
Abstract
Basement membranes are thin connective tissue structures composed of organ-specific assemblages of collagens, laminins, proteoglycan-like perlecan, nidogens, and other components. Traditionally, basement membranes are thought of as structures which primarily function to anchor epithelial, endothelial, or parenchymal cells to underlying connective tissues. While this role is important, other functions such as the modulation of growth factors and cytokines that regulate cell proliferation, migration, differentiation, and fibrosis are equally important. An example of this is the critical role of both the epithelial basement membrane and Descemet’s basement membrane in the cornea in modulating myofibroblast development and fibrosis, as well as myofibroblast apoptosis and the resolution of fibrosis. This article compares the ultrastructure and functions of key basement membranes in several organs to illustrate the variability and importance of these structures in organs that commonly develop fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Basement membranes (BM) are specialized extracellular matrix protein complexes found in every organ of the human body. They have specific structures that provide adhesion for epithelium, endothelium, or parenchymal cells and separate them from connective tissues, nerves, and muscles. BMs delineate boundaries and compartmentalize tissues in organs while providing scaffolds that guide morphogenesis and tissue repair. BM-mediated cell signaling events and cellular behavior are altered by tissue-specific BM composition and structure. BMs are best detected with transmission electron microscopy (TEM) or immunohistochemical staining. The four major components most BMs have in common are nidogens, perlecan, laminins, and collagen type IV (Fig. 1). However, even though there is great heterogeneity of the primary components, other components also are commonly present and provide specificity of function. Basement membrane components are key players in specialized extracellular matrices and changes in BM composition play significant roles in facilitating the development of various diseases in different organs (Kruegel and Miosge 2012).

Schematic diagram of typical components found in basement membranes, using skin as an example. A basal keratinocyte adheres to the underlying basement membrane and dermis via the focal adhesions that transmit mechanical force and regulatory signals that consist of numerous interacting components such as the hemidesmosome with bullous pemphigoid antigen (BPAG), integrin a6b4, laminin 332, perlecan, anchoring fibrils, and dozens of other components that vary depending on the organ and the status (homeostasis, post-injury, etc.) of the tissues. Many of these components extend into, and are part of, the lamina lucida of the basement membrane. The underling lamina densa of the basement membrane is composed of collagen type IV, nidogens, perlecan, laminin 332, that directly interact with each other, and other components. Lamina lucida is not as wide naturally as it is drawn here for clarity reasons. Illustration by David Schumick, BS, CMI. Reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography© 2018. All Rights Reserved
BM proteins were first discovered in mouse yolk sac tumors which produce typical extracellular matrix (ECM) proteins (Chung et al. 1977; Kleinman and Martin 2005; Orkin et al. 1977). Further analysis showed that laminins (Chung et al. 1979; Timpl et al. 1979), nidogens (Carlin et al. 1981; Timpl 1989), perlecan (Carlin et al. 1981), and collagen type IV (Kleinman et al. 1982) are large multi-domain proteins that self-polymerize, bind to other proteins to augment function and promote stability of the tissue (Timpl and Brown 1996).
Laminins are alpha1, beta1, and gamma1 heterotrimeric glycoproteins with more than 15 trimer combinations identified that contribute to tissue specificity of BMs (Miner and Yurchenco 2004). The laminin nomenclature has been simplified to refer to the alpha, beta, and gamma chains that comprise a specific laminin—such as laminin 332 (Aumailley et al. 2005). Laminins initiate the BM self-polymerization process during development, repair, and regeneration following injury, and other BM components bind to and assemble the mature BM (Miner and Yurchenco 2004; Smyth et al. 1999). Collagen type IV has six genetically different α-chains that assemble into three linear collagen type IV heterotrimers (Khoshnoodi et al. 2008). The stability of the BM structure is attributable primarily to the network formed by laminins and collagen type IV along with other linker components (Halfter et al. 2015; Timpl 1989; Yurchenco et al. 1986).
Nidogen-1 and nidogen-2 are sulfated monomeric glycoproteins (Ho et al. 2008) with the molecular mass of ~ 150 kDa that specifically interact with other BM components such as laminins and collagens to organize and stabilize the BM. Perlecan, also known as basement membrane-specific heparan sulfate proteoglycan core protein (HSPG) or heparan sulfate proteoglycan 2 (HSPG2), is a large multi-domain HSPG that also interacts with other BM components (Hassell et al. 1980). Unlike laminin and collagen type IV, nidogen and perlecan form irregular polymers using their multiple binding sites. They bridge these scaffolds for laminin and collagen type IV, as well as for each other, and hence they are called as bridging molecules (Aumailley et al. 1993; Ettner et al. 1998; Fox et al. 1991). Complete perlecan deficiency is lethal for mouse embryos at the mid-gestational stage (Arikawa-Hirasawa et al. 1999; Costell et al. 1999) and the deletion of both nidogens is prenatally lethal (Bader et al. 2005). Although nidogen-1 and nidogen-2 are present in all tissues, nidogen-2 alone show more restricted expression patterns and tissue specificity (Kimura et al. 1998). In vitro, both proteins interact with laminins and collagen type IV and play a critical role in assembly of the mature BM (Fox et al. 1991; Salmivirta et al. 2002). Perlecan establishes a high negative charge in the BM because of its three heparan sulfate side chains. Therefore, perlecan plays a major role in regulatory processes of BMs by providing a barrier for some regulatory molecules in addition to serving as an anchoring port and connecting bridges in BMs (Behrens et al. 2012; Yurchenco et al. 1986).
In general, BMs have at least one component from the four major proteins and the tissue specificity depends on the differential expression of the respective isoforms and inclusion of other tissue-specific BM components. Thus, the main structural elements, collagen type IV and laminin form a highly-organized network which is non-covalently interconnected by nidogen and perlecan (Paulsson 1988; Timpl 1989; Yurchenco et al. 1986). Laminin gamma 3 chain binds specifically to nidogen (Gersdorff et al. 2005). In vivo, laminin is necessary for the initial steps involved in the BM assembly (Miner and Yurchenco 2004; Smyth et al. 1999) but the stability of the entire BM is determined by the collagen type IV network (Poschl et al. 2004) forming structured polymers (Fig. 1). Gene deletion analysis of the several mutant phenotypes demonstrate the numerous tissue-specific roles of the four major BM components (LeBleu et al. 2007). Additional components such as fibrillin (Tiedemann et al. 2005), collagen type V (Bonod-Bidaud et al. 2012) and BM-associated collagen type XV and type XVIII may also be involved in these complexes depending on the specific tissue (Breitkreutz et al. 2013; Miosge et al. 2003). Agrin is a major proteoglycan component in some BMs, such as the glomerular basement membrane (Denzer et al. 1995; Tsen et al. 1995). Collagen type XVIII has also been found to be BM heparan sulfate proteoglycan that is important in retinal BM (Halfter et al. 1998; Saarela et al. 1998).
Epithelial, endothelial, and parenchymal cells adhere to the BM via a large family of transmembrane cell adhesion proteins called integrins, which are commonly tissue-specific in distribution, and are receptors that tie the matrix to the cell’s cytoskelatin (Boudreau and Jones 1999). There are also other cell-associated receptors that bind BM besides integrins (Boudreau and Jones 1999). The binding of cell surface receptors to BM proteins initiates’ intracellular signaling pathway that influence cellular functions such as migration, proliferation, differentiation, and maintenance of the BM.
BMs have many biological functions ranging from tissue organization to functions as depositors for very active molecule such as growth or differentiation factors, including TGFβ and PDGF that can alter the cellularity and composition of underlying tissue such as the stroma in the cornea (Schubert and Kimura 1991; Torricelli et al. 2013b). The binding of such growth factors to BM is a very efficient way to regulate the signaling of these growth factors and differentiation factors that can, for example, trigger fibrotic wound healing changes in tissues underlying BMs. The following sections will highlight some of the organ-specific structures and functions of a few BMs.
BM in cornea
In the cornea, the epithelial BM is present between the basal epithelial cells and the underlying stroma composed primarily of extracellular matrix and fibroblastic keratocytes (Fig. 2). The functions of the epithelial BM include anchoring of the epithelium to the stroma, bi-directional regulation of the passage, and therefore functions, of growth factors and cytokines that modulate functions such as cell proliferation, migration, and differentiation in the epithelium and stroma, as well as the production of chemokines, metalloproteinases, and collagenases (Torricelli et al. 2013b). The composition of corneal BM is different from other organ BMs due to their heterogeneity. Corneal epithelial BM components include laminins, collagen type IV and other collagens, heparan sulfate proteoglycans, and nidogens (Tuori et al. 1996). Descemet’s membrane (DM) is the basement membrane of the posterior cornea that lies between the corneal endothelium and posterior stroma. It has functions similar to the epithelial BM but contributes to the “leaky” barrier function of the endothelial-Descemet’s membrane complex important to corneal function (Murphy et al. 1984). Descemet’s membrane, in contrast to the epithelial BM in the cornea, increases in thickness during both prenatal development (striated BM) and post-natal during the life of the animal by addition of non-striated, non-lamellar extracellular matrix (Murphy et al. 1984). The structure and function of Descemet’s layer is altered in corneal diseases such as Fuchs’ endothelial dystrophy and bullous keratopathy (Johnson et al. 1982; Zhang and Patel 2015).

