
Abstract
The epithelial-mesenchymal transition (EMT) describes the global process by which stationary epithelial cells undergo phenotypic changes, including the loss of cell-cell adhesion and apical-basal polarity, and acquire mesenchymal characteristics that confer migratory capacity. EMT and its converse, MET (mesenchymal-epithelial transition), are integral stages of many physiologic processes and, as such, are tightly coordinated by a host of molecular regulators. Converging lines of evidence have identified EMT as a component of cutaneous wound healing, during which otherwise stationary keratinocytes (the resident skin epithelial cells) migrate across the wound bed to restore the epidermal barrier. Moreover, EMT plays a role in the development of scarring and fibrosis, as the matrix-producing myofibroblasts arise from cells of the epithelial lineage in response to injury but are pathologically sustained instead of undergoing MET or apoptosis. In this review, we summarize the role of EMT in physiologic repair and pathologic fibrosis of tissues and organs. We conclude that further investigation into the contribution of EMT to the faulty repair of fibrotic wounds might identify components of EMT signaling as common therapeutic targets for impaired healing in many tissues.

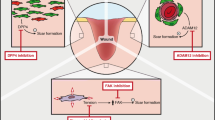
Model for injury-triggered EMT activation in physiologic wound repair (left) and fibrotic wound healing (right)
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The epithelial-mesenchymal transition (EMT) is a process during which epithelial cells gradually transform into mesenchymal-like cells and lose their epithelial functionality and characteristics. Converging lines of evidence suggest that EMT plays a role in both physiologic and pathologic healing. In this review, we summarize findings from animal and human wound-healing models supporting the importance of the proper execution of EMT in achieving successful tissue repair following injury. For instance, during cutaneous wound healing, epidermal keratinocytes undergo EMT by losing their adherent epithelial phenotype to become motile cells that have a mesenchymal phenotype and that migrate across the wound bed (Yan et al. 2010). We discuss several growth factors common to both wound healing and EMT, such as fibroblast growth factor (FGF), hepatocyte growth factor (HGF), epidermal growth factor (EGF), and transforming growth factor-beta (TGFβ), and highlight shared signaling pathways.
Whereas EMT is necessary for proper re-epithelialization and extracellular matrix (ECM) deposition, an uncontrolled continued transition from epithelial cells to myofibroblasts can result in fibrosis. We discuss the role of EMT in generating myofibroblasts from resident epithelial cells during the maturation phase of wound healing. We summarize evidence that sustained EMT is a key mechanism underlying the fibrotic pathology of multiple organs including the skin. The role of EMT in the pathophysiology of renal, pulmonary, cardiac, and liver fibrosis, cutaneous scleroderma, and impaired wound healing are also discussed.
Global features of EMT
EMT is often divided by biological context into three subtypes: Type I, which occurs during embryogenesis; Type II, which takes place during tissue repair; and Type III, which is involved in the metastatic spread of cancer. The three types of EMT have a shared outcome: the production of motile cells with a mesenchymal phenotype from otherwise classically adherent epithelial cells with apical-basal polarity (Kalluri and Neilson 2003). However, in contrast to Types I and III, Type II EMT is instigated exclusively by damage and inflammation (Volk et al. 2013).
The first step of EMT is the loss of epithelial cell markers, one of the most notable of which is the decreased expression of E-cadherin (Whiteman et al. 2008). E-cadherin is responsible for maintaining the lateral contacts of the epithelial cells via adherens junctions and for the cell adhesion and relative immobility in the tissue (Huang et al. 2012; Moreno-Bueno et al. 2008; Qin et al. 2005). E-cadherin downregulation is also mediated through the upregulation of vimentin, an intermediate filament that decreases E-cadherin trafficking to the cell surface (Mendez et al. 2010). The cell then progresses towards a mesenchymal phenotype by gaining mesenchymal markers and capabilities (Lee et al. 2006). This change is orchestrated by the temporally regulated expression of proteins, including neural cadherin (N-cadherin), vimentin, integrin, fibronectin, and matrix metalloproteinases (MMPs; Huang et al. 2012; Thiery and Sleeman 2006; Wheelock et al. 2008). Integrins that interact with ECM components such as fibronectin are then upregulated to increase motility (Maschler et al. 2005; Yang et al. 2009). A driving force behind this motility is the loss of the polarized cytoskeleton in epithelial cells, and the development of lamellipodia in the advancing edge of the transitioning mesenchymal cells (Takenawa and Suetsugu 2007). Notably, the EMT process may not always be complete. In some instances, cells lie along a gradient on which incomplete transition occurs, and both epithelial and mesenchymal characteristics are exhibited by the same cell (Jordan et al. 2011).
EMT in physiologic tissue repair
Wound healing exhibits EMT-like features
Converging lines of evidence indicate that EMT is an essential component of physiologic tissue repair. The majority of studies have been conducted in models of cutaneous wound healing.
Wound healing consists of several overlapping phases that involve an injury-induced inflammatory response that is associated with cellular proliferation, migration, and ECM remodeling (Eming et al. 2014; Martin 1997). Of these processes, the one most reminiscent of EMT is the process of re-epithelialization, which has been termed “partial EMT” (Arnoux et al. 2005). As discussed above, a hallmark of EMT is cell-cell dissociation and acquisition of motility, and during re-epithelialization, keratinocytes at the wound edge lose their intercellular adhesions and migrate across the wound (Coulombe 2003). Specifically, these keratinocytes undergo changes in junctional complexes including a reduction in desmosomes and adherens junctions, a disruption of intermediate filaments, and cytoskeletal reorganization that results in the creation of intercellular gaps (Baum and Arpey 2005; Santoro and Gaudino 2005). These changes enable the keratinocytes to shift morphologically from cuboidal and stationary to flattened and migratory, with extended lamellipodia (Baum and Arpey 2005; Santoro and Gaudino 2005).
Evidence is also available that myofibroblasts, the key players in the remodeling and maturation phase of wound healing, are derived from resident epithelial cells that have transformed through EMT to synthesize ECM components and to contract the wound bed, enabling an approximation of the injured edges (Iwano et al. 2002; Radisky et al. 2007; Wynn and Ramalingam 2012).
EMT has been implicated in animal and human models of cutaneous wound healing
Evidence from in vitro, in vivo, and ex vivo animal and human models supports the importance of the proper execution of EMT in achieving successful wound repair following cutaneous injury.
To start with, the EMT transcription factor Slug has been implicated in the process of re-epithelialization in numerous studies. Healing of excisional wounds is impaired in Slug knockout mice almost twofold in comparison with wild-type controls (Hudson et al. 2009), and epidermal keratinocytes from these mice display defects in migration (Savagner et al. 2005). In ex vivo skin explants from Slug null mice, epithelial cell outgrowth is also severely impaired, again indicating compromised motility (Savagner et al. 2005; Kusewitt et al. 2009). Indeed, Slug expression is elevated in wild-type keratinocytes at the edges of murine wounds in vivo (Shirley et al. 2010; Savagner et al. 2005), and its expression specifically increases in actively migrating mouse keratinocytes (Savagner et al. 2005).
