The jasmonate pathway. Science 296:1649–1650
Litvak ME, Monson RK (1998) Patterns of induced and constitutive monoterpene production in conifer needles in relation to insect herbivory. Oecologia 114:531–540
Lowman MD, Heatwole H (1987) The impact of defoliating insects on the growth of eucalypt saplings. Aust J Ecol 12:175–181
Maarse H, Kepner RE (1970) Changes in composition of volatile terpenes in Douglas fir needles during maturation. J Agric Food Chem 18:1095–1101
Marsh KJ, Foley WJ, Cowling A, Wallis IR (2003) Differential susceptibility to Eucalyptus secondary compounds explains feeding by the common ringtail (Pseudocheirus peregrinus) and common brushtail possum (Thichosurus vulpecula). J Comp Physiol B Biochem Syst Environ Physiol 173:69–78
Martin D, Tholl D, Gershenzon J, Bohlmann J (2002) Methyl jasmonate induces traumatic resin ducts, terpenoid resin biosynthesis, and terpenoid accumulation in developing xylem of Norway spruce stems. Plant Physiol 129:1003–1018
Martin DM, Gershenzon J, Bohlmann J (2003) Induction of volatile terpene biosynthesis and diurnal emission by methyl jasmonate in foliage of Norway spruce. Plant Physiol 132:1586–1599
Mattson WJ, Lawrence RK, Haack RA, Herms DA, Charles P-J (1988) Defensive strategies of woody plants against different insect-feeding guilds in relation to plant ecological strategies and intimacy of association with insects. In: Mattson WJ, Levieux J, Bernard-Dagan C (eds) Mechanisms of woody plant defenses against insects. Springer, New York, pp 3–38
Moore BD, Foley WJ, Wallis IR, Cowling A, Handasyde KA (2005) Eucalyptus foliar chemistry explains selective feeding by koalas. Biol Lett 1:64–67
Morrow PA, Fox LR (1980) Effects of variation in Eucalyptus essential oil yield on insect growth and grazing damage. Oecologia 45:209–219
Mumm R, Hilker M (2006) Direct and indirect chemical defence of pine against folivorous insects. Trends Plant Sci 11:351–358
Neuvonen S, Haukioja E (1991) The effects of inducible resistance in host foliage on birch-feeding herbivores. In: Tallamy DW, Raupp MJ (eds) Phytochemical induction by herbivores. Wiley, New York
Neuvonen S, Hanhimaki S, Suomela J, Haukioja E (1988) Early season damage to birch foliage affects the performance of a late season herbivore. J Appl Entomol 105:182–189
Nykanen H, Koricheva J (2004) Damage-induced changes in woody plants and their effects on insect herbivore performance: a meta-analysis. Oikos 104:247–268
Ohmart CP, Larsson S (1989) Evidence for absorption of essential oils by Paropsis atomaria Oliver (Coleoptera: Chrysomelidae). J Aust Entomol Soc 28:201–205
Osier TL, Lindroth RL (2001) Effects of genotype, nutrient availability, and defoliation on aspen phytochemistry and insect performance. J Chem Ecol 27:1289–1313
Osier TL, Hwang SY, Lindroth RL (2000) Effects of phytochemical variation in quaking aspen Populus tremuloides clones on gypsy moth Lymantria dispar performance in the field and laboratory. Ecol Entomol 25:197–207
Ozawa R, Arimura G, Takabayashi J, Shimoda T, Nishioka T (2000) Involvement of jasmonate- and salicylate-related signaling pathways for the production of specific herbivore-induced volatiles in plants. Plant Cell Physiol 41:391–398
Patterson KC, Clarke AR, Raymond CA, Zalucki MP (1996) Performance of first instar Chrysophtharta bimaculata larvae (Coleoptera: Chrysomelidae) on nine families of Eucalyptus regnans (Myrtaceae). Chemoecology 7:94–106
Raffa KF, Smalley EB (1995) Interaction of pre-attack and induced monoterpene concentrations in host conifer defense against bark beetle fungal complexes. Oecologia 102:285–295
Reymond P, Farmer EE (1998) Jasmonate and salicylate as global signals for defense gene expression. Curr Opin Plant Biol 1:404–411
Rhoades DF, Cates RG (1976) Toward a general theory of plant antiherbivore chemistry. Recent Adv Phytochem 10:168–213
Rodriguez-Saona C, Crafts-Brander SJ, Pare PW, Henneberry TJ (2001) Exogenous methyl jasmonate induces volatile emissions in cotton plants. J Chem Ecol 27:679–695
Schutze MK, Mather PB, Clarke AR (2006) Species status and population structure of the Australian Eucalyptus pest Paropsis atomaria Olivier (Coleoptera: Chrysomelidae). Agric For Entomol 8:323–332
Simmons D, Parsons RF (1987) Seasonal variation in the volatile leaf oils of two Eucalyptus species. Biochem Syst Ecol 15:209–215
Snyder MJ, Glendinning JI (1996) Causal connection between detoxification enzyme activity and consumption of a toxic plant compound. J Comp Physiol A Sens Neural Behav Physiol 179:255–261
Stone C, Bacon PE (1994) Relationships among moisture stress, insect herbivory, foliar cineole content and the growth of river red gum Eucalyptus camaldulensis. J Appl Ecol 31:604–612
Tellam RL (1996) The peritrophic matrix. In: Lehane MJ, Billingsley PF (eds) Biology of the insect midgut. Chapman and Hall, London, pp 86–114
Tremacoldi CR, Pascholati SF (2002) Detection of trypsin inhibitor in seeds of Eucalyptus urophylla and its influence on the in vitro growth of the fungi Pisolithus tinctorius and Rhizoctonja solani. Braz J Microbiol 33:281–286
Wallis IR, Foley WJ (2005) The rapid determination of sideroxylonals in Eucalyptus foliage by extraction with sonication followed by HPLC. Phytochem Anal 16:49–54
Wallis IR, Watson ML, Foley WJ (2002) Secondary metabolites in Eucalyptus melliodora: field distribution and laboratory feeding choices by a generalist herbivore, the common brushtail possum. Aust J Zool 50:507–519
Acknowledgments
We thank Ulrike Mathesius for assistance with the protease inhibition assays and Tony Ashton for advice on protein extraction from eucalypts. The clonal eucalypts were propagated and grown by Helen Smith and Chris Moran at Forests NSW, Tree Improvement Group, Grafton. Jeff Wood assisted with statistical analyses. Martin Henery was financially supported by the Australian Research Council and Forests NSW. The work was funded by a grant from the Australian Research Council to WJF. We declare that the experiments described above comply with the current laws of Australia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Andrea Polle.
Rights and permissions
About this article
Cite this article
Henery, M.L., Wallis, I.R., Stone, C. et al. Methyl jasmonate does not induce changes in Eucalyptus grandis leaves that alter the effect of constitutive defences on larvae of a specialist herbivore. Oecologia 156, 847–859 (2008). https://doi.org/10.1007/s00442-008-1042-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00442-008-1042-x