Transmission electron micrograph from central rabbit cornea of the epithelial basement membrane (EBM) at ×36,000 magnification. In the cornea, the EBM functions to adhere basal epithelial cells (e) to the underlying stroma and to modulate growth factor-mediated communications between the epithelium and the keratocytes within the stroma (S). As artifacts of fixation, the lamina lucida (arrows) and lamina densa (arrowhead) can be seen. Although this morphology is an artifact of fixation, it signifies the presence of a mature BM. The regular packing of the collagen fibrils, visible as uniform diameter circles, in the stroma contributes to corneal transparency
Initially, it was reported that laminin-111 and laminin-332 were the major laminins in the epithelial BM of the human cornea (Ljubimov et al. 1995; Tuori et al. 1996). Later, however, Filenius et al. found that the BM of human cornea contain only laminin-332 and laminin-511 but not laminin-111. Laminin-332 is produced by the epithelial cells (Filenius et al. 2001) and laminin-511 is produced by the keratocytes (Hassell et al. 1992) and epithelial cells (Saikai and Wilson, unpublished data, 2018). Corneal epithelial cells adhere to the laminins in the epithelial BM through integrins. Human corneal epithelial cells have been shown to express integrins α6β4, α3β1, and α2β1 involved in these interactions (Tervo et al. 1991; Virtanen et al. 1992). Epithelial BM contains collagen type IV, as well as collagen types VII, XVII, and XVIII (Michelacci 2003). Several investigators identified collagen type IV in the epithelial BM, but some were not able to identify this molecule in the BM of the central cornea in some species (Cameron et al. 1991). Collagen type IV is known to be present in the human corneal BM as early as 8 weeks of gestation and throughout life (Ben-Zvi et al. 1986). After injury, collagen type IV is present in the re-synthesized BM and it is involved in the binding of the basal surface of the epithelial cells to the BM. In vitro, collagen type IV has been shown to promote migration and adhesion of corneal epithelial cells (Cameron et al. 1991). Thus, collagen type IV is one of the native components in BM involved in the development, maintenance, and wound healing process in the cornea. Collagen type XVII and α6β4 integrin are present in the hemi-desmosomes, the stud-like structures present in basal corneal epithelial cells that adhere the epithelium to the underlying stroma via anchoring fibrils (Gipson et al. 1988). In vitro, it has been shown that collagen type XVII interacts with the β3 chain of the laminin-332 to support cell binding (Torricelli et al. 2013a).
Collagen type XVIII is the only known BM component with heparan sulfate glycosaminoglycan side chains (Dong et al. 2003). In cornea, collagen type XVIII is localized in the epithelial BM and Descemet membrane (Lin et al. 2001). Knockout of collagen type XVIII does not result in a known corneal phenotype but is known to cause other eye abnormalities (Fukai et al. 2002; Maatta et al. 2007), including pigment granule release, massive disorganization of retinal pigment epithelium, and photoreceptor and iris abnormalities (Marneros et al. 2004; Marneros and Olsen 2003). In humans with Knobloch syndrome, a rare disorder with retinal degeneration and high myopia, have mutations in the gene encoding the α1 chain or deficiency of collagen type XVIII (Menzel et al. 2004; Nystrom et al. 2017; Suzuki et al. 2002).
Perlecan is a key component of the corneal epithelial BM which interacts with other basement membrane components to establish the epithelial barrier function and epithelial morphology. A thinner corneal epithelium and microphthalmos were observed in perlecan-deficient mice (Inomata et al. 2012). Pseudomonas aeruginosa is a bacterium that can produce corneal ulcers and perlecan is known to serve as a binding site for these bacteria. Chen and Hazlett (2001) showed that anti-perlecan antibody can decrease binding of P. aeruginosa to corneal epithelial cells in the human cornea. After epithelial scrape injury in humans that damages the corneal epithelial basement membrane, stromal keratocytes were shown to produce high levels of perlecan and nidogen-2 and, therefore, contribute to epithelial BM regeneration (Torricelli et al. 2015).
Nidogens are present in the epithelial BM, stroma, and Descemet’s membrane of the cornea. Nidogen-1 and nidogen-2 bind to various BM-associated proteins and they are known to be a connecting element between laminin and the collagen network in BM (Kabosova et al. 2007). Keratocytes and myofibroblasts have been shown to produce nidogen-1 and nidogen-2 in vitro (Santhanam et al. 2015). In nidogen-1 knockout mice, minimal pathological changes were observed in the anterior segment of the eye, including the epithelial BM (May 2012). These changes were not seen in the nidogen-2 knockout mice (May 2012).
In addition to epithelial BM, cornea possesses another basement membrane called Descemet’s membrane (DM) that lies between the corneal endothelium and posterior stroma (Fig. 3) that participates in the “leaky barrier function” of the corneal endothelium (Zhang and Patel 2015; Kapoor et al. 1986). For example, the endothelial-DM complex allows critical nutrients to pass into the stroma but restricts the passage of transforming growth factor beta from the aqueous humor into the stroma in the absence of endothelial-DM injury (Marino et al. 2017a). The thickness of DM increases with age, with it having approximately 3 μm of thickness in children and 10 um in adults (Chi et al. 1958; Johnson et al. 1982). DM is composed of two layers: an anterior banded layer which is composed of collagen lamellae and proteoglycans and a posterior non-banded layer which is continually synthesized and thickens over decades (Kefalides et al. 1976). Freeze-fracture, deep-etch replica method has clearly showed that the lattice of the DM is constructed of mainly four components (Sawada 1982): (1) round densities forming the nodes of the lattice, (2) rod-like structures connecting the nodes, (3) fine filaments two-dimensionally distributed in the interstices, and (4) amorphous materials. Biochemical studies of DM revealed similarities with other BM in major molecular components, including collagen type IV, fibronectin, laminins, and heparan sulfate proteoglycan (Carlson et al. 1981; Kefalides and Denduchis 1969) and nidogens (Medeiros et al. 2018). In contrast to other BMs, collagen type VIII is a major constituent in DM, which forms ladder-like structure visible with electron microscopy (Labermeier and Kenney 1983). The finding that corneal endothelial cells in vitro synthesized collagen type VIII supports the presence of type VIII collagen in DM (Benya and Padilla 1986; Sage et al. 1981). Collagen type VIII is a hetero-trimer composed of two distinct alpha chains, α1 and α2, each with molecular weight of about 60,000 Da (Benya and Padilla 1986; Shuttleworth 1997). The hexagonal lattice structure creates a matrix that can resist compression and maintains the open porous structure that allows nutrients to pass in to stroma (Shuttleworth 1997), an important function of the Descemet’s membrane-endothelial complex. Fuchs’ endothelial corneal dystrophy (FECD) has typical pathological changes that include progressive loss of endothelial cells, thickening of the DM, and deposition of anomalous extracellular matrix in the form of guttae (Chi et al. 1958). FECD is likely a group of genetic disorders affecting the corneal endothelium and DM.

Rabbit cornea Descemet’s basement membrane at ×12,600 magnification. Notice the impressive thickness (greater than 6 μm) of Descemet’s basement membrane (DM) in a rabbit only 14 weeks old. Descemet’s basement membrane continues to increase in thickness throughout life. Descemet’s basement membrane provides adhesion for the monolayered corneal endothelial cells (e) that modulate corneal hydration critical to corneal transparency, allows passage of nutrients from the aqueous humor into the stroma, and modulates the passage of TGFβ from the aqueous humor into the corneal stroma that would drive keratocyte differentiation into myofibroblasts and trigger fibrosis. S is the stroma that makes up greater than 90% of the corneal thickness. A stromal fibroblastic cell referred to as a “keratocyte” is indicated by the arrow
BM in skin
Skin consists of two compartments, epidermis and dermis (Fig. 4). Epidermis serves as the first line of defense between the external environment and the animal’s internal organs, and it is connected to the dermis compartment by the BM (Breitkreutz et al. 2013). Apart from structural properties, the BM controls keratinocyte adhesion, traffic of cells, and diffusion of molecules such as growth factors and cytokines, including keratinocyte and platelet-derived growth factor, that regulate both keratinocyte and dermal fibroblast functions through regulation of activation and release (Breitkreutz et al. 2013). In addition, BM plays an important role during the remodeling process after injury and damage to BM by cancer leads to cell activation in the stroma (Mueller and Fusenig 2004).

Transmission electron micrograph of rabbit inner thigh skin basement membrane (BM) at ×40,000 magnification. The overlying basal keratinocyte adheres to the dermis (D) via the basement membrane (BM) composed of lamina lucida (arrows) and lamina densa (arrowhead). The BM in skin also regulates growth factor-mediated interactions between the epithelium and skin fibroblasts in the dermis. Note that the basal epithelial cell membrane in skin is much more prominent than in the cornea
Skin BM components, such as perlecan, collagen types IV and VII, are produced by dermal fibroblasts and epidermal keratinocytes. Other components, like laminin-511 and laminin-332, are primarily synthesized by keratinocytes, although the main source of nidogens is thought to be dermal fibroblasts (Bechtel et al. 2012; Fleischmajer et al. 1995). Additionally, dermal fibroblasts transiently synthesize laminin-211 during wound healing in adult skin (Sugawara et al. 2008).
The upper layer epidermis is connected to the BM by hemidesmosomes containing plectin and bullous pemphigoid antigen 1 (BPAG1) proteins (Sterk et al. 2000). These proteins are linked to alpha 6/beta 4 (Sonnenberg et al. 1991), CD151 (Sterk et al. 2002), and collagen type XVII (Qiao et al. 2009). Integrin alpha6/beta 4 also binds to laminin-332, the only integrin associated with keratins (Aumailley et al. 2005). The BM is connected to the dermis beneath by loop structures of collagen type VII and anchoring fibrils. Also, anchoring fibril-collagen type VII tightly binds to collagen types I and III fibrils in the dermis (Villone et al. 2008). Together, these bridges are essential to maintenance of the structural and functional integrity of skin. Defects in the skin BM or BM-associated molecules is often associated with severe or lethal disease (Sterk et al. 2002; Aumailley et al. 2005).