Mechanistically, Slug regulates keratinocyte motility during re-epithelialization by repressing E-cadherin, leading to decreased cell-cell adhesion (Savagner 2001). It also drives intercellular desmosomal disruption at the wound edge (Savagner et al. 2005). Finally, the EGF receptor (EGFR) signaling pathway, which is integral to re-epithelialization in physiologic wound healing, might be the master regulator of EMT/Slug-mediated effects, since EGFR ligands stimulate the expression of Slug and the subsequent migration of keratinocytes (Kusewitt et al. 2009) in a process that is mediated by Erk5 (Arnoux et al. 2008). Indeed, in the absence of Slug, EGFR ligands are unable to stimulate the migration of skin explants in the ex vivo model of physiologic re-epithelialization (Kusewitt et al. 2009).
Work in additional mammalian models provides further evidence for EMT involvement in skin repair. Treatment of rat mucosal keratinocytes with EGFR ligands and inflammatory cytokines TGFβ or interleukin 1 beta (IL1β) induces EMT-associated MMP9 and MMP13, together with EMT-like changes in cell morphology (Lyons et al. 1993). The N-acetylglucosaminyltransferase V transgenic (GnT-V Tg) mouse, which features aberrant structural modifications of oligosaccharides, carries an enhanced EMT-like phenotype that culminates in rapid re-epithelialization in vivo, in part attributable to the differential glycosylation of EGFR and the subsequent amplification of signaling that leads to increased migration (Terao et al. 2011). Specifically, wounded GnT-V keratinocytes exhibit spindle-like morphology, increased expression of EMT factors N-cadherin, Snail and Twist, and enhanced migration (Terao et al. 2011). Foxn1, a potent mammalian wound healing factor, also appears to be involved in EMT-driven re-epithelialization during repair, as evidenced by studies of Foxn1 transgenic mice. In these mice, the induction of EMT post-wounding has been demonstrated through the upregulation of the EMT transcriptional regulator Snail1, the increased MMP9 expression, and the presence of vimentin+/E-cadherin+ cells, and migratory keratinocytes at the wound edge expressing Foxn1, which co-localizes with Snail (Gawronska-Kozak et al. 2016). Finally, zebrafish keratocytes in explant culture, which serves as a well-studied model of epithelial wound healing, display evidence of EMT (McDonald et al. 2013). During injury-triggered migration, keratocytes feature the loss of epithelial keratins and E-cadherin accompanied by the gain of mesenchymal markers, namely vimentin and N-cadherin. Moreover, explanted zebrafish keratocytes exhibit EMT-like morphologic changes including actin cytoskeletal rearrangements, disassembly of cellular sheets, and flattened cells. Interestingly, cell motility in this model appears to be driven in part by TGFβ1 (Tan et al. 2011), which is a known trigger of EMT.
In the in vitro models of human wound healing, immortalized HaCaT keratinocytes with forced overexpression of the EMT transcription factor Slug feature enhanced migration and disruption of desmosomes at the wound margin, recapitulating the effects of Slug in wounded skin of animal models in vivo (Savagner et al. 2005). Similarly, antimicrobial peptides shown to enhance wound healing concurrently induce Slug at the edge of wounded HaCaTs (Carretero et al. 2008). Heparin-binding EGF (HB-EGF), a keratinocyte-expressed ligand that activates EGFR during human wound healing (Mathay et al. 2008; McCarthy et al. 1996; Stoll et al. 1997), triggers a migratory phenotype that is reminiscent of EMT. Specifically, the expression of HB-EGF in human keratinocytes decreases epithelial keratins and E-cadherin, increases vimentin expression, and increases EMT factors SNAIL1 and ZEB1. HB-EGF also increases COX2 and MMP1, which are additional markers of cellular motility (Stoll et al. 2012). However, perhaps the most compelling evidence for the involvement of EMT in human cutaneous wound healing originates from a study by Yan et al. (2010) who demonstrated what they termed “partial EMT” in wound healing in vitro, ex vivo, and in vivo. Basal keratinocytes in the migrating tongue of re-epithelializing human acute wounds gained the expression of the mesenchymal markers fibroblast-specific protein 1 (FSP1) and/or vimentin, whereas the basement membrane zone displayed collagen disassembly, reflecting EMT-associated degradation of the ECM. Furthermore, the treatment of ex vivo human skin with inflammatory cytokines tumor necrosis factor- alpha (TNFα) and TGFβ induced an EMT-positive cell population. Primary keratinocytes treated similarly displayed morphologic cellular elongation and an enhanced migratory phenotype that was reversible following the removal of the cytokine stimuli. As such, injury-inducible mobilization of epithelial cells involving TNFα and bone morphogenetic protein (BMP)-2 produced a mesenchymal phenotype in migrating keratinocytes (Yan et al. 2010).
Role of EMT in extra-cutaneous organ repair
Additional evidence exists for EMT occurring during the repair of organs other than the skin. During in vitro healing of a breast (mammary) epithelial cell line, time-lapse microscopy indicated that EMT-associated vimentin was expressed in a migration-dependent fashion, such that vimentin was exclusively induced in actively migrating cells at the leading wound edge, an event that was accompanied by actin filament reorganization. Vimentin expression subsequently disappeared once wound closure was achieved (Gilles et al. 1999). Similarly, in a murine model of lacrimal gland injury, inflammation induced by interleukin-1 (IL-1) injection triggered the generation and migration of cells with mesenchymal features to the site of injury, but these cells subsequently reverted to an epithelial phenotype once repair was complete (You et al. 2012). These cells initially expressed EMT markers Snail1 and vimentin during the repair phase, the levels of which decreased after injury resolution, indicating a reversible or “partial” EMT. Finally, EMT is a key feature of cardiac development during embryogenesis, and accumulating evidence in zebrafish and other models of myocardial injury indicates that a subpopulation of epicardial cells undergo EMT to regenerate the damaged epithelial cover and to help the establishment of new vasculature (Lepilina et al. 2006; Krainock et al. 2016).
Wound healing and EMT share central signaling pathways
Notably, a complex signaling network involving numerous growth factors activated during wound healing are also involved in the initiation and regulation of the EMT, supporting a global role for EMT in epithelial barrier restoration following injury (Fig. 1). The common growth factors indispensable for both processes include FGF, EGF, HGF, and TGFβ (Akhurst and Derynck 2001; Camenisch et al. 2002; Jechlinger et al. 2006; Kim et al. 2007; Murillo et al. 2005; Nawshad and Hay 2003). FGF, EGF, and HGF function as ligands for the corresponding receptors, namely tyrosine kinase transmembrane proteins, resulting in their dimerization and autophosphorylation, the phosphorylation of downstream target proteins, and the activation of the signaling cascades (Lemmon and Schlessinger 2010; Tsai and Yang 2013). Thus, ERK MAPK, p38 MAPK, and JNK are among the activated pathways that ultimately upregulate EMT transcription factors such as SNAIL, Slug, and ZEB (Tsai and Yang 2013) on the one hand, while triggering wound healing processes on the other (Castilho et al. 2013; Zhang et al. 2015).

Common growth factor signals initiate and regulate essential EMT and wound-healing processes (FGF fibroblast growth factor, EGF epidermal growth factor, TGFβ transforming growth factor beta, HGF hepatocyte growth factor)
FGF signaling
The FGF family comprises 23 members, with the three crucial FGFs for the wound healing process being FGF-2, FGF-7, and FGF-10 (Golinko et al. 2009). FGF-2 (or basic FGF) increases in the acute wound and plays a role in granulation tissue formation, epithelialization, and tissue remodeling (Powers et al. 2000). In vitro studies have shown that the activation of the FGF receptor by FGF-2 increases keratinocyte and fibroblast motility (Di Vita et al. 2006; Sogabe et al. 2006) and stimulates fibroblasts to produce collagenase (Sasaki 1992). The FGF family is also induced during EMT (Smith and Bhowmick 2016), with the role of ensuring that epithelial cells adopt a mesenchymal phenotype through classic effects such as the downregulation of E-cadherin and catenins and the induction of mesenchymal MMPs (Ciruna et al. 1997; Strutz et al. 2002). In particular, FGF-2 is important in repair-associated EMT (Ciruna et al. 1997; Sun et al. 1999). Other FGF family members (e.g., FGF-1) instigate EMT in carcinomas, prompting an increase in the EMT transcription factor Slug, the downregulation of desmosomal components, and the upregulation of MMPs and integrins, all of which are essential for cell motility (Billottet et al. 2008; Savagner et al. 1997; Valles et al. 1996).
EGF signaling
The EGF family represents the best-characterized growth factor family in wound healing and includes a wide variety of ligands such as EGF, HB-EGF, TGFα, Cripto-1, epiregulin, amphiregulin, betacellulin, epigen, and neuregulins (NRG) 1–6 (Barrientos et al. 2008, 2014). Ultimately, EGF signaling leads to the activation of a number of converging signaling pathways promoting keratinocyte migration and proliferation (Omenetti et al. 2008). EGF also helps to accomplish EMT by downregulating E-cadherin via E-cadherin internalization, upregulating SNAIL1 and/or TWIST, and increasing cell motility through MMP-directed ECM degradation (Ahmed et al. 2006; Lo et al. 2007; Lu et al. 2003). In murine mammary epithelial cell tumors, the upregulation of Cripto1, an EGF family member, results in enhanced mesenchymal characteristics, such as increased expression of N-cadherin, vimentin, and Snail1 (Rangel et al. 2012; Strizzi et al. 2004; Tao et al. 2005).
HGF signaling
HGF signaling is an additional example of the wound healing/EMT crosstalk. HGF, mainly produced by fibroblasts, exerts its function by binding to its tyrosine kinase receptor c-Met (mesenchymal epithelial transition factor or HGFR), which is expressed on the surface of keratinocytes (Toyoda et al. 2001). Both HGF and c-Met are upregulated during wound healing and promote granulation tissue formation and neoangiogenesis (Toyoda et al. 2001; Wang et al. 2009; Yoshida et al. 2003). Furthermore, c-Met plays an important role in re-epithelialization through the activation of PI3K/AKT, ERK1/2, Gab1 (Grb2-associated-binding protein 1), and PAK1/2 (p21-activated protein kinase) signaling (Chmielowiec et al. 2007). HGF and its receptor also clearly induce various changes in the EMT process, depending on the specific cell type expressing c-Met (Grotegut et al. 2006; Savagner et al. 1997). To begin with, HGF can regulate master EMT transcription factor SNAIL1 (which decreases E-cadherin) and Slug (which decreases desmoplakins) aiding the breakdown of intercellular adhesions (Grotegut et al. 2006; Savagner et al. 1997). Additionally, the c-Met-PI3K/AKT pathway influences the cell cycle, proliferation, and quiescence (King et al. 2015), and PI3K-activated mTORC2 is one of the driving factors for the phenotypic transition in EMT, whereas mTORC1 encourages cell growth and movement (Lamouille et al. 2012; Lamouille and Derynck 2007). Since one of the roles of AKT is to phosphorylate and inactivate GSK3β, which itself is an inhibitor of SNAIL1 expression, the inhibition of AKT can cause the downregulation of SNAIL activity in the cell and impede EMT (Lamouille et al. 2012; Zhou et al. 2004). The resultant decrease in MMP production and non-inhibited production of E-cadherin makes EMT and subsequent movement difficult for the cell to achieve (Lamouille et al. 2012).
TGFβ signaling in wound healing, EMT, and fibrosis
The TGFβ pathway is well studied not only in wound healing (Ramirez et al. 2014), but also in all three types of EMT (Akhurst and Derynck 2001; Camenisch et al. 2002; Nawshad and Hay 2003). TGFβ progresses via two pathways: SMAD-dependent and SMAD-independent (Xu et al. 2000). In SMAD-dependent pathways, the TGFβ cell surface receptors (known as TGFβ receptors type II) are activated by ligand and phosphorylate the transmembrane kinase (TGFβ receptor type I), which then forms a SMAD complex; this complex can enter the nucleus, subsequently activating or inhibiting transcription factors important for either wound healing or EMT (Derynck and Zhang 2003; Ramirez et al. 2014). In wound healing, TGFβ1 plays important roles in inflammation, angiogenesis, re-epithelialization, and connective tissue regeneration (Ramirez et al. 2014). TGFβ and SMAD complexes induce SNAIL1 expression and are themselves potent downregulators of E-cadherin, occludin, and other epithelial phenotypic markers, while promoting mesenchymal markers such as vimentin and N-cadherin (Vincent et al. 2009). SMAD3-SMAD4 complexes can also activate TWIST and ZEB transcription factors, via the MAPK signaling route, one of the SMAD-independent pathways (Javelaud and Mauviel 2005). Another major SMAD-independent pathway is the PI3K/AKT pathway, whose importance in both EMT and wound healing is discussed above.
EMT in scarring and fibrosis
EMT-derived myofibroblasts, TGFβ, and fibrosis
During physiologic repair, tissue integrity must be restored not only through re-epithelialization, but also through the formation of a stress-resistant scar. The cellular orchestrator of this remodeling process is the contractile myofibroblast, which secretes large amounts of ECM proteins and aids in the mechanical closure of the wound (Gabbiani et al. 1971; Hinz and Gabbiani 2003). In normal wound healing, many myofibroblasts undergo apoptosis and disappear once re-epithelialization is complete (Desmouliere et al. 1995; Gabbiani 2003). However, pathologically prolonged myofibroblast activity results in fibrogenesis. Indeed, persistent myofibroblast activation is a shared feature of fibrotic diseases. As such, the dysregulation of injury-triggered EMT is believed to contribute to fibrosis of multiple organs.
Although the myofibroblast can be derived from a variety of sources (Abe et al. 2001; Direkze et al. 2003; Ebihara et al. 2006; Frid et al. 2002; Higashiyama et al. 2011; Wynn and Ramalingam 2012), a large body of evidence supports that a proportion of them arise through EMT during organ fibrosis. Moreover, TGFβ1, a critical regulator of EMT signaling and physiologic wound healing (as discussed above), is also the major driver of fibrosis (Border and Noble 1994; Roberts et al. 1986), in part through its role in sustaining myofibroblast activation (Desmouliere et al. 1993; Gabbiani 2003; Hong et al. 2007; Ronnov-Jessen and Petersen 1993; Serini and Gabbiani 1999). This section focuses on evidence implicating EMT in the fibrogenesis of various tissues; this fibrogenesis arises as a pathological response to injury.