Mice lacking nidogen-1 and nidogen-2 live to birth and have skin that appear grossly normal (Bader et al. 2005). But the ultrastructure of the skin reveals abnormal basal cells with micro-blistering, microvascular aberrations, BM duplications, and leakiness of small vessels (Mirancea et al. 2007). These mice die from lung and heart abnormalities that are directly related to BM defects, but kidney BMs appear normal (Bader et al. 2005).
Co-cultures of epidermal keratinocytes and dermal fibroblasts have been investigated extensively to study on skin physiology and repair. However, these approaches have major drawbacks that limit communications between two cell types that occur in vivo. Therefore, organotypic co-cultures have been used to provide a better understanding of cellular interactions and BM generation in skin (Fleischmajer et al. 1995; Smola et al. 1998). Thus, in these models, normal epidermal phenotype and BM structure is generated with cells from different sources and with several combinations of epithelial and fibroblastic cells. For example, normal BM structure and epidermal phenotype, including hair follicles, can be generated in organotypic co-culture (Limat et al. 1996; Stark et al. 1999). These systems serve as alternative approaches to study the functions of mutated BM components (Di Nunzio et al. 2008; Fritsch et al. 2009; Murauer et al. 2011).
Transplantation models offer other strategies to study the regeneration of skin and BM in mice. Cultured mouse keratinocytes regain full differentiated function, including the production of BM, when transplanted on the backs of C3H mice (Breitkreutz et al. 1984). Similarly, HaCat cells or keratinocytes from human skin transplanted on nude mice generate normal epidermal tissue and BMs when examined with immunohistochemistry for BM components and ultrastructure examined with TEM. In these studies, the first BM component to appear is laminin-332, followed by nidogens, laminin-511, and collagen type IV (Breitkreutz et al. 1997, 1998).
Skin BMs from histological specimens, transplantation models, and organotypic co-cultures appear structurally and functionally the closest in morphology to corneal epithelial BM (compared to other imaged organs), although at high magnification after identical fixation and processing, a difference in morphology appears obvious and could relate to transparency in the cornea (compare Figs. 2, 3, 4).
There are a number of skin disorders associated with skin blistering, including epidermolysis bullosa affecting at least 18 genes associated with the epithelial basement membrane (EBM) and adhesion to the EBM (Uitto et al. 2017), pemphigus, and bullous pemphigoid (Hammers and Stanley 2016). Pemphigus and bullous pemphigoid are auto-antibody-mediated blistering diseases of the skin. In pemphigus, keratinocytes in epidermis and mucous membranes lose cell-cell adhesion, and in pemphigoid, the basal keratinocytes lose adhesion to their basement membrane. Detailed discussion of these disorders is beyond the scope of this review, but they have been instrumental in understanding the specific functions of many BM components.
BM in kidney
The glomerular basement membrane (GBM) lies between the glomerular endothelial cells and the podocytes (Fig. 5) and functions in the removal of waste and other molecules from blood plasma into the urine without the release of other plasma components such as albumin. The podocytes, which adhere to the GBM, play an active role in preventing plasma proteins from entering the urinary ultrafiltrate by providing a barrier comprising filtration slits between the podocyte foot processes (Fig. 5) (Reiser and Altintas 2016). Unlike other BMs, the GBM is unusually thick and composed primarily of collagen type IV and laminins (Timpl 1989). Mutations in these components cause filtration defects and result in severe renal disease (Miner 2012). Agrin is the major heparan sulfate proteoglycan in GBM (Timpl 1989). This structure allows the passage of plasma water and small waste solutes but limits the flow of large plasma proteins such as albumin. Defects in one of these layers will result in high levels of albumin in the urine.

Transmission electron micrograph of glomerular basement membrane (BM) in rabbit kidney at ×45,000 magnification. The BM that functions in the excretion of waste molecules from capillaries into the urine is a “double” BM that provides adhesion of capillary endothelial cells (endo) with fenestrations (arrowheads) on one side and podocyte foot plates (PFP) of podocytes on the other
In adult GBM, laminin-521 is the major laminin. During GBM formation and maturation, however, laminins go through a transition from laminin-111 to laminin-511 and then to laminin-521 (Miner et al. 1997; Miner and Sanes 1994) and genetic defects in this transition results in GBM breakdown. For example, in mice, a mutation in laminin α5 inhibits the laminin-111 to laminin-511 transition and results in the failure of glomerular vascularization (Miner and Li 2000). Mutation in laminin β2 in humans or in mice results in a congenital nephrotic syndrome with neurological manifestations and it is known as Pierson syndrome (in humans) (Matejas et al. 2010).
Collagen type IV plays a critical role in BM stability (Poschl et al. 2004). Collagen also undergo developmental transitions in GBM during glomerulogenesis. Initially, GBM contains an α1/α2 network but when the glomerular capillaries begin to function, the podocytes secrete α3α4α5 trimers. Then, these components polymerize to form a collagen type IV network characteristic of the fully mature GBM (Abrahamson 2009). Mutations in genes encoding any one of the collagen chains can cause defects in the GBM resulting in mild to severe disease. For example, “thin basement membrane disease” has been found in 40 to 50% of patients having mutations in COL4A3 or COL4A4, which encode the α3 and α4 chains of collagen type IV, respectively (Voskarides et al. 2007). Alport syndrome is a severe basement membrane disease, which eventually leads to kidney failure along with deafness and ocular abnormalities. The X-linked form is the most common version of Alport syndrome and it is caused by mutations in COL4A5 encoding the α5 chain of collagen type IV (Heidet et al. 2000).
In mice, deletion of both nidogen-1 and nidogen-2 genes results in perinatal lethality. Nidogen-1 binds to both laminins and collagen type IV and, therefore, is an important component for BM formation. However, BM can form in the absence of both nidogens and the GBM can appear ultrastructurally normal. However, renal dysgenesis or hydronephrosis can be noted in the fully developed kidney (Bechtel et al. 2012; Miosge et al. 2002). The absence of one or both nidogens does not alter basement membrane composition in adult murine kidney (Gersdorff et al. 2007).
Agrin is the major heparan sulfate proteoglycan of the GBM (Groffen et al. 1998; Timpl 1989). As a heparan sulfate proteoglycan, and also due to the presence of sulfated glycosaminoglycan side chains, agrin has a high net negative charge. All BM, and particularly the GBM, have a net negative charge. Perlecan and agrin are thought to be the most important contributors to this negative charge (Kanwar et al. 2007). It is thought that the net negative charge of the GBM is crucial for function, including the filtration of molecules by the glomerulus. Thus, studies show that molecules that are positively charged cross the filtration barrier more easily than the neutral molecules, which in turn cross more easily than the negatively charged molecules. For example, plasma albumin, which is negatively charged, is repelled by the GBM. Defects in GBM allow albumin to pass the filtration barrier and results in high albumin content in urine. However, selective knockout of agrin had no effect on the glomerular filtration barrier in one study (Harvey et al. 2007).
There are a number of diseases that affect the glomerular BM. In mature GBM, the major collagen type IV molecule is the alpha-3 alpha-4 alpha-5 isoform, associated with laminin-521 (alpha-5 beta-2 gamma-1), nidogen and agrin heparan sulfate proteoglycans. Several hereditary glomerular diseases are linked to structural anomalies of GBM tissue-specific components; for example, the alpha-3 alpha-4 alpha-5 isoform of collagen type IV in Alport syndrome and thin basement membrane nephropathy (benign familial hematuria), and laminin in Pierson syndrome (Gubler 2008). Tumor necrosis factor-α has been shown to drive Alport glomerulosclerosis in mice by promoting podocyte apoptosis (Ryu et al. 2012). Another example is the Goodpasture’s antigen associated with Goodpasture’s syndrome that is the NC1 domain of the alpha-3 chain of collagen type IV found in the glomerular BM (Derry and Pusey 1994).
BM in lung
The BMs of the alveolus functions in cell adhesion for alveolar epithelial and endothelial cells, to facilitate gas exchange between the alveolar space and the alveolar capillaries (West and Mathieu-Costello 1999), to regulate cytokine and growth factor functions, and other alveolar cellular processes in the lung (Sannes and Wang 1997). The polarity of the lung is maintained by the BMs and they act as physical barriers between epithelium, endothelium and mesenchymal tissues.
Pulmonary alveoli have been shown to have a thinner side and a thicker side (Vaccaro and Brody 1981; Weibel 1973). The thinner side consists of alveolar epithelium and capillary endothelium separated only by a common fused BM that is thought to facilitate gas exchange. The thicker side consists of alveolar epithelium and capillary endothelium, each with their respective BMs (alveolar BM (Fig. 6) and capillary BM, respectively) separated by connective tissues within the interstitial space (Vaccaro and Brody 1981; Weibel 1973). These two lung BMs appear to have similar ultrastructure when examined with standard TEM techniques. However, staining with ruthenium red demonstrated that the two lung BMs have different ultrastructural characteristics and that the type and distribution of proteoglycans differs between alveolar BM and capillary BM (Vaccaro and Brody 1981). Otherwise, the component differences between these BMs have not been fully characterized. The integrity of the BMs maintains the normal lung architecture and the alveolar BM is crucial for restoration of alveolar epithelial homoeostasis following lung injury (Strieter and Mehrad 2009).

Transmission electron micrograph of alveolar basement membrane (BM) of rabbit lung at ×46,000. Shown is the “thicker side” of the alveolus where the alveolar BM and capillary BM are separated by an interstitial space containing collagen fibrils and other extracellular matrix materials. The alveolar epithelial (AE) type 1 cell rests on the BM with lamina lucida (arrows) and lamina densa (arrowhead). On the “thinner side” of the alveolus (not shown) the alveolar BM and capillary BM fuse, at least focally, to form a single BM separating alveolar epithelial type 1 cells and capillary endothelial cells—a BM morphological variation that is thought to facilitate gas exchange between the alveolar space and the alveolar capillaries (Vaccaro and Brody 1981)
Loss of alveolar BM/capillary BM integrity has been observed in idiopathic pulmonary fibrosis (IPF) (Chen et al. 2016). Mechanisms underlying this disruption have not been well defined. Specific alveolar BM components have been described, including the Goodpasture’s antigen associated with Goodpasture’s syndrome that is the NC1 domain of the alpha-3 chain of collagen type IV, which is also found in the glomerular BM (Derry and Pusey 1994). The absence of the basement membrane component nidogen-2, but not of nidogen-1, has been shown to result in increased lung metastases in mice (Mokkapati et al. 2012).
BM in the liver
BMs are found in blood and lymphatic vessels and around the bile ducts in human liver (Hahn et al. 1980; Mak and Mei 2017). Their presence in the tubular regions, particularly between sinusoids lining cells and hepatocytes, is still controversial (Schaffner and Poper 1963). Lack of a typical BM in the perisinusoidal space in normal liver (Fig. 7) is thought to allow the intimate contact between blood and parenchymal cells necessary for normal hepatocyte function. However, the appearance of a continuous perisinusoidal BM in experimental liver injury and in human liver fibrosis has been reported (Bucher 1963; Mak and Mei 2017). These disease-related changes may severely restrict the normal functions of the liver.

Transmission electron micrograph of rabbit liver hepatocyte, sinusoids and spaces of Disse at ×30,000 magnification. Hepatocytes are organized into plates separated by the space of Disse (D) from vascular channels termed sinusoids. Hepatocyte processes (arrowheads) extend into the space of Disse. Sinusoids have a discontinuous, fenestrated endothelial cell lining. Of note, there is no basement membrane between either hepatocytes or endothelial cells and the space of Disse—allowing direct cellular contact that is thought to facilitate hepatocyte functions such as detoxification, modification, and excretion of exogenous and endogenous substances
Thus, liver hepatocytes lack the typical electron-dense structure of BM in other organs and contains non-BM constituents such as collagen type I and fibronectin, in addition to some typical BM constituents (Martinez-Hernandez and Amenta 1995; Matsumoto et al. 1999). Sinusoidal endothelial cells in liver can secrete collagen type IV, laminin, nidogen, and perlecan (Wells 2008). Collagen type IV, laminin, and perlecan are also produced by perisinusoidal hepatic stellate cells (HSCs) (Wells 2008). In the portal tracts, biliary epithelial cells are the principal cells producing collagen type IV, laminin, and perlecan, while portal fibroblasts and myofibroblasts, when generated, also contribute to their production. Expression of collagen type IV along with increased deposition of laminin in the space of Disse results in the formation of a perisinusoidal BM in liver fibrosis (Mak et al. 2013). Laminin expression is not detected in the parenchyma of normal human liver, only in liver fibrogenesis, where β2 laminin chain may be deposited in the space Disse, along with collagen type IV and perlecan, forming a continuous basement membrane beneath the endothelium of liver sinusoids (Mak and Mei 2017).
BM in other organs
Most other organs in a human or animal, including brain, heart, gut (with variation in the different segments of the small intestine and the large intestine), pancreas, gall bladder, and testis, have BMs that are similar in overall composition to those that have been described for cornea, skin, lung, and kidney, but with tissue-specific alterations in BM components that facilitate the specific functions of these organs. A specific description of the differences in BM composition between these many different organs is beyond the scope of this review.
EBM injury, regeneration and fibrosis in different organs
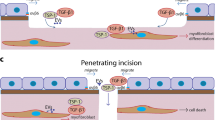
Corneal EBM injury and regeneration, and its relationship to organ fibrosis, is one of the best-characterized systems. Whether the injured cornea heals with transparency or with fibrosis and transparency depends on the type and level of injury (Torricelli et al. 2013b). Corneal stromal keratocytes are fibroblastic cells that are normally relatively quiescent and function to maintain the precise structure of the stromal extracellular matrix associated with transparency (Chaurasia et al. 2009; Hassell and Birk 2010; Ishizaki et al. 1993; Jester et al. 1995, 1999; Kaur et al. 2009). Corneal injuries “activate” keratocytes at the site of injury and in the proximate stroma to “corneal fibroblasts” that participate in the healing response and can differentiate into myofibroblasts when exposed to sustained transforming growth factor β1 or β2 (Jester et al. 1987; Kaur et al. 2009; Wilson 2012). In vitro cell culture experiments have identified several key growth factors such as TGFβ1, TGFβ2, and PDGF that play critical roles in mediating keratocyte differentiation to wound healing corneal fibroblast and myofibroblast phenotypes (Jester et al. 1987, 2002; Tuli et al. 2006; Wilson 2012). In addition, after corneal injury, bone marrow-derived fibrocytes penetrate the corneal stroma and differentiate into myofibroblasts when TGFβ and PDGF are present in the stroma at sufficient and sustained levels (Barbosa et al. 2010). It has been well documented that these key growth factors are produced in high levels by corneal epithelial cells but their penetration into the stroma is negligible when the EBM is intact (Fini 1999; Torricelli et al. 2013b; Wilson 2012). The corneal epithelium, like other epithelial layers in animals, is continuously subjected to physical, chemical, and biological insults. If an insult is sufficiently severe, the EBM is also injured, allowing the penetration of pro-fibrotic TGFβ, PDGF, and possibly other growth factors and cytokines, into the corneal stroma to initiate the development of corneal fibroblasts and myofibroblasts from local (keratocyte) and bone marrow-derived (fibrocyte) precursors (Torricelli et al. 2013b; Wilson 2012). If the EBM is promptly repaired, for example, after most simple corneal abrasions, the penetration of TGFβ and PDGF into the stroma is consequently cut off and the developing myofibroblast precursors undergo IL-1-mediated apoptosis (Kaur et al. 2009) before they become mature vimentin+ alpha-smooth muscle actin+ desmin+ myofibroblasts (that secrete large amounts disordered extracellular matrix) (Chaurasia et al. 2009), keratocytes repopulate the anterior stroma and transparency of the corneal stroma is maintained (Fig. 8a–c). If, however, repair of the EBM is sufficiently delayed, then TGFβ and PDGF continue to penetrate the stroma at high levels, resulting in the development of large numbers of stromal myofibroblasts, and the prodigious amounts disordered extracellular matrix they produce, results in fibrosis and loss of transparency that is crucial for corneal function (Fig. 8d–f) (Torricelli et al. 2013b; Wilson 2012). Delayed regeneration of EBM can result from mechanical factors such as corneal stromal surface irregularity produced by injury, surgery, infection, or disease (Netto et al. 2006). Another mechanism for delayed EBM regeneration, however, is likely insufficient stromal keratocyte contributions of basement membrane components needed for full restoration of EBM structure and function (Santhanam et al. 2015, 2017; Torricelli et al. 2015). Thus, keratocytes produce laminins, nidogen-1, nidogen-2, perlecan, and possibly other EBM components. The working hypothesis is that after corneal injury, the healed corneal epithelium lays down a self-polymerizing laminin nascent EBM and that this layer produces a barrier to the penetration of more posterior EBM components that must be provided, at least in part, by keratocytes (Santhanam et al. 2017; Wilson et al. 2017). If the original injury is sufficiently severe, resulting in substantial loss of adjacent keratocytes by apoptosis and/or necrosis (Marino et al. 2017a; Mohan et al. 2003; Wilson et al. 1996) and, therefore, there are diminished keratocyte contributions of components to EBM repair, then defective regeneration of the EBM promotes the development and persistence of myofibroblasts via ongoing penetration of TGFβ and PDGF into the stroma. These persistent myofibroblasts produce the fibrosis in the anterior subepithelial stroma. After a period of time, importantly without recurrent injury and typically measured for the cornea in many months to years, the normal mature EBM may be regenerated—likely via keratocyte penetration of the layer of myofibroblasts and their disordered extracellular matrix—where the keratocytes coordinate with the overlying epithelium to facilitate EBM regeneration. Once the EBM is fully repaired, myofibroblasts, deprived of their ongoing source of TGFβ, undergo apoptosis (Wilson et al. 2007). Subsequently, the anterior stroma is repopulated by keratocytes, which remove and reorganize the disordered extracellular matrix and restore corneal stromal transparency (Marino et al. 2017b; Wilson et al. 2017).