Renal fibrosis
Progressive chronic kidney disease characterized by interstitial fibrosis can lead to tubular atrophy, loss of kidney function, and end-stage renal failure (Liu 2011). Numerous studies have provided evidence that EMT-derived myofibroblasts originating from tubular epithelia contribute to renal fibrosis. These studies have involved animal models, human kidney biopsies, staining techniques for epithelial and fibroblast cell lineage markers, lineage tags, and the activation of various transcriptional signals known to stimulate the EMT program (Higgins et al. 2007; Humphreys et al. 2010; Inoue et al. 2009; Iwano et al. 2002; Nishitani et al. 2005; Rastaldi et al. 2002; Strutz et al. 2002; Zeisberg et al. 2003). Although conflicting at times, a series of genetic-lineage tracking and fate-mapping studies have provided support for the existence of EMT-derived myofibroblasts in renal fibrosis (Humphreys et al. 2010). In one experimental murine model, fibroblasts expressing the mesenchymal EMT marker FSP1 have been shown to be derived from both the bone marrow and local EMT during renal fibrogenesis (Iwano et al. 2002). In vivo evidence for EMT in renal fibrosis has also been reported in human biopsy studies (Inoue et al. 2009; Nishitani et al. 2005; Rastaldi et al. 2002). In a patient with fibrosis-inducing obstructive nephropathy, obstructed tubular epithelial cells expressed FSP1 (Okada et al. 1997), and some adopted an EMT-like fibroblast morphology (Inoue et al. 2009; Nishitani et al. 2005). FSP1 has also been shown to be a prognostic marker in renal fibrosis in humans (Nishitani et al. 2005). Another study of 133 biopsies from various renal fibrosis conditions has demonstrated that tubular epithelia cells produce a variety of ECM proteins characteristic of a mesenchymal phenotype, the levels of which correlate clinically with elevated serum creatinine levels and indices of renal dysfunction and the histologic extent of interstitial fibrotic damage (Rastaldi et al. 2002).
TGFβ1 is the main inducer of EMT in renal tubular epithelial cells (Fan et al. 1999; Strutz et al. 2002). The expression of FSP1 in transitioning tubular epithelium is induced by TGFβ (Okada et al. 2000), and tubular basement membrane disintegration leads to TGFβ1 upregulation by mouse proximal tubular epithelial cells contributing to EMT during renal fibrosis (Zeisberg et al. 2001). Interestingly, TGFβ1-induced EMT in tubular epithelial cells can be reversed by BMP7 by inducing E-cadherin in a SMAD-dependent manner in vitro, and the systemic administration of recombinant human BMP-7 leads to the repair of damaged renal tubular epithelial cells in a murine model of fibrotic chronic renal injury (Zeisberg et al. 2003), indicating that the TGFβ-EMT axis represents a therapeutic target for injury-induced fibrosis.
Pulmonary fibrosis
Lung epithelial cells responding to repeated injury experience persistent inflammation and sustained EMT, leading to fibrosis (Chapman 2011; Crosby and Waters 2010). Although the origin of myofibroblasts in lung fibrosis is not certain, some studies have reported the occurrence of EMT in lung fibrosis, partly mediated through TGFβ signaling (Chen et al. 2015; Kim et al. 2006; Mubarak et al. 2012; Willis et al. 2005; Zhou et al. 2009; Zolak et al. 2013). Alveolar epithelial cells (AECs) undergo EMT and contribute to pulmonary fibrosis pathology induced by TGFβ (Kim et al. 2006; Willis et al. 2005; Zhou et al. 2009). Moreover, in a TGFβ1 murine model of pulmonary fibrosis, the beta-galactosidase (β-gal)-expressing epithelial cells also expressed mesenchymal markers within injured lungs, indicating epithelial cells as the progenitors for the fibroblasts. Primary AECs cultured on provisional matrix components, fibronectin, or fibrin undergo EMT via the integrin-dependent activation of endogenous latent TGFβ1 indicating that the ECM acts as a regulator in the EMT process during fibrogenesis (Kim et al. 2006). Exposure of TGFβ to rat primary AECs increased the expression of mesenchymal cell markers and a fibroblastic-phenotype, an effect accelerated by TNFα. In vivo, AECs co-expressed epithelial markers and α-smooth muscle actin in lung tissue samples from patients with idiopathic pulmonary fibrosis (IPF; Willis et al. 2005). Studies have also demonstrated that pleural mesothelial cells (PMCs) are capable of transitioning into myofibroblasts, a process thought to be driven by TGFβ (Chen et al. 2015; Zolak et al. 2013). PMCs are seen in lung tissue of IPF patients, and labeled PMCs injected into mice travel to IPF lungs and display myofibroblast phenotypic markers in response to TGFβ; the numbers of PMCs correlate with the degree of fibrosis and IPF disease severity (Mubarak et al. 2012). Increased production of type I collagen and mesenchymal phenotypic markers and decreased epithelial phenotypic markers are features of PMCs in the bleomycin animal model of injury-triggered pulmonary fibrosis, which is phenotypically similar to human IPF. Moreover, in this model, PMC migration is mediated both in vivo and in vitro by TGFβ1-SMAD2/3 signaling (Chen et al. 2015).
Cardiac fibrosis
Following cardiac injury, EMT appears to play a role in regeneration or fibrosis to produce mesenchymal cells with both stem cell and myofibroblast characteristics (Limana et al. 2007). Adult epicardium-derived cells have been shown to reactivate post myocardial injury, undergo EMT, and migrate into the injured myocardium where they produce various cell types in vivo, including cardiac interstitial fibroblasts and coronary smooth muscle cells that aid the cardiac repair process (Limana et al. 2007; Mikawa and Fischman 1992; Mikawa and Gourdie 1996; Poelmann et al. 1993; Smart et al. 2013; Winter et al. 2007). Evidence also supports the positive regulation of epicardial cell transformation and smooth muscle differentiation by TGFβ, as human adult epicardial cells with an epithelial-like phenotype expressing the cell surface marker vascular cell adhesion marker (VCAM-1) spontaneously undergo EMT and adopt a smooth-muscle-like phenotype in vitro when activated by TGFβ1 receptor signaling and inhibited by VCAM-1 (Moore et al. 1999). Furthermore, in epicardium explant studies, both TGFβ1 and TGFβ2 induce the loss of epithelial cell markers cytokeratin and membrane-associated Zonula Occludens-1 from epicardial cells and trigger the gain of smooth muscle markers calponin and caldesmon; this is dependent upon ALK5 kinase activity, culminating in the induction of epicardial cell EMT and invasion (Compton et al. 2006).
Hepatic fibrosis
Chronic liver disease gives rise to hepatic fibrosis, but the origin of the activated myofibroblasts is still under debate, and various epithelial cells undergoing EMT may serve as the sources. Hepatic stellate cells (HSCs) are one of the cellular candidates for activated myofibroblasts (Friedman et al. 1985), adopting a spindle-shaped phenotype and expressing α-smooth muscle actin and type I collagen (Gressner and Weiskirchen 2006; Lee et al. 1995). Lineage tracing experiments in mice have demonstrated that HSCs contribute to 82-96 % of myofibroblasts mediating fibrogenesis (Mederacke et al. 2013). Epithelial hepatocytes and cholangiocytes are also likely candidates for contributing to the myofibroblast population in liver fibrosis. Interestingly, mouse cholangiocytes, co-cultured with myofibroblastic HSCs undergo EMT in vitro, exhibiting increased cell migration, reduced epithelial markers, and induced mesenchymal markers (Omenetti et al. 2008).