Regenerative vs. fibrotic repair of the rabbit cornea after injury. At 1 month after minor injuries to the rabbit cornea, such as epithelial abrasion or − 4.5 diopter photorefractive keratectomy (PRK) that is shown (a–c), in which the EBM and a small amount of the anterior stroma is ablated with the excimer laser and relatively few stromal keratocytes die by apoptosis or necrosis, transmission electron microscopy (TEM) shows that the EBM regenerates normally (a ×22,000 mag., arrows are lamina lucida and arrowheads are lamina densa), keratocytes repopulate the anterior stroma (b ×400 mag., arrows) and few, and in this case no, myofibroblasts are detected by staining for the alpha-smooth muscle actin (SMA) myofibroblast marker (b ×400 mag. showing DAPI stained keratocytes in the stroma (s). The cornea overlying the pupil (arrows) is transparent and iris details are clear when photographed with a slit lamp at 1 month after − 4.5D PRK (c ×20 mag.). After a more severe injury (such as high correction − 9 diopter PRK) (d–f), the EBM is not regenerated at 1 month after surgery and no lamina lucida or lamina densa is detected (d ×22,000 mag., arrows note no EBM beneath the epithelium) and myofibroblasts (arrowheads) with large amounts of rough endoplasmic reticulum fill the anterior stroma of the cornea. d The disorganization of the collagen in the stroma surrounding the myofibroblasts compared to a, where the collagen fibrils are uniform diameter and regularly packed—an important contributor to the transparency of the normal corneal stroma. After this level of injury (e ×400 mag.) the anterior stroma beneath the epithelium (the ongoing source of TGFβ that penetrates the stroma to maintain the viability of the myofibroblasts in the absence of normal EBM) has a layer of SMA+ myofibroblasts (arrows). A slit lamp photograph of the cornea at 1 month after surgery shows fibrosis (f ×20 mag., arrows delineate area of fibrosis that is also called haze) in the area of the previous PRK surgery. e = epithelium and s = stroma (a–e)
The importance of the coordination and interplay between the epithelial cells, stromal cells, bone marrow-derived cells, and the EBM in modulating transparency and fibrosis in the cornea at every stage of the corneal wound healing process, as well as in homeostasis in the normal uninjured cornea is remarkable and likely relevant to the interactions between epithelial cells, parenchymal cells, fibroblasts, endothelial cells, bone marrow-derived cells, and basement membranes that occur in other organs during homeostasis in normal tissues, and after injuries in which fibrosis may occur. Fibroblasts and other non-epithelial and non-parenchymal cells have been shown to produce basement membrane components in many other organs (El Ghalbzouri et al. 2005; El Ghalbzouri and Ponec 2004; Fleischmajer et al. 1995; Fox et al. 1991; Furuyama et al. 1997; Marinkovich et al. 1993; Simon-Assmann et al. 1998; Smola et al. 1998). Thus, keratinocyte-fibroblast interactions have been shown to be important in basement membrane generation in organotypic skin cultures (Smola et al. 1998). Similarly, assembly of the alveolar basement membrane after lung injury is likely orchestrated by cooperation between alveolar epithelial cells and pulmonary fibroblasts (Furuyama et al. 1997). In addition, fibrosis has been shown to resolve in other organs after removal of sources of chronic injury. For example, bleomycin-induced lung fibrosis in mice can reverse spontaneously after removal of the inciting agent (Cabrera et al. 2013; Lawson et al. 2005; Li et al. 2011). In humans, skin fibrosis associated with systemic sclerosis can at least partially resolve following neutralization of the antifibrinolytic function of plasminogen activator inhibitor 1 (Lemaire et al. 2016). Further research should be directed at fully understanding these critical cellular and extracellular matrix interactions that likely lie at the core of the development and resolution of fibrotic diseases that occur in many organs.
References
Abrahamson DR (2009) Development of kidney glomerular endothelial cells and their role in basement membrane assembly. Organogenesis 5:275–287
Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y (1999) Perlecan is essential for cartilage and cephalic development. Nat Genet 23:354–358
Aumailley M, Battaglia C, Mayer U, Reinhardt D, Nischt R, Timpl R, Fox JW (1993) Nidogen mediates the formation of ternary complexes of basement membrane components. Kidney Int 43:7–12
Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD (2005) A simplified laminin nomenclature. Matrix Biol 24:326–332
Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, Murshed M, Nischt R (2005) Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol 25:6846–6856
Barbosa FL, Chaurasia SS, Cutler A, Asosingh K, Kaur H, de Medeiros FW, Agrawal V, Wilson SE (2010) Corneal myofibroblast generation from bone marrow-derived cells. Exp Eye Res 91:92–96
Bechtel M, Keller MV, Bloch W, Sasaki T, Boukamp P, Zaucke F, Paulsson M, Nischt R (2012) Different domains in nidogen-1 and nidogen-2 drive basement membrane formation in skin organotypic cocultures. FASEB J 26:3637–3648
Behrens DT, Villone D, Koch M, Brunner G, Sorokin L, Robenek H, Bruckner-Tuderman L, Bruckner P, Hansen U (2012) The epidermal basement membrane is a composite of separate laminin- or collagen IV-containing networks connected by aggregated perlecan, but not by nidogens. J Biol Chem 287:18700–18,709
Benya PD, Padilla SR (1986) Isolation and characterization of type VIII collagen synthesized by cultured rabbit corneal endothelial cells. A conventional structure replaces the interrupted-helix model. J Biol Chem 261:4160–4169
Ben-Zvi A, Rodrigues MM, Krachmer JH, Fujikawa LS (1986) Immunohistochemical characterization of extracellular matrix in the developing human cornea. Curr Eye Res 5:105–117
Bonod-Bidaud C, Roulet M, Hansen U, Elsheikh A, Malbouyres M, Ricard-Blum S, Faye C, Vaganay E, Rousselle P, Ruggiero F (2012) In vivo evidence for a bridging role of a collagen V subtype at the epidermis-dermis interface. J Invest Dermatol 132:1841–1849
Boudreau NJ, Jones PL (1999) Extracellular matrix and integrin signaling: the shape of things to come. Biochem J 339(Pt 3):481–488
Breitkreutz D, Bohnert A, Herzmann E, Bowden PE, Boukamp P, Fusenig NE (1984) Differentiation specific functions in cultured and transplanted mouse keratinocytes: environmental influences on ultrastructure and keratin expression. Differentiation 26:154–169
Breitkreutz D, Stark HJ, Mirancea N, Tomakidi P, Steinbauer H, Fusenig NE (1997) Integrin and basement membrane normalization in mouse grafts of human keratinocytes—implications for epidermal homeostasis. Differentiation 61:195–209
Breitkreutz D, Schoop VM, Mirancea N, Baur M, Stark HJ, Fusenig NE (1998) Epidermal differentiation and basement membrane formation by HaCaT cells in surface transplants. Eur J Cell Biol 75:273–286
Breitkreutz D, Koxholt I, Thiemann K, Nischt R (2013) Skin basement membrane: the foundation of epidermal integrity-BM functions and diverse roles of bridging molecules nidogen and perlecan. Biomed Res Int 2013:179784
Bucher NL (1963) Regeneration of mammalian liver. Int Rev Cytol 15:245–300
Cabrera S, Selman M, Lonzano-Bolanos A, Konishi K, Richards TJ, Kaminski N, Pardo A (2013) Gene expression profiles reveal molecular mechanisms involved in the progression and resolution of bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol 304:L593–L601
Cameron JD, Skubitz AP, Furcht LT (1991) Type IV collagen and corneal epithelial adhesion and migration. Effects of type IV collagen fragments and synthetic peptides on rabbit corneal epithelial cell adhesion and migration in vitro. Invest Ophthalmol Vis Sci 32:2766–2773
Carlin B, Jaffe R, Bender B, Chung AE (1981) Entactin, a novel basal lamina-associated sulfated glycoprotein. J Biol Chem 256:5209–5214
Carlson EC, Meezan E, Brendel K, Kenney MC (1981) Ultrastructural analyses of control and enzyme-treated isolated renal basement membranes. Anat Rec 200:421–436
Chaurasia SS, Kaur H, de Medeiros FW, Smith SD, Wilson SE (2009) Dynamics of the expression of intermediate filaments vimentin and desmin during myofibroblast differentiation after corneal injury. Exp Eye Res 89:133–139
Chen L, Hazlett LD (2001) Human corneal epithelial extracellular matrix perlecan serves as a site for Pseudomonas aeruginosa binding. Curr Eye Res 22:19–27
Chen H, Qu J, Huang X, Kurundkar A, Zhu L, Yang N, Venado A, Ding Q, Liu G, Antony VB, Thannickal VJ, Zhou Y (2016) Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat Commun 7:12564