As in the kidney and lung, TGFβ might be involved in the induction of the EMT phenotype in liver fibrosis. In one study, EMT was induced in hepatocytes in vitro via the activation of the TGFβ1/SMAD pathway (Kaimori et al. 2007). Additional lineage-tracing experiments on transgenic mice demonstrated that TGFβ1 induced hepatocytes to undergo EMT and contributed to the population of FSP1-positive fibroblasts in the CCl4-induced model of liver fibrosis, an effect that could be blocked by BMP-7 administration. Moreover, human cultured intrahepatic epithelial cells treated with TGFβ were shown to undergo EMT-like changes, adopting an invasive fibroblast-type phenotype with the loss of cytokeratin-7 and the gain of SMAD2/3, S100A4, and α-smooth muscle actin expression. In the same study, TGFβ mRNA and nuclear phospho-SMAD2/3 were highly expressed in damaged ducts of chronic diseased liver tissue that also expressed S100A4, vimentin, and MMP-2. Finally, the co-expression of epithelial and mesenchymal markers in biliary epithelial cells and cholangiocytes of chronic liver disease patients also supports an in vivo role for TGFβ-induced EMT in human hepatic fibrosis (Diaz et al. 2008; Rygiel et al. 2008).
Scleroderma and skin fibrosis
Scleroderma (Sc) is a systemic disorder characterized by autoimmunity, chronic inflammation, vasculopathy, and extensive skin and organ fibrosis of unknown etiology (Gazi et al. 2007). In Sc, early vascular injury precedes fibrosis, and as with renal fibrosis, the persistently activated myofibroblasts drive TGFβ-induced gene expression and increase pro-fibrotic cytokine and protease production (Postlethwaite et al. 2004). Although the origin of the myofibroblasts in Sc fibrotic skin is unknown, studies have once again indicated that the EMT process is one possible source (Postlethwaite et al. 2004). Indeed, the increased nuclear translocation of myocardin-related transcription factor-A (MRTF-A), a key mechano-responsive transcription factor that signals EMT, has been observed in Sc epidermis (O’Connor and Gomez 2013; Shiwen et al. 2015).
Increased levels of TGFβ1 and TGFβ receptors and enhanced TGFβ signaling has been reported in Sc (Dong et al. 2002; Leask et al. 2002) thus supporting a role for this cytokine in myofibroblast activation and in the pathogenesis in the fibrosis observed in Sc (Xu et al. 2009). In one murine model, active TGFβ signaling was enhanced, leading to skin fibrosis that resembled the biochemical, clinical, and histologic features of human Sc (Sonnylal et al. 2007). In Sc epidermis, keratinocytes have been shown to adopt an activated phenotype associated with active SMAD/TGFβ signaling and to display increased expression of pro-fibrotic factors, namely connective tissue growth factor (CTGF) and SNAIL1 (Nikitorowicz-Buniak et al. 2015). Sc keratinocytes stimulate fibroblasts to increase ECM contractility and growth factor expression, the effects of which are dependent on elevated levels of IL-1α expression by epidermal cells and the induction of endothelin-1 and TGFβ in fibroblasts (Aden et al. 2010). In vitro, Sc fibroblasts display enhanced collagen deposition and ECM contraction and remodeling (Jimenez et al. 1986).
Less is known regarding the contribution of EMT processes to fibrotic skin conditions other than scleroderma. High expression of the mesenchymal marker FSP1 has been found in the epidermis and dermis of human hypertrophic scars; this is accompanied by increased levels of inflammatory cytokines, fibrotic markers, and EMT-related Slug and TWIST. Thus, a link has been demonstrated between unresolved inflammation and the development EMT characteristics during fibrogenesis in hypertrophic scar tissue in vivo (Yan et al. 2010).
Concluding remarks and future directions
Injury triggers the inflammatory wound healing cascade, and pathologically sustained inflammation is tightly associated with fibrogenesis. This review has summarized evidence that EMT plays a role in physiologic tissue repair, and that sustained EMT is a key mechanism underlying the fibrotic pathology of multiple organs. Given the fundamental parallels between the regulation and signaling of EMT and the critical wound-healing processes, we consider it quite conceivable that early and prolonged activation of EMT in the context of the response to injury promotes inflammation and fibrogenesis that culminates in non-healing wounds of many epithelial tissues (Fig. 2). In investigating this hypothesis further, we need to keep in mind that EMT is a dynamic and reversible process, and that cells cannot always be classified as purely epithelial or mesenchymal, especially in vivo, as they may carry features of each. Loss-of-epithelial and gain-of-mesenchymal features can also occur simultaneously. Nevertheless, an assessment of the presence of the classic EMT biomarkers in non-healing tissues and organs in vivo will be critical to define the role of EMT in initiating and sustaining a poor healing response and may represent a way forward to the potential targeting of EMT as a novel and global therapeutic approach for difficult-to-treat wounds.

Model for injury-triggered EMT activation in physiologic wound repair (left) and fibrotic wound healing (right)
References
Abe R, Donnelly SC, Peng T, Bucala R, Metz CN (2001) Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol 166:7556–7562
Aden N, Nuttall A, Shiwen X, de Winter P, Leask A, Black CM, Denton CP, Abraham DJ, Stratton RJ (2010) Epithelial cells promote fibroblast activation via IL-1alpha in systemic sclerosis. J Invest Dermatol 130:2191–2200
Ahmed N, Maines-Bandiera S, Quinn MA, Unger WG, Dedhar S, Auersperg N (2006) Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. Am J Physiol Cell Physiol 290:C1532–C1542
Akhurst RJ, Derynck R (2001) TGF-beta signaling in cancer—a double-edged sword. Trends Cell Biol 11:S44–S51
Arnoux V, Come C, Kusewitt D, Hudson L, Savagner P (2005) Cutaneous wound reepithelialization: a partial and reversible EMT. In: Savagner P (ed) Rise and fall of epithelial phenotype: concepts of epithelial-mesenchymal transition. Springer, Berlin, pp 111–134
Arnoux V, Nassour M, L’Helgoualc’h A, Hipskind RA, Savagner P (2008) Erk5 controls Slug expression and keratinocyte activation during wound healing. Mol Biol Cell 19:4738–4749
Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M (2008) Growth factors and cytokines in wound healing. Wound Repair Regen 16:585–601
Barrientos S, Brem H, Stojadinovic O, Tomic-Canic M (2014) Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen 22:569–578
Baum CL, Arpey CJ (2005) Normal cutaneous wound healing: clinical correlation with cellular and molecular events. Dermatol Surg 31:674–686. discussion 686
Billottet C, Tuefferd M, Gentien D, Rapinat A, Thiery JP, Broet P, Jouanneau J (2008) Modulation of several waves of gene expression during FGF-1 induced epithelial-mesenchymal transition of carcinoma cells. J Cell Biochem 104:826–839
Border WA, Noble NA (1994) Transforming growth factor beta in tissue fibrosis. N Engl J Med 331:1286–1292
Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, Klewer SE (2002) Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol 248:170–181
Carretero M, Escamez MJ, Garcia M, Duarte B, Holguin A, Retamosa L, Jorcano JL, Rio MD, Larcher F (2008) In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J Invest Dermatol 128:223–236
Castilho RM, Squarize CH, Gutkind JS (2013) Exploiting PI3K/mTOR signaling to accelerate epithelial wound healing. Oral Dis 19:551–558
Chapman HA (2011) Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol 73:413–435
Chen LJ, Ye H, Zhang Q, Li FZ, Song LJ, Yang J, Mu Q, Rao SS, Cai PC, Xiang F, Zhang JC, Su Y, Xin JB, Ma WL (2015) Bleomycin induced epithelial-mesenchymal transition (EMT) in pleural mesothelial cells. Toxicol Appl Pharmacol 283:75–82
Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W (2007) c-Met is essential for wound healing in the skin. J Cell Biol 177:151–162
Ciruna BG, Schwartz L, Harpal K, Yamaguchi TP, Rossant J (1997) Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a role for FGFR1 in morphogenetic movement through the primitive streak. Development 124:2829–2841
Compton LA, Potash DA, Mundell NA, Barnett JV (2006) Transforming growth factor-beta induces loss of epithelial character and smooth muscle cell differentiation in epicardial cells. Dev Dyn 235:82–93
Coulombe PA (2003) Wound epithelialization: accelerating the pace of discovery. J Invest Dermatol 121:219–230
Crosby LM, Waters CM (2010) Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 298:L715–L731
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577–584
Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G (1993) Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122:103–111
Desmouliere A, Redard M, Darby I, Gabbiani G (1995) Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 146:56–66
Di Vita G, Patti R, D’Agostino P, Caruso G, Arcara M, Buscemi S, Bonventre S, Ferlazzo V, Arcoleo F, Cillari E (2006) Cytokines and growth factors in wound drainage fluid from patients undergoing incisional hernia repair. Wound Repair Regen 14:259–264
Diaz R, Kim JW, Hui JJ, Li Z, Swain GP, Fong KS, Csiszar K, Russo PA, Rand EB, Furth EE, Wells RG (2008) Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum Pathol 39:102–115
Direkze NC, Forbes SJ, Brittan M, Hunt T, Jeffery R, Preston SL, Poulsom R, Hodivala-Dilke K, Alison MR, Wright NA (2003) Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells 21:514–520
Dong C, Zhu S, Wang T, Yoon W, Li Z, Alvarez RJ, ten Dijke P, White B, Wigley FM, Goldschmidt-Clermont PJ (2002) Deficient Smad7 expression: a putative molecular defect in scleroderma. Proc Natl Acad Sci U S A 99:3908–3913
Ebihara Y, Masuya M, Larue AC, Fleming PA, Visconti RP, Minamiguchi H, Drake CJ, Ogawa M (2006) Hematopoietic origins of fibroblasts. II. In vitro studies of fibroblasts, CFU-F, and fibrocytes. Exp Hematol 34:219–229
Eming SA, Martin P, Tomic-Canic M (2014) Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med 6:265sr266
Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY (1999) Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 56:1455–1467
Frid MG, Kale VA, Stenmark KR (2002) Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res 90:1189–1196
Friedman SL, Roll FJ, Boyles J, Bissell DM (1985) Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A 82:8681–8685
Gabbiani G (2003) The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 200:500–503
Gabbiani G, Ryan GB, Majne G (1971) Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27:549–550
Gawronska-Kozak B, Grabowska A, Kur-Piotrowska A, Kopcewicz M (2016) Foxn1 transcription factor regulates wound healing of skin through promoting epithelial-mesenchymal transition. PLoS One 11:e0150635
Gazi H, Pope JE, Clements P, Medsger TA, Martin RW, Merkel PA, Kahaleh B, Wollheim FA, Baron M, Csuka ME, Emery P, Belch JF, Hayat S, Lally EV, Korn JH, Czirjak L, Herrick A, Voskuyl AE, Bruehlmann P, Inanc M, Furst DE, Black C, Ellman MH, Moreland LW, Rothfield NF, Hsu V, Mayes M, McKown KM, Krieg T, Siebold JR (2007) Outcome measurements in scleroderma: results from a delphi exercise. J Rheumatol 34:501–509
Gilles C, Polette M, Zahm JM, Tournier JM, Volders L, Foidart JM, Birembaut P (1999) Vimentin contributes to human mammary epithelial cell migration. J Cell Sci 112:4615–4625
Golinko MS, Joffe R, de Vinck D, Chandrasekaran E, Stojadinovic O, Barrientos S, Vukelic S, Tomic-Canic M, Brem H (2009) Surgical pathology to describe the clinical margin of debridement of chronic wounds using a wound electronic medical record. J Am Coll Surg 209:254–260
Gressner AM, Weiskirchen R (2006) Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med 10:76–99
Grotegut S, von Schweinitz D, Christofori G, Lehembre F (2006) Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J 25:3534–3545
Higashiyama R, Nakao S, Shibusawa Y, Ishikawa O, Moro T, Mikami K, Fukumitsu H, Ueda Y, Minakawa K, Tabata Y, Bou-Gharios G, Inagaki Y (2011) Differential contribution of dermal resident and bone marrow-derived cells to collagen production during wound healing and fibrogenesis in mice. J Invest Dermatol 131:529–536
Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH (2007) Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 117:3810–3820
Hinz B, Gabbiani G (2003) Cell-matrix and cell-cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb Haemost 90:993–1002
Hong KM, Belperio JA, Keane MP, Burdick MD, Strieter RM (2007) Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J Biol Chem 282:22910–22920
Huang RY, Guilford P, Thiery JP (2012) Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci 125:4417–4422
Hudson LG, Newkirk KM, Chandler HL, Choi C, Fossey SL, Parent AE, Kusewitt DF (2009) Cutaneous wound reepithelialization is compromised in mice lacking functional Slug (Snai2). J Dermatol Sci 56:19–26
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176:85–97
Inoue T, Okada H, Takenaka T, Watanabe Y, Suzuki H (2009) A case report suggesting the occurrence of epithelial-mesenchymal transition in obstructive nephropathy. Clin Exp Nephrol 13:385–388
Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG (2002) Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110:341–350
Javelaud D, Mauviel A (2005) Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene 24:5742–5750
Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, Donovan M, Cordon-Cardo C, Beug H, Grunert S (2006) Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest 116:1561–1570
Jimenez SA, Feldman G, Bashey RI, Bienkowski R, Rosenbloom J (1986) Co-ordinate increase in the expression of type I and type III collagen genes in progressive systemic sclerosis fibroblasts. Biochem J 237:837–843
Jordan NV, Johnson GL, Abell AN (2011) Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 10:2865–2873
Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E, Koteish A (2007) Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem 282:22089–22101
Kalluri R, Neilson EG (2003) Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112:1776–1784
Kim HJ, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, Lewis MT, Gottardis MM, Wong TW, Attar RM, Carboni JM, Lee AV (2007) Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol 27:3165–3175
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA (2006) Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A 103:13180–13185
King D, Yeomanson D, Bryant HE (2015) PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol 37:245–251
Krainock M, Toubat O, Danopoulos S, Beckham A, Warburton D, Kim R (2016) Epicardial epithelial-to-mesenchymal transition in heart development and disease. J Clin Med 5:27
Kusewitt DF, Choi C, Newkirk KM, Leroy P, Li Y, Chavez MG, Hudson LG (2009) Slug/Snai2 is a downstream mediator of epidermal growth factor receptor-stimulated reepithelialization. J Invest Dermatol 129:491–495