Chi HH, Teng CC, Katzin HM (1958) Histopathology of primary endothelial-epithelial dystrophy of the cornea. Am J Ophthalmol 45:518–535
Chung AE, Freeman IL, Braginski JE (1977) A novel extracellular membrane elaborated by a mouse embryonal carcinoma-derived cell line. Biochem Biophys Res Commun 79:859–868
Chung AE, Jaffe R, Freeman IL, Vergnes JP, Braginski JE, Carlin B (1979) Properties of a basement membrane-related glycoprotein synthesized in culture by a mouse embryonal carcinoma-derived cell line. Cell 16:277–287
Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R (1999) Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol 147:1109–1122
Denzer AJ, Gesemann M, Schumacher B, Ruegg MA (1995) An amino-terminal extension is required for the secretion of chick agrin and its binding to extracellular matrix. J Cell Biol 131:1547–1560
Derry CJ, Pusey CD (1994) Tissue-specific distribution of the Goodpasture antigen demonstrated by 2-D electrophoresis and western blotting. Nephrol Dial Transplant 9:355–361
Di Nunzio F, Maruggi G, Ferrari S, Di Iorio E, Poletti V, Garcia M, Del Rio M, De Luca M, Larcher F, Pellegrini G, Mavilio F (2008) Correction of laminin-5 deficiency in human epidermal stem cells by transcriptionally targeted lentiviral vectors. Mol Ther 16:1977–1985
Dong S, Cole GJ, Halfter W (2003) Expression of collagen XVIII and localization of its glycosaminoglycan attachment sites. J Biol Chem 278:1700–1707
El Ghalbzouri A, Ponec M (2004) Diffusible factors released by fibroblasts support epidermal morphogenesis and deposition of basement membrane components. Wound Repair Regen 12:359–367
El Ghalbzouri A, Jonkman MF, Dijkman R, Ponec M (2005) Basement membrane reconstruction in human skin equivalents is regulated by fibroblasts and/or exogenously activated keratinocytes. J Invest Dermatol 124:79–86
Ettner N, Gohring W, Sasaki T, Mann K, Timpl R (1998) The N-terminal globular domain of the laminin alpha1 chain binds to alpha1beta1 and alpha2beta1 integrins and to the heparan sulfate-containing domains of perlecan. FEBS Lett 430:217–221
Filenius S, Hormia M, Rissanen J, Burgeson RE, Yamada Y, Araki-Sasaki K, Nakamura M, Virtanen I, Tervo T (2001) Laminin synthesis and the adhesion characteristics of immortalized human corneal epithelial cells to laminin isoforms. Exp Eye Res 72:93–103
Fini ME (1999) Keratocyte and fibroblast phenotypes in the repairing cornea. Prog Retin Eye Res 18:529–551
Fleischmajer R, Schechter A, Bruns M, Perlish JS, Macdonald ED, Pan TC, Timpl R, Chu ML (1995) Skin fibroblasts are the only source of nidogen during early basal lamina formation in vitro. J Invest Dermatol 105:597–601
Fox JW, Mayer U, Nischt R, Aumailley M, Reinhardt D, Wiedemann H, Mann K, Timpl R, Krieg T, Engel J et al (1991) Recombinant nidogen consists of three globular domains and mediates binding of laminin to collagen type IV. EMBO J 10:3137–3146
Fritsch A, Spassov S, Elfert S, Schlosser A, Gache Y, Meneguzzi G, Bruckner-Tuderman L (2009) Dominant-negative effects of COL7A1 mutations can be rescued by controlled overexpression of normal collagen VII. J Biol Chem 284:30248–30256
Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, Niemela M, Ilves M, Li E, Pihlajaniemi T, Olsen BR (2002) Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J 21:1535–1544
Furuyama A, Kimata K, Mochitate K (1997) Assembly of basement membrane in vitro by cooperation between alveolar epithelial cells and pulmonary fibroblasts. Cell Struct Funct 22:603–614
Gersdorff N, Kohfeldt E, Sasaki T, Timpl R, Miosge N (2005) Laminin gamma3 chain binds to nidogen and is located in murine basement membranes. J Biol Chem 280:22146–22153
Gersdorff N, Otto S, Roediger M, Kruegel J, Miosge N (2007) The absence of one or both nidogens does not alter basement membrane composition in adult murine kidney. Histol Histopathol 22:1077–1084
Gipson IK, Spurr-Michaud SJ, Tisdale AS (1988) Hemidesmosomes and anchoring fibril collagen appear synchronously during development and wound healing. Dev Biol 126:253–262
Groffen AJ, Ruegg MA, Dijkman H, van de Velden TJ, Buskens CA, van den Born J, Assmann KJ, Monnens LA, Veerkamp JH, van den Heuvel LP (1998) Agrin is a major heparan sulfate proteoglycan in the human glomerular basement membrane. J Histochem Cytochem 46:19–27
Gubler MC (2008) Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol 4:24–37
Hahn E, Wick G, Pencev D, Timpl R (1980) Distribution of basement membrane proteins in normal and fibrotic human liver: collagen type IV, laminin, and fibronectin. Gut 21:63–71
Halfter W, Dong S, Schurer B, Cole GJ (1998) Collagen XVIII is a basement membrane heparan sulfate proteoglycan. J Biol Chem 273:25404–25,412
Halfter W, Oertle P, Monnier CA, Camenzind L, Reyes-Lua M, Hu H, Candiello J, Labilloy A, Balasubramani M, Henrich PB, Plodinec M (2015) New concepts in basement membrane biology. FEBS J 282:4466–4479
Hammers CM, Stanley JR (2016) Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol 11:175–197
Harvey SJ, Jarad G, Cunningham J, Rops AL, van der Vlag J, Berden JH, Moeller MJ, Holzman LB, Burgess RW, Miner JH (2007) Disruption of glomerular basement membrane charge through podocyte-specific mutation of agrin does not alter glomerular permselectivity. Am J Pathol 171:139–152
Hassell JR, Birk DE (2010) The molecular basis of corneal transparency. Exp Eye Res 91:326–335
Hassell JR, Robey PG, Barrach HJ, Wilczek J, Rennard SI, Martin GR (1980) Isolation of a heparan sulfate-containing proteoglycan from basement membrane. Proc Natl Acad Sci U S A 77:4494–4498
Hassell JR, Schrecengost PK, Rada JA, SundarRaj N, Sossi G, Thoft RA (1992) Biosynthesis of stromal matrix proteoglycans and basement membrane components by human corneal fibroblasts. Invest Ophthalmol Vis Sci 33:547–557
Heidet L, Cai Y, Guicharnaud L, Antignac C, Gubler MC (2000) Glomerular expression of type IV collagen chains in normal and X-linked Alport syndrome kidneys. Am J Pathol 156:1901–1910
Ho MS, Bose K, Mokkapati S, Nischt R, Smyth N (2008) Nidogens-extracellular matrix linker molecules. Microsc Res Tech 71:387–395
Inomata T, Ebihara N, Funaki T, Matsuda A, Watanabe Y, Ning L, Xu Z, Murakami A, Arikawa-Hirasawa E (2012) Perlecan-deficient mutation impairs corneal epithelial structure. Invest Ophthalmol Vis Sci 53:1277–1284
Ishizaki M, Zhu G, Haseba T, Shafer SS, Kao WW (1993) Expression of collagen I, smooth muscle alpha-actin, and vimentin during the healing of alkali-burned and lacerated corneas. Invest Ophthalmol Vis Sci 34:3320–3328
Jester JV, Rodrigues MM, Herman IM (1987) Characterization of avascular corneal wound healing fibroblasts. New insights into the myofibroblast. Am J Pathol 127:140–148
Jester JV, Petroll WM, Barry PA, Cavanagh HD (1995) Temporal, 3-dimensional, cellular anatomy of corneal wound tissue. J Anat 186(Pt 2):301–311
Jester JV, Huang J, Barry-Lane PA, Kao WW, Petroll WM, Cavanagh HD (1999) Transforming growth factor(beta)-mediated corneal myofibroblast differentiation requires actin and fibronectin assembly. Invest Ophthalmol Vis Sci 40:1959–1967
Jester JV, Huang J, Petroll WM, Cavanagh HD (2002) TGFbeta induced myofibroblast differentiation of rabbit keratocytes requires synergistic TGFbeta, PDGF and integrin signaling. Exp Eye Res 75:645–657
Johnson DH, Bourne WM, Campbell RJ (1982) The ultrastructure of Descemet’s membrane. II Aphakic bullous keratopathy. Arch Ophthalmol 100:1948–1951
Kabosova A, Azar DT, Bannikov GA, Campbell KP, Durbeej M, Ghohestani RF, Jones JC, Kenney MC, Koch M, Ninomiya Y, Patton BL, Paulsson M, Sado Y, Sage EH, Sasaki T, Sorokin LM, Steiner-Champliaud MF, Sun TT, Sundarraj N, Timpl R, Virtanen I, Ljubimov AV (2007) Compositional differences between infant and adult human corneal basement membranes. Invest Ophthalmol Vis Sci 48:4989–4999
Kanwar YS, Danesh FR, Chugh SS (2007) Contribution of proteoglycans towards the integrated functions of renal glomerular capillaries: a historical perspective. Am J Pathol 171:9–13
Kapoor R, Bornstein P, Sage EH (1986) Type VIII collagen from bovine Descemet’s membrane: structural characterization of a triple-helical domain. Biochemistry 25:3930–3937
Kaur H, Chaurasia SS, Agrawal V, Suto C, Wilson SE (2009) Corneal myofibroblast viability: opposing effects of IL-1 and TGF beta1. Exp Eye Res 89:152–158
Kefalides NA, Denduchis B (1969) Structural components of epithelial and endothelial basement membranes. Biochemistry 8:4613–4621
Kefalides NA, Cameron JD, Tomichek EA, Yanoff M (1976) Biosynthesis of basement membrane collagen by rabbit corneal endothelium in vitro. J Biol Chem 251:730–733
Khoshnoodi J, Pedchenko V, Hudson BG (2008) Mammalian collagen IV. Microsc Res Tech 71:357–370
Kimura N, Toyoshima T, Kojima T, Shimane M (1998) Entactin-2: a new member of basement membrane protein with high homology to entactin/nidogen. Exp Cell Res 241:36–45
Kleinman HK, Martin GR (2005) Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol 15:378–386
Kleinman HK, McGarvey ML, Liotta LA, Robey PG, Tryggvason K, Martin GR (1982) Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry 21:6188–6193
Kruegel J, Miosge N (2012) Basement membrane components are key players in specialized extracellular matrices. Cell Mol Life Sci 67:2879–2895
Labermeier U, Kenney MC (1983) The presence of EC collagen and type IV collagen in bovine Descemet’s membranes. Biochem Biophys Res Commun 116:619–625
Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, Collins RD, Hull WM, Glasser SW, Whitsett JA, Blackwell TS (2005) Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol 167:1267–1277
LeBleu VS, Macdonald B, Kalluri R (2007) Structure and function of basement membranes. Exp Biol Med 232:1121–1129
Lemaire R, Burwell T, Sun H, Delaney T, Bakken J, Cheng L, Rebelatto MC, Czapiga M, de-Mendez I, Coyle AJ, Herbst R, Lafyatis R, Connor J (2016) Resolution of skin fibrosis by neutralization of the antifibrinolytic function of plasminogen activator inhibitor 1. Arthritis Rheumatol 68:473–483
Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, Minoo P (2011) Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 121:277–287
Limat A, Stockhammer E, Breitkreutz D, Schaffner T, Egelrud T, Salomon D, Fusenig NE, Braathen LR, Hunziker T (1996) Endogeneously regulated site-specific differentiation of human palmar skin keratinocytes in organotypic cocultures and in nude mouse transplants. Eur J Cell Biol 69:245–258
Lin HC, Chang JH, Jain S, Gabison EE, Kure T, Kato T, Fukai N, Azar DT (2001) Matrilysin cleavage of corneal collagen type XVIII NC1 domain and generation of a 28-kDa fragment. Invest Ophthalmol Vis Sci 42:2517–2524
Ljubimov AV, Burgeson RE, Butkowski RJ, Michael AF, Sun TT, Kenney MC (1995) Human corneal basement membrane heterogeneity: topographical differences in the expression of type IV collagen and laminin isoforms. Lab Invest 72:461–473
Maatta M, Heljasvaara R, Pihlajaniemi T, Uusitalo M (2007) Collagen XVIII/endostatin shows a ubiquitous distribution in human ocular tissues and endostatin-containing fragments accumulate in ocular fluid samples. Graefe’s archive for clinical and experimental ophthalmology. Albrecht Von Graefes Arch Klin Exp Ophthalmol 245:74–81
Mak KM, Mei R (2017) Basement membrane type IV collagen and laminin: an overview of their biology and value as fibrosis biomarkers of liver disease. Anat Rec (Hoboken) 300:1371–1390
Mak KM, Chen LL, Lee TF (2013) Codistribution of collagen type IV and laminin in liver fibrosis of elderly cadavers: immunohistochemical marker of perisinusoidal basement membrane formation. Anat Rec (Hoboken) 296:953–964
Marinkovich MP, Keene DR, Rimberg CS, Burgeson RE (1993) Cellular origin of the dermal-epidermal basement membrane. Dev Dyn 197:25,5–25,2267
Marino GK, Santhiago MR, Santhanam A, Lassance L, Thangavadivel S, Medeiros CS, Bose K, Tam KP, Wilson SE (2017a) Epithelial basement membrane injury and regeneration modulates corneal fibrosis after pseudomonas corneal ulcers in rabbits. Exp Eye Res 161:101–105
Marino GK, Santhiago MR, Santhanam A, Torricelli AAM, Wilson SE (2017b) Regeneration of defective epithelial basement membrane and restoration of corneal transparency after photorefractive keratectomy. J Refract Surg 33:337–346
Marneros AG, Olsen BR (2003) Age-dependent iris abnormalities in collagen XVIII/endostatin deficient mice with similarities to human pigment dispersion syndrome. Invest Ophthalmol Vis Sci 44:2367–2372
Marneros AG, Keene DR, Hansen U, Fukai N, Moulton K, Goletz PL, Moiseyev G, Pawlyk BS, Halfter W, Dong S, Shibata M, Li T, Crouch RK, Bruckner P, Olsen BR (2004) Collagen XVIII/endostatin is essential for vision and retinal pigment epithelial function. EMBO J 23:89–99
Martinez-Hernandez A, Amenta PS (1995) The extracellular matrix in hepatic regeneration. FASEB J 9:1401–1410
Matejas V, Hinkes B, Alkandari F, Al-Gazali L, Annexstad E, Aytac MB, Barrow M, Blahova K, Bockenhauer D, Cheong HI, Maruniak-Chudek I, Cochat P, Dotsch J, Gajjar P, Hennekam RC, Janssen F, Kagan M, Kariminejad A, Kemper MJ, Koenig J, Kogan J, Kroes HY, Kuwertz-Broking E, Lewanda AF, Medeira A, Muscheites J, Niaudet P, Pierson M, Saggar A, Seaver L, Suri M, Tsygin A, Wuhl E, Zurowska A, Uebe S, Hildebrandt F, Antignac C, Zenker M (2010) Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat 31:992–1002
Matsumoto S, Yamamoto K, Nagano T, Okamoto R, Ibuki N, Tagashira M, Tsuji T (1999) Immunohistochemical study on phenotypical changes of hepatocytes in liver disease with reference to extracellular matrix composition. Liver 19:32–38
May CA (2012) Distribution of nidogen in the murine eye and ocular phenotype of the nidogen-1 knockout mouse. ISRN Ophthalmol 2012:378641
Medeiros CS, Lassance L, Saikia P, Wilson SE (2018) Posterior stromal keratocyte apoptosis triggered by mechanical endothelial injury and nidogen-1 production in the cornea. Exp Eye Res 172:30–35
Menzel O, Bekkeheien RC, Reymond A, Fukai N, Boye E, Kosztolanyi G, Aftimos S, Deutsch S, Scott HS, Olsen BR, Antonarakis SE, Guipponi M (2004) Knobloch syndrome: novel mutations in COL18A1, evidence for genetic heterogeneity, and a functionally impaired polymorphism in endostatin. Hum Mutat 23:77–84
Michelacci YM (2003) Collagens and proteoglycans of the corneal extracellular matrix. Braz J Med Biol Res 36:1037–1046
Miner JH (2012) The glomerular basement membrane. Exp Cell Res 318:973–978
Miner JH, Li C (2000) Defective glomerulogenesis in the absence of laminin alpha5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol 217:278–289
Miner JH, Sanes JR (1994) Collagen IV alpha 3, alpha 4, and alpha 5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J Cell Biol 127:879–891
Miner JH, Yurchenco PD (2004) Laminin functions in tissue morphogenesis. Annu Rev. Cell Dev Biol 20:255–284
Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR (1997) The laminin alpha chains: expression, developmental transitions, and chromosomal locations of alpha1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel alpha3 isoform. J Cell Biol 137:685–701
Miosge N, Sasaki T, Timpl R (2002) Evidence of nidogen-2 compensation for nidogen-1 deficiency in transgenic mice. Matrix Biol 21:611–621
Miosge N, Simniok T, Sprysch P, Herken R (2003) The collagen type XVIII endostatin domain is co-localized with perlecan in basement membranes in vivo. J Histochem Cytochem 51:285–296
Mirancea N, Hausser I, Metze D, Stark HJ, Boukamp P, Breitkreutz D (2007) Junctional basement membrane anomalies of skin and mucosa in lipoid proteinosis (hyalinosis cutis et mucosae). J Dermatol Sci 45:175–185
Mohan RR, Hutcheon AE, Choi R, Hong J, Lee J, Mohan RR, Ambrosio R Jr, Zieske JD, Wilson SE (2003) Apoptosis, necrosis, proliferation, and myofibroblast generation in the stroma following LASIK and PRK. Exp Eye Res 76:71–87
Mokkapati S, Bechtel M, Reibetanz M, Miosge N, Nischt R (2012) Absence of the basement membrane component nidogen 2, but not of nidogen 1, results in increased lung metastasis in mice. J Histochem Cytochem 60:280–289
Mueller MM, Fusenig NE (2004) Friends or foes—bipolar effects of the tumor stroma in cancer. Nat Rev Cancer 4:839–849
Murauer EM, Gache Y, Gratz IK, Klausegger A, Muss W, Gruber C, Meneguzzi G, Hintner H, Bauer JW (2011) Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Invest Dermatol 131:74–83
Murphy C, Alvarado J, Juster R (1984) Prenatal and postnatal growth of the human Descemet’s membrane. Invest Ophthalmol Vis Sci 25:1402–1415
Netto MV, Mohan RR, Sinha S, Sharma A, Dupps W, Wilson SE (2006) Stromal haze, myofibroblasts, and surface irregularity after PRK. Exp Eye Res 82:788–797
Nystrom A, Bornert O, Kuhl T (2017) Cell therapy for basement membrane-linked diseases. Matrix Biol 57–58:124–139
Orkin RW, Gehron P, McGoodwin EB, Martin GR, Valentine T, Swarm R (1977) A murine tumor producing a matrix of basement membrane. J Exp Med 145:204–220
Paulsson M (1988) The role of Ca2+ binding in the self-aggregation of laminin-nidogen complexes. J Biol Chem 263:5425–5430
Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U (2004) Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 131:1619–1628
Qiao H, Shibaki A, Long HA, Wang G, Li Q, Nishie W, Abe R, Akiyama M, Shimizu H, McMillan JR (2009) Collagen XVII participates in keratinocyte adhesion to collagen IV, and in p38MAPK-dependent migration and cell signaling. J Invest Dermatol 129:2288–2295