Lamouille S, Derynck R (2007) Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 178:437–451
Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R (2012) TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci 125:1259–1273
Leask A, Abraham DJ, Finlay DR, Holmes A, Pennington D, Shi-Wen X, Chen Y, Venstrom K, Dou X, Ponticos M, Black C, Bernabeu C, Jackman JK, Findell PR, Connolly MK (2002) Dysregulation of transforming growth factor beta signaling in scleroderma: overexpression of endoglin in cutaneous scleroderma fibroblasts. Arthritis Rheum 46:1857–1865
Lee JM, Dedhar S, Kalluri R, Thompson EW (2006) The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172:973–981
Lee KS, Buck M, Houglum K, Chojkier M (1995) Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest 96:2461–2468
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141:1117–1134
Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, Poss KD (2006) A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127:607–619
Limana F, Zacheo A, Mocini D, Mangoni A, Borsellino G, Diamantini A, De Mori R, Battistini L, Vigna E, Santini M, Loiaconi V, Pompilio G, Germani A, Capogrossi MC (2007) Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ Res 101:1255–1265
Liu Y (2011) Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7:684–696
Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC (2007) Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res 67:9066–9076
Lu Z, Ghosh S, Wang Z, Hunter T (2003) Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 4:499–515
Lyons JG, Birkedal-Hansen B, Pierson MC, Whitelock JM, Birkedal-Hansen H (1993) Interleukin-1 beta and transforming growth factor-alpha/epidermal growth factor induce expression of M(r) 95,000 type IV collagenase/gelatinase and interstitial fibroblast-type collagenase by rat mucosal keratinocytes. J Biol Chem 268:19143–19151
Martin P (1997) Wound healing—aiming for perfect skin regeneration. Science 276:75–81
Maschler S, Wirl G, Spring H, Bredow DV, Sordat I, Beug H, Reichmann E (2005) Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene 24:2032–2041
Mathay C, Giltaire S, Minner F, Bera E, Herin M, Poumay Y (2008) Heparin-binding EGF-like growth factor is induced by disruption of lipid rafts and oxidative stress in keratinocytes and participates in the epidermal response to cutaneous wounds. J Invest Dermatol 128:717–727
McCarthy DW, Downing MT, Brigstock DR, Luquette MH, Brown KD, Abad MS, Besner GE (1996) Production of heparin-binding epidermal growth factor-like growth factor (HB-EGF) at sites of thermal injury in pediatric patients. J Invest Dermatol 106:49–56
McDonald TM, Pascual AS, Uppalapati CK, Cooper KE, Leyva KJ, Hull EE (2013) Zebrafish keratocyte explant cultures as a wound healing model system: differential gene expression and morphological changes support epithelial-mesenchymal transition. Exp Cell Res 319:1815–1827
Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF (2013) Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 4:2823
Mendez MG, Kojima S, Goldman RD (2010) Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J 24:1838–1851
Mikawa T, Fischman DA (1992) Retroviral analysis of cardiac morphogenesis: discontinuous formation of coronary vessels. Proc Natl Acad Sci U S A 89:9504–9508
Mikawa T, Gourdie RG (1996) Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol 174:221–232
Moore AW, McInnes L, Kreidberg J, Hastie ND, Schedl A (1999) YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 126:1845–1857
Moreno-Bueno G, Portillo F, Cano A (2008) Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 27:6958–6969
Mubarak KK, Montes-Worboys A, Regev D, Nasreen N, Mohammed KA, Faruqi I, Hensel E, Baz MA, Akindipe OA, Fernandez-Bussy S, Nathan SD, Antony VB (2012) Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary fibrosis. Eur Respir J 39:133–140
Murillo MM, del Castillo G, Sanchez A, Fernandez M, Fabregat I (2005) Involvement of EGF receptor and c-Src in the survival signals induced by TGF-beta1 in hepatocytes. Oncogene 24:4580–4587
Nawshad A, Hay ED (2003) TGFbeta3 signaling activates transcription of the LEF1 gene to induce epithelial mesenchymal transformation during mouse palate development. J Cell Biol 163:1291–1301
Nikitorowicz-Buniak J, Denton CP, Abraham D, Stratton R (2015) Partially evoked epithelial-mesenchymal transition (EMT) is associated with increased TGFbeta signaling within lesional scleroderma skin. PLoS One 10:e0134092
Nishitani Y, Iwano M, Yamaguchi Y, Harada K, Nakatani K, Akai Y, Nishino T, Shiiki H, Kanauchi M, Saito Y, Neilson EG (2005) Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 68:1078–1085
O’Connor JW, Gomez EW (2013) Cell adhesion and shape regulate TGF-beta1-induced epithelial-myofibroblast transition via MRTF-A signaling. PLoS One 8:e83188
Okada H, Danoff TM, Kalluri R, Neilson EG (1997) Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 273:F563–F574
Okada H, Ban S, Nagao S, Takahashi H, Suzuki H, Neilson EG (2000) Progressive renal fibrosis in murine polycystic kidney disease: an immunohistochemical observation. Kidney Int 58:587–597
Omenetti A, Porrello A, Jung Y, Yang L, Popov Y, Choi SS, Witek RP, Alpini G, Venter J, Vandongen HM, Syn WK, Baroni GS, Benedetti A, Schuppan D, Diehl AM (2008) Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest 118:3331–3342
Poelmann RE, Gittenberger-de Groot AC, Mentink MM, Bokenkamp R, Hogers B (1993) Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ Res 73:559–568
Postlethwaite AE, Shigemitsu H, Kanangat S (2004) Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr Opin Rheumatol 16:733–738
Powers CJ, McLeskey SW, Wellstein A (2000) Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 7:165–197
Qin Y, Capaldo C, Gumbiner BM, Macara IG (2005) The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J Cell Biol 171:1061–1071
Radisky DC, Kenny PA, Bissell MJ (2007) Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 101:830–839
Ramirez H, Patel SB, Pastar I (2014) The role of TGFbeta signaling in wound epithelialization. Adv Wound Care 3:482–491
Rangel MC, Karasawa H, Castro NP, Nagaoka T, Salomon DS, Bianco C (2012) Role of Cripto-1 during epithelial-to-mesenchymal transition in development and cancer. Am J Pathol 180:2188–2200
Rastaldi MP, Ferrario F, Giardino L, Dell’Antonio G, Grillo C, Grillo P, Strutz F, Muller GA, Colasanti G, D’Amico G (2002) Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int 62:137–146
Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Fauci AS (1986) Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A 83:4167–4171
Ronnov-Jessen L, Petersen OW (1993) Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest 68:696–707
Rygiel KA, Robertson H, Marshall HL, Pekalski M, Zhao L, Booth TA, Jones DE, Burt AD, Kirby JA (2008) Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest 88:112–123
Santoro MM, Gaudino G (2005) Cellular and molecular facets of keratinocyte reepithelization during wound healing. Exp Cell Res 304:274–286
Sasaki T (1992) The effects of basic fibroblast growth factor and doxorubicin on cultured human skin fibroblasts: relevance to wound healing. J Dermatol 19:664–666
Savagner P (2001) Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays 23:912–923
Savagner P, Yamada KM, Thiery JP (1997) The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol 137:1403–1419