Reiser J, Altintas MM (2016) Podocytes. F1000Res :5. https://doi.org/10.12688/f1000research.7255.1
Ryu M, Mulay SR, Miosge N, Gross O, Anders HJ (2012) Tumor necrosis factor-α drives Alport glomerulosclerosis in mice by promoting podocyte apoptosis. J Pathol 226:120–131
Saarela J, Ylikarppa R, Rehn M, Purmonen S, Pihlajaniemi T (1998) Complete primary structure of two variant forms of human type XVIII collagen and tissue-specific differences in the expression of the corresponding transcripts. Matrix Biol 16:319–328
Sage H, Pritzl P, Bornstein P (1981) Secretory phenotypes of endothelial cells in culture: comparison of aortic, venous, capillary, and corneal endothelium. Arteriosclerosis 1:427–442
Salmivirta K, Talts JF, Olsson M, Sasaki T, Timpl R, Ekblom P (2002) Binding of mouse nidogen-2 to basement membrane components and cells and its expression in embryonic and adult tissues suggest complementary functions of the two nidogens. Exp Cell Res 279:188–201
Sannes PL, Wang J (1997) Basement membranes and pulmonary development. Exp Lung Res 23:101–108
Santhanam A, Torricelli AA, Wu J, Marino GK, Wilson SE (2015) Differential expression of epithelial basement membrane components nidogens and perlecan in corneal stromal cells in vitro. Mol Vis 21:1318–1327
Santhanam A, Marino GK, Torricelli AA, Wilson SE (2017) EBM regeneration and changes in EBM component mRNA expression in stromal cells after corneal injury. Mol Vis 23:39–51
Sawada H (1982) The fine structure of the bovine Descemet’s membrane with special reference to biochemical nature. Cell Tissue Res 226:241–255
Schaffner F, Poper H (1963) Capillarization of hepatic sinusoids in man. Gastroenterology 44:239–242
Schubert D, Kimura H (1991) Substratum-growth factor collaborations are required for the mitogenic activities of activin and FGF on embryonal carcinoma cells. J Cell Biol 114:841–846
Shuttleworth CA (1997) Type VIII collagen. Int J Biochem Cell Biol 29:1145–1148
Simon-Assmann P, Lefebvre O, Bellissent-Waydelich A, Olsen J, Orian-Rousseau V, De Arcangelis A (1998) The laminins: role in intestinal morphogenesis and differentiation. Ann N Y Acad Sci 859:46–64
Smola H, Stark HJ, Thiekotter G, Mirancea N, Krieg T, Fusenig NE (1998) Dynamics of basement membrane formation by keratinocyte-fibroblast interactions in organotypic skin culture. Exp Cell Res 239:399–410
Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D (1999) Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol 144:151–160
Sonnenberg A, Calafat J, Janssen H, Daams H, van der Raaij-Helmer LM, Falcioni R, Kennel SJ, Aplin JD, Baker J, Loizidou M et al (1991) Integrin alpha 6/beta 4 complex is located in hemidesmosomes, suggesting a major role in epidermal cell-basement membrane adhesion. J Cell Biol 113:907–917
Stark HJ, Baur M, Breitkreutz D, Mirancea N, Fusenig NE (1999) Organotypic keratinocyte cocultures in defined medium with regular epidermal morphogenesis and differentiation. J Invest Dermatol 112:681–691
Sterk LM, Geuijen CA, Oomen LC, Calafat J, Janssen H, Sonnenberg A (2000) The tetraspan molecule CD151, a novel constituent of hemidesmosomes, associates with the integrin alpha6beta4 and may regulate the spatial organization of hemidesmosomes. J Cell Biol 149:969–982
Sterk LM, Geuijen CA, van den Berg JG, Claessen N, Weening JJ, Sonnenberg A (2002) Association of the tetraspanin CD151 with the laminin-binding integrins alpha3beta1, alpha6beta1, alpha6beta4 and alpha7beta1 in cells in culture and in vivo. J Cell Sci 115:1161–1173
Strieter RM, Mehrad B (2009) New mechanisms of pulmonary fibrosis. Chest 136:1364–1370
Sugawara K, Tsuruta D, Ishii M, Jones JC, Kobayashi H (2008) Laminin-332 and -511 in skin. Exp Dermatol 17:473–480
Suzuki OT, Sertie AL, Der Kaloustian VM, Kok F, Carpenter M, Murray J, Czeizel AE, Kliemann SE, Rosemberg S, Monteiro M, Olsen BR, Passos-Bueno MR (2002) Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. Am J Hum Genet 71:1320–1329
Tervo K, Tervo T, van Setten GB, Virtanen I (1991) Integrins in human corneal epithelium. Cornea 10:461–465
Tiedemann K, Sasaki T, Gustafsson E, Gohring W, Batge B, Notbohm H, Timpl R, Wedel T, Schlotzer-Schrehardt U, Reinhardt DP (2005) Microfibrils at basement membrane zones interact with perlecan via fibrillin-1. J Biol Chem 280:11404–11,412
Timpl R (1989) Structure and biological activity of basement membrane proteins. Eur J Biochem 180:487–502
Timpl R, Brown JC (1996) Supramolecular assembly of basement membranes. Bioessays 18:123–132
Timpl R, Rohde H, Robey PG, Rennard SI, Foidart JM, Martin GR (1979) Laminin—a glycoprotein from basement membranes. J Biol Chem 254:9933–9937
Torricelli AA, Singh V, Agrawal V, Santhiago MR, Wilson SE (2013a) Transmission electron microscopy analysis of epithelial basement membrane repair in rabbit corneas with haze. Invest Ophthalmol Vis Sci 54:4026–4033
Torricelli AA, Singh V, Santhiago MR, Wilson SE (2013b) The corneal epithelial basement membrane: structure, function, and disease. Invest Ophthalmol Vis Sci 54:6390–6400
Torricelli AA, Marino GK, Santhanam A, Wu J, Singh A, Wilson SE (2015) Epithelial basement membrane proteins perlecan and nidogen-2 are up-regulated in stromal cells after epithelial injury in human corneas. Exp Eye Res 134:33–38
Tsen G, Halfter W, Kroger S, Cole GJ (1995) Agrin is a heparan sulfate proteoglycan. J Biol Chem 270:3392–3399
Tuli SS, Liu R, Chen C, Blalock TD, Goldstein M, Schultz GS (2006) Immunohistochemical localization of EGF, TGF-alpha, TGF-beta, and their receptors in rat corneas during healing of excimer laser ablation. Curr Eye Res 31:709–719
Tuori A, Uusitalo H, Burgeson RE, Terttunen J, Virtanen I (1996) The immunohistochemical composition of the human corneal basement membrane. Cornea 15:286–294
Uitto J, Has C, Vahidnezhad H, Youssefian L, Bruckner-Tuderman L (2017) Molecular pathology of the basement membrane zone in heritable blistering diseases: the paradigm of epidermolysis bullosa. Matrix Biol 57–58:76–85
Vaccaro CA, Brody JS (1981) Structural features of alveolar wall basement membrane in the adult rat lung. J Cell Biol 91:427–437
Villone D, Fritsch A, Koch M, Bruckner-Tuderman L, Hansen U, Bruckner P (2008) Supramolecular interactions in the dermo-epidermal junction zone: anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J Biol Chem 283:24506–24,513
Virtanen I, Tervo K, Korhonen M, Paallysaho T, Tervo T (1992) Integrins as receptors for extracellular matrix proteins in human cornea. Acta Ophthalmol Suppl:18–21
Voskarides K, Damianou L, Neocleous V, Zouvani I, Christodoulidou S, Hadjiconstantinou V, Ioannou K, Athanasiou Y, Patsias C, Alexopoulos E, Pierides A, Kyriacou K, Deltas C (2007) COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol: JASN 18:3004–3016
Weibel ER (1973) Morphological basis of alveolar-capillary gas exchange. Physiol Rev 53:419–495
Wells RG (2008) Cellular sources of extracellular matrix in hepatic fibrosis. Clin Liver Dis 12:759–768 viii
West JB, Mathieu-Costello O (1999) Structure, strength, failure, and remodeling of the pulmonary blood-gas barrier. Annu Rev Physiol 61:543–572
Wilson SE (2012) Corneal myofibroblast biology and pathobiology: generation, persistence, and transparency. Exp Eye Res 99:78–88
Wilson SE, He YG, Weng J, Li Q, McDowall AW, Vital M, Chwang EL (1996) Epithelial injury induces keratocyte apoptosis: hypothesized role for the interleukin-1 system in the modulation of corneal tissue organization and wound healing. Exp Eye Res 62:325–327
Wilson SE, Chaurasia SS, Medeiros FW (2007) Apoptosis in the initiation, modulation and termination of the corneal wound healing response. Exp Eye Res 85:305–311
Wilson SE, Marino GK, Torricelli AAM, Medeiros CS (2017) Injury and defective regeneration of the epithelial basement membrane in corneal fibrosis: a paradigm for fibrosis in other organs? Matrix Biol 64:17–26
Yurchenco PD, Tsilibary EC, Charonis AS, Furthmayr H (1986) Models for the self-assembly of basement membrane. J Histochem Cytochem 34:93–102
Zhang J, Patel DV (2015) The pathophysiology of Fuchs’ endothelial dystrophy—a review of molecular and cellular insights. Exp Eye Res 130:97–105
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported in part by US Public Health Service grants RO1EY10056 (SEW) and P30-EY025585 from the National Eye Institute, National Institutes of Health, Bethesda, MD.
Rights and permissions
About this article

Cite this article
Saikia, P., Medeiros, C.S., Thangavadivel, S. et al. Basement membranes in the cornea and other organs that commonly develop fibrosis. Cell Tissue Res 374, 439–453 (2018). https://doi.org/10.1007/s00441-018-2934-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-018-2934-7