Savagner P, Kusewitt DF, Carver EA, Magnino F, Choi C, Gridley T, Hudson LG (2005) Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J Cell Physiol 202:858–866
Serini G, Gabbiani G (1999) Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res 250:273–283
Shirley SH, Hudson LG, He J, Kusewitt DF (2010) The skinny on Slug. Mol Carcinog 49:851–861
Shiwen X, Stratton R, Nikitorowicz-Buniak J, Ahmed-Abdi B, Ponticos M, Denton C, Abraham D, Takahashi A, Suki B, Layne MD, Lafyatis R, Smith BD (2015) A role of myocardin related transcription factor-A (MRTF-A) in scleroderma related fibrosis. PLoS One 10:e0126015
Smart N, Dube KN, Riley PR (2013) Epicardial progenitor cells in cardiac regeneration and neovascularisation. Vasc Pharmacol 58:164–173
Smith BN, Bhowmick NA (2016) Role of EMT in metastasis and therapy resistance. J Clin Med 5:17
Sogabe Y, Abe M, Yokoyama Y, Ishikawa O (2006) Basic fibroblast growth factor stimulates human keratinocyte motility by Rac activation. Wound Repair Regen 14:457–462
Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, Vanpelt CS, Geng YJ, Deng JM, Behringer RR, de Crombrugghe B (2007) Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum 56:334–344
Stoll S, Garner W, Elder J (1997) Heparin-binding ligands mediate autocrine epidermal growth factor receptor activation in skin organ culture. J Clin Invest 100:1271–1281
Stoll SW, Rittie L, Johnson JL, Elder JT (2012) Heparin-binding EGF-like growth factor promotes epithelial-mesenchymal transition in human keratinocytes. J Invest Dermatol 132:2148–2157
Strizzi L, Bianco C, Normanno N, Seno M, Wechselberger C, Wallace-Jones B, Khan NI, Hirota M, Sun Y, Sanicola M, Salomon DS (2004) Epithelial mesenchymal transition is a characteristic of hyperplasias and tumors in mammary gland from MMTV-Cripto-1 transgenic mice. J Cell Physiol 201:266–276
Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Muller GA, Neilson EG (2002) Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int 61:1714–1728
Sun X, Meyers EN, Lewandoski M, Martin GR (1999) Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev 13:1834–1846
Takenawa T, Suetsugu S (2007) The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 8:37–48
Tan B, Pascual A, de Beus A, Cooper K, Hull E (2011) TGFbeta (transforming growth factor beta) and keratocyte motility in 24 h zebrafish explant cultures. Cell Biol Int 35:1131–1139
Tao Q, Yokota C, Puck H, Kofron M, Birsoy B, Yan D, Asashima M, Wylie CC, Lin X, Heasman J (2005) Maternal wnt11 activates the canonical wnt signaling pathway required for axis formation in Xenopus embryos. Cell 120:857–871
Terao M, Ishikawa A, Nakahara S, Kimura A, Kato A, Moriwaki K, Kamada Y, Murota H, Taniguchi N, Katayama I, Miyoshi E (2011) Enhanced epithelial-mesenchymal transition-like phenotype in N-acetylglucosaminyltransferase V transgenic mouse skin promotes wound healing. J Biol Chem 286:28303–28311
Thiery JP, Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7:131–142
Toyoda M, Takayama H, Horiguchi N, Otsuka T, Fukusato T, Merlino G, Takagi H, Mori M (2001) Overexpression of hepatocyte growth factor/scatter factor promotes vascularization and granulation tissue formation in vivo. FEBS Lett 509:95–100
Tsai JH, Yang J (2013) Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 27:2192–2206
Valles AM, Boyer B, Tarone G, Thiery JP (1996) Alpha 2 beta 1 integrin is required for the collagen and FGF-1 induced cell dispersion in a rat bladder carcinoma cell line. Cell Adhes Commun 4:187–199
Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J (2009) A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol 11:943–950
Volk SW, Iqbal SA, Bayat A (2013) Interactions of the extracellular matrix and progenitor cells in cutaneous wound healing. Adv Wound Care 2:261–272
Wang Y, Weil BR, Herrmann JL, Abarbanell AM, Tan J, Markel TA, Kelly ML, Meldrum DR (2009) MEK, p38, and PI-3K mediate cross talk between EGFR and TNFR in enhancing hepatocyte growth factor production from human mesenchymal stem cells. Am J Physiol Cell Physiol 297:C1284–C1293
Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR (2008) Cadherin switching. J Cell Sci 121:727–735
Whiteman EL, Liu CJ, Fearon ER, Margolis B (2008) The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene 27:3875–3879
Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z (2005) Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol 166:1321–1332
Winter EM, Grauss RW, Hogers B, van Tuyn J, van der Geest R, Lie-Venema H, Steijn RV, Maas S, Deruiter MC, DeVries AA, Steendijk P, Doevendans PA, van der Laarse A, Poelmann RE, Schalij MJ, Atsma DE, Gittenberger-de Groot AC (2007) Preservation of left ventricular function and attenuation of remodeling after transplantation of human epicardium-derived cells into the infarcted mouse heart. Circulation 116:917–927
Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18:1028–1040
Xu J, Lamouille S, Derynck R (2009) TGF-beta-induced epithelial to mesenchymal transition. Cell Res 19:156–172
Xu L, Chen YG, Massague J (2000) The nuclear import function of Smad2 is masked by SARA and unmasked by TGFbeta-dependent phosphorylation. Nat Cell Biol 2:559–562
Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, Han YP (2010) Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol 176:2247–2258
Yang X, Pursell B, Lu S, Chang TK, Mercurio AM (2009) Regulation of beta 4-integrin expression by epigenetic modifications in the mammary gland and during the epithelial-to-mesenchymal transition. J Cell Sci 122:2473–2480
Yoshida S, Yamaguchi Y, Itami S, Yoshikawa K, Tabata Y, Matsumoto K, Nakamura T (2003) Neutralization of hepatocyte growth factor leads to retarded cutaneous wound healing associated with decreased neovascularization and granulation tissue formation. J Invest Dermatol 120:335–343
You S, Avidan O, Tariq A, Ahluwalia I, Stark PC, Kublin CL, Zoukhri D (2012) Role of epithelial-mesenchymal transition in repair of the lacrimal gland after experimentally induced injury. Invest Ophthalmol Vis Sci 53:126–135
Zeisberg M, Bonner G, Maeshima Y, Colorado P, Muller GA, Strutz F, Kalluri R (2001) Renal fibrosis: collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am J Pathol 159:1313–1321
Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R (2003) BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9:964–968
Zhang YM, Zhang ZQ, Liu YY, Zhou X, Shi XH, Jiang Q, Fan DL, Cao C (2015) Requirement of Galphai1/3-Gab1 signaling complex for keratinocyte growth factor-induced PI3K-AKT-mTORC1 activation. J Invest Dermatol 135:181–191
Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC (2004) Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 6:931–940
Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI (2009) Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol 297:L1120–L1130
Zolak JS, Jagirdar R, Surolia R, Karki S, Oliva O, Hock T, Guroji P, Ding Q, Liu RM, Bolisetty S, Agarwal A, Thannickal VJ, Antony VB (2013) Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am J Pathol 182:1239–1247
Author information
Authors and Affiliations
Corresponding author
Additional information
Our research is supported by the National Institutes of Health grants NR015649, NR013881, and DK098055.
Rights and permissions
About this article

Cite this article
Stone, R.C., Pastar, I., Ojeh, N. et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res 365, 495–506 (2016). https://doi.org/10.1007/s00441-016-2464-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-016-2464